DOI:
10.1039/C9RA08142A
(Paper)
RSC Adv., 2019,
9, 39924-39931
Ultrasound-induced reactive oxygen species generation and mitochondria-specific damage by sonodynamic agent/metal ion-doped mesoporous silica†
Received
8th October 2019
, Accepted 15th November 2019
First published on 3rd December 2019
Abstract
Designing tumor microenvironment (TME)-specific active nanoparticles with minimum side effects for synergistic cancer therapy has become a hot topic in the recent decades. Aiming at further enhancing the therapeutic efficacy, an in situ-induced mitochondrial dysfunction is a very promising strategy. To achieve these goals, a nano-sono-chemodynamic agent denoted as TPP-Cu@HMS, which integrated hematoporphyrin monomethyl ether (HMME), mPEG-NHS, triphenylphosphonium (TPP)-decorated mesoporous silica (MS) and coordinatively bound Cu2+ ions for mitochondria-specific sonodynamic-chemodynamic therapy (SDT-CDT) of cancer, was designed. Upon the ultrasound (US) treatment, TPP-Cu@HMS can specifically target mitochondria and in situ generate 1O2 against cancer cells. Specifically, to overcome the short lifespan of 1O2, the released Cu2+ ions from TPP-Cu@HMS could act as a Fenton-like agent to convert endogenous H2O2 to ·OH in the acidic environment of cancer cells, disrupt the mitochondrial membrane potential and lead to mitochondrial disintegration, which could systematically enhance the therapeutic efficiency of SDT. Therefore, we highlight the current strategy as a promising prospect for cancer therapy.
Introduction
Mitochondria as cellular power components play an important part in cell energy metabolism and survival in response to external stresses such as nutrient deprivation, ischemia and genotoxic agents.1–4 Damaged mitochondria can also induce a mitochondrial membrane potential change and subsequently cause mitochondria swelling/disintegration, cytochrome c and pro-apoptotic protein release, and eventually induce cell apoptosis/death.5,6 To control mitochondria-specific damage, stimuli-responsive probes and smart nano-carrier systems have been designed recently, which are sensitive to endogenous (pH, redox chemistry and enzymes) and exogenous stimuli (NIR lasers and magnetic field).7–13 Among the various stimuli, ultrasound (mechanical waves with a high penetrative power) as a new exogenous stimulus not only penetrates the depth of tumor tissues but also minimizes the side effects on the surrounding normal tissues.14 Moreover, ultrasound sonodynamic therapy (SDT) as a promising non-invasive approach results in apoptosis in a variety of cancer cells including lymphocytes, myeloid leukemia and sarcoma cells through the activation of SDT agents to generate reactive oxygen species (ROS), cavitation, gas bubbles and hyperthermia. Especially in mitochondria-specific damage, ROS (1O2, ·OH, etc.) can lead to cell apoptosis/death efficiently.15 Nevertheless, the heterogeneity of TME can set several biological barriers for SDT, such as lower pH and hypoxia conditions, which are attributed to the disorganized vasculature inside tumors and the anaerobic respiration of tumor cells,16–18 increasing the risk of tumor relapse after SDT.
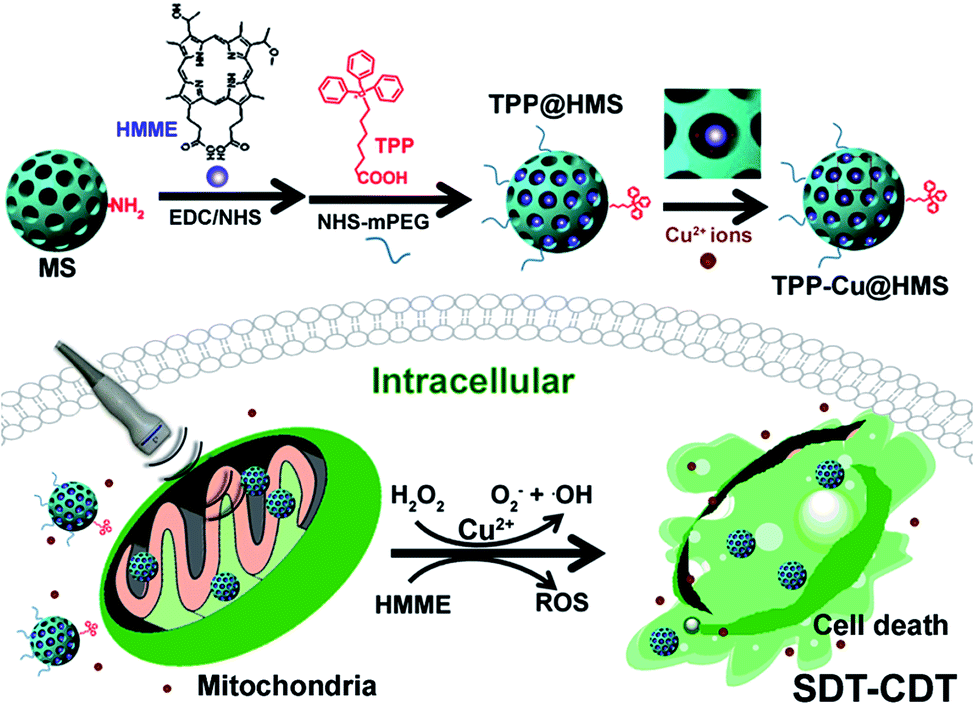
Recently, Fenton-like agent (such as Cu2+, Mn2+, and Fe2+)-based chemodynamic therapy (CDT) has been demonstrated for cancer therapy via the generation of ·OH from endogenous H2O2 in the most important physiological buffers to effectively induce DNA, protein and lipid damage, subsequently leading to cell apoptosis.18–21 Noteworthily, CDT is an oxygen-independent therapy that can overcome the hypoxia in TME and be easily combined with other therapeutic approaches such as PTT for enhanced cancer therapy according to our previous works.19,21 In this study, we introduced a new strategy combining SDT and CDT for the mitochondria-specific therapy of cancer cells (Scheme 1). To achieve this goal, we used the FDA-approved SDT agent hematoporphyrin monomethyl ether (HMME) to covalently conjugate with triphenylphosphonium (TPP)-modified mesoporous silica as the SDT nanoagent (TPP@HMS) and subsequently coordinated it with Cu2+ (denoted as TPP-Cu@HMS) for the in situ generation of 1O2 and ·OH in mitochondria upon H2O2/US for SDT-CDT.
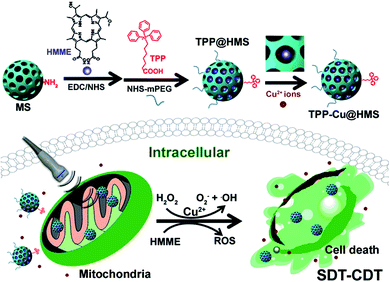 |
| | Scheme 1 Mitochondria-specific TPP-Cu@HMS with ROS burst for SDT-CDT synergy therapy. TPP-Cu@HMS consists of TPP-COOH, Cu(II), HMME and MS. TPP-Cu@HMS can specifically target mitochondria and in situ generate 1O2 and ·OH upon exposure to ultrasound and H2O2 conditions for enhanced cancer therapy. | |
Experimental
Materials
Mesoporous silica (MS) with an average diameter of 71 ± 2.1 nm and pore diameter of 13.3 nm, triphenylphosphonium (TPP-COOH) and succinimidyl propionic acid PEG (NHS-mPEG) were purchased from Xi'an Ruixi Company. N-Hydroxysuccinimide (NHS), hematoporphyrin monomethyl ether (HMME), N-(3-dimethylamino-propyl)-N-ethylcarbodiimide hydrochloride (EDC) and CuCl2 were obtained from J&K Scientific. The Cell Counting Kit (CCK-8) and LIVE/DEAD Viability/Cytotoxicity Kit were obtained from Dojindo China Co., Ltd. 3-Aminopropyltrimethoxysilane (APTES) and 2,7-dichlorodihydrofluoresceindiacetate (DCFH-DA) were purchased from Sigma-Aldrich. Deionized water with a resistivity of 18.2 MΩ cm was obtained from a Milli-Q Gradient System (Millipore, Bedford, MA, U.S.).
Cell culture
The liver cancer cells line (HepG2 cells) was obtained from ATCC (Manassas, VA). The cells were cultured in RPMI 1640 medium (contained 10% FBS) and penicillin–streptomycin (100 IU mL−1, Cellgro, Manassas, VA) at 37 °C in a humidified incubator containing 5% CO2.
Synthesis of the TPP-Cu@HMS
Initially, 10 mg of MS in ethanol was mixed with APTES (50 μL) under reflux for 24 h. Afterwards, MS-NH2 was centrifuged and 1 mg of HMME and 2 mg of TPP-COOH were added to the above mixture solution containing EDC/NHS (10 mg mL−1) for 24 h. Then, the resulting products TPP@HMS was centrifuged and washed 3 times with ethanol. Afterwards, TPP@HMS was mixed with NHS-mPEG for 24 h, and purified by centrifugation, and washed 3 times with DI water. The dose of HMME in TPP@HMS was calculated by subtracting the absorbance at 400 nm of HMME from its standard curve for concentrations ranging from 1–14 μg mL−1 (y = 0.141x + 0.118, R2 = 0.996). After that, the TPP@HMS solution was mixed with CuCl2 (pH 7.4, 10 mg mL−1) and stirred for 12 h. The final product of TPP-Cu@HMS was obtained by centrifugation, and it was stored at 4 °C for further use.
Characterization of TPP-Cu@HMS
Scanning transmission electron microscopy (STEM) with high-angle annular dark-field and transmission electron microscopy (TEM) were conducted on a JEM-2010 electron microscope (JEOL, Japan). UV-vis spectra of TPP@HMS were investigated using a Spectro Max M5e (Germany). Fluorescence spectra were measured using an Agilent Cary Eclipse fluorescence spectrophotometer (Santa Clara, USA).
The intracellular location of TPP@HMS
To evaluate the subcellular location of TPP@HMS, HepG2 cells were first seeded in 35 mm glass bottom Petri dishes with 5 × 104 cells at 37 °C in a 5% CO2 atmosphere for 24 h incubation. Then, HepG2 cells containing TPP@HMS were co-incubated for 6 h, and washed with PBS solution (pH 7.4) to remove the free TPP@HMS. Afterwards, the treated cells were co-stained with MitoTracker Red (500 nM) for 15 min and washed with PBS buffer solution. Subsequently, the cells were imaged via CLSM at 405 nm for HMME (TPP@HMS) and 561 nm for MitoTracker Red. Moreover, to investigate the morphology and mitochondrial membrane potential, the cells were then treated with or without US (0.75 W cm−2, 1 WHz) for 3 min and then treated with the JC-1 dye detector kit according to the commercial protocol, and real-time imaged by CLSM.
The intracellular ROS generation and cytotoxicity of TPP-Cu@HMS
The ROS generation by TPP-Cu@HMS with or without US treatment was detected using DCFH-DA as the ROS indicator. Firstly, HepG2 cells were seeded in a 96-well plate with a density of 1 × 105 per well and co-incubated a humid atmosphere containing 5% CO2 for 24 h. Then, the above medium was refreshed with a new culture including TPP-Cu@HMS and co-incubated for 6 h. Next, the cancer cells were treated with ROS detector (100 μM, DCFH-DA) for 30 min. Subsequently, the cells were washed with PBS buffer and then treated with or without US (0.75 W cm−2, 1 WHz) for 3 min, and imaged by CLSM. To evaluate the SDT killing effects of TPP-Cu@HMS, HepG2 cells were seeded into 35 mm glass bottom Petri dishes at a density of 5 × 104 cells at 37 °C in a 5% CO2 atmosphere for 24 h. Then, the HepG2 cells were co-incubated with TPP@HMS for 6 h, then washed with PBS buffer (pH 7.4) to remove the free TPP@HMS, and finally treated with 1 WHz US (0.75 W cm−2, 1 WHz) for 3 min. The treated cells were stained by propidium iodide (PI) and imaged using a fluorescence microscope. Next, to quantitative measure the cytotoxicity of TPP@HMS upon US treatment, the CCK8 assay was performed according to our previously published protocols.18–21
Statistical analysis
Statistical analysis of CCK8 data was investigated through the two-tailed paired Student's t-test or one-way of variance (ANOVA) protocol, where *p < 0.05, **p < 0.01, ***p < 0.001. The data is presented as the mean ± SD from at least three experiments.
Results and discussion
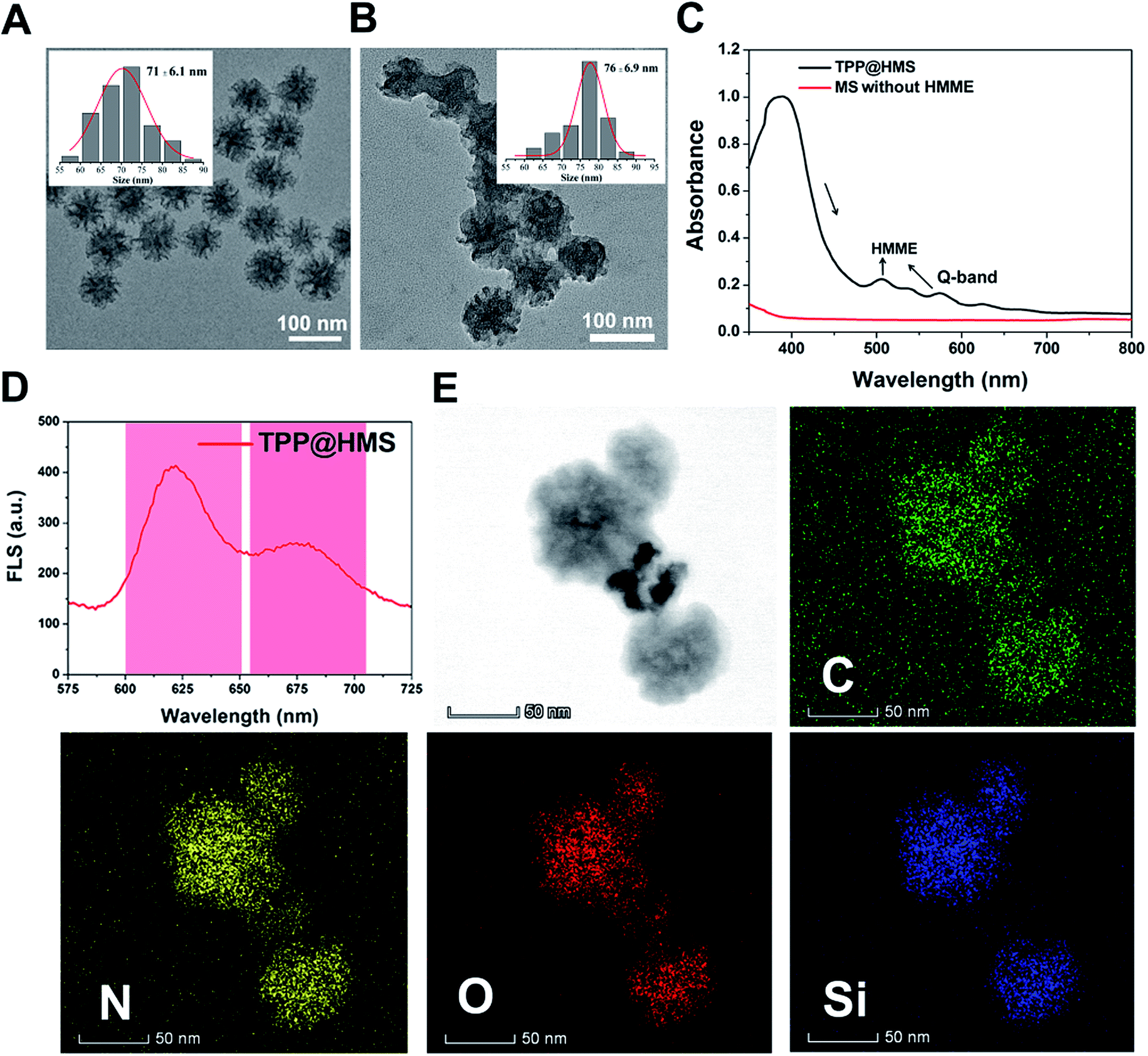
Hemoporfin® is a commonly used PDT agent due to its satisfactory optical properties and ROS quantum yield (approximately 0.6).21 However, the metabolism of hydrophilic Hemoporfin® in clinic is very fast in the body, and thus it always suffers from a short retention time in tumors.21,22 In this study, we chose the hydrophobic HMME to covalently conjugate with MS as a new SDT nanoagent and decorated mPEG for enhancement in its circulation half-time. Firstly, the obtained MS with an average diameter of 71 ± 2.1 nm and pore diameter of 13.3 nm was modified with 3-aminopropyltrimethoxysilane (APTES) to gain the amino (–NH2) group on the surface of MS. The morphology of MS-NH2 was evaluated via transmission electron microscopy (TEM). As shown in Fig. 1A and the inset picture, MS-NH2 showed good monodispersity and a spherical shape, with an average diameter of 71 ± 6.1 nm. After modification with APTES, HMME was then covalently conjugated to MS-NH2 via the amido bond to form a new SDT agent (named HMS). Afterwards, the mitochondria-specific ligand of TPP-COOH and NHS-mPEG were conjugated with the surface of HMS through amido bonds (refer to TPP@HMS). The TEM image of TPP@HMS showed the typical spherical shape and the average diameter of 76 ± 6.9 nm (Fig. 1B). The UV-vis-NIR spectrum obtained on a Spectro-Max M5e was further investigated to confirm the components of the final product. As shown in Fig. 1C, four distinct peaks in the wavelength range of 450–750 nm at 506 nm, 544 nm, 576 nm and 622 nm (Q-band) were observed clearly compared to MS-NH2 without HMME conjugation, and the results are consistent with the typical absorption peaks of HMME.22 The emission spectrum of TPP@HMS demonstrates that its fluorescence emission is long in the NIR range (600 to 700 nm) (excited by 391 nm). To further confirm its components, STEM with high-angle annular dark-field was performed. The final data suggests the presence of N, O, C and Si homogenously distributed throughout TPP@HMS (Fig. 1E).
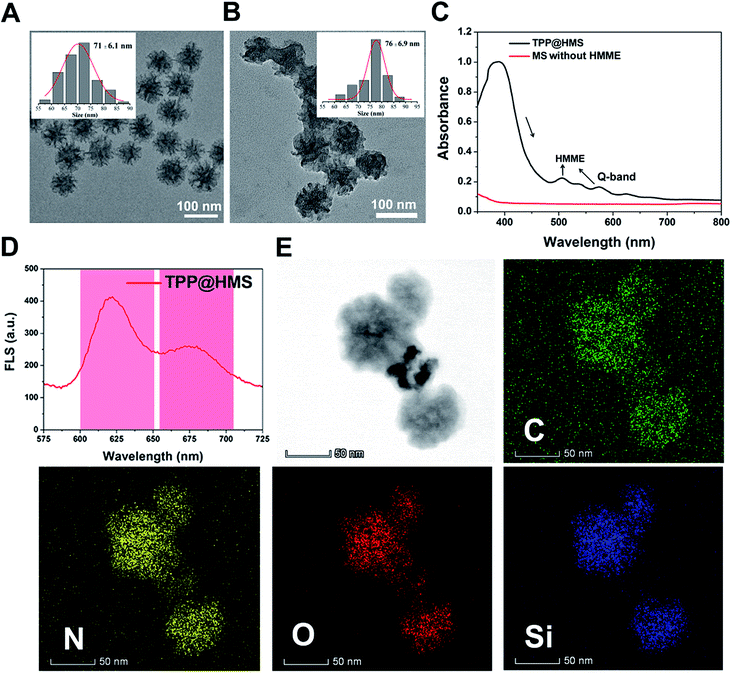 |
| | Fig. 1 (A) TEM image of MS-NH2 and (B) TPP@HMS, and the inset image is the size distribution of MS-NH2 and TPP@HMS, respectively. (C) UV-vis-NIR spectra of TPP@HMS and MS-NH2. (D) Fluorescence spectrum of TPP@HMS (ex 391 nm). (E) STEM elemental mapping of C, N, O and Si edge signals of TPP@HMS. | |
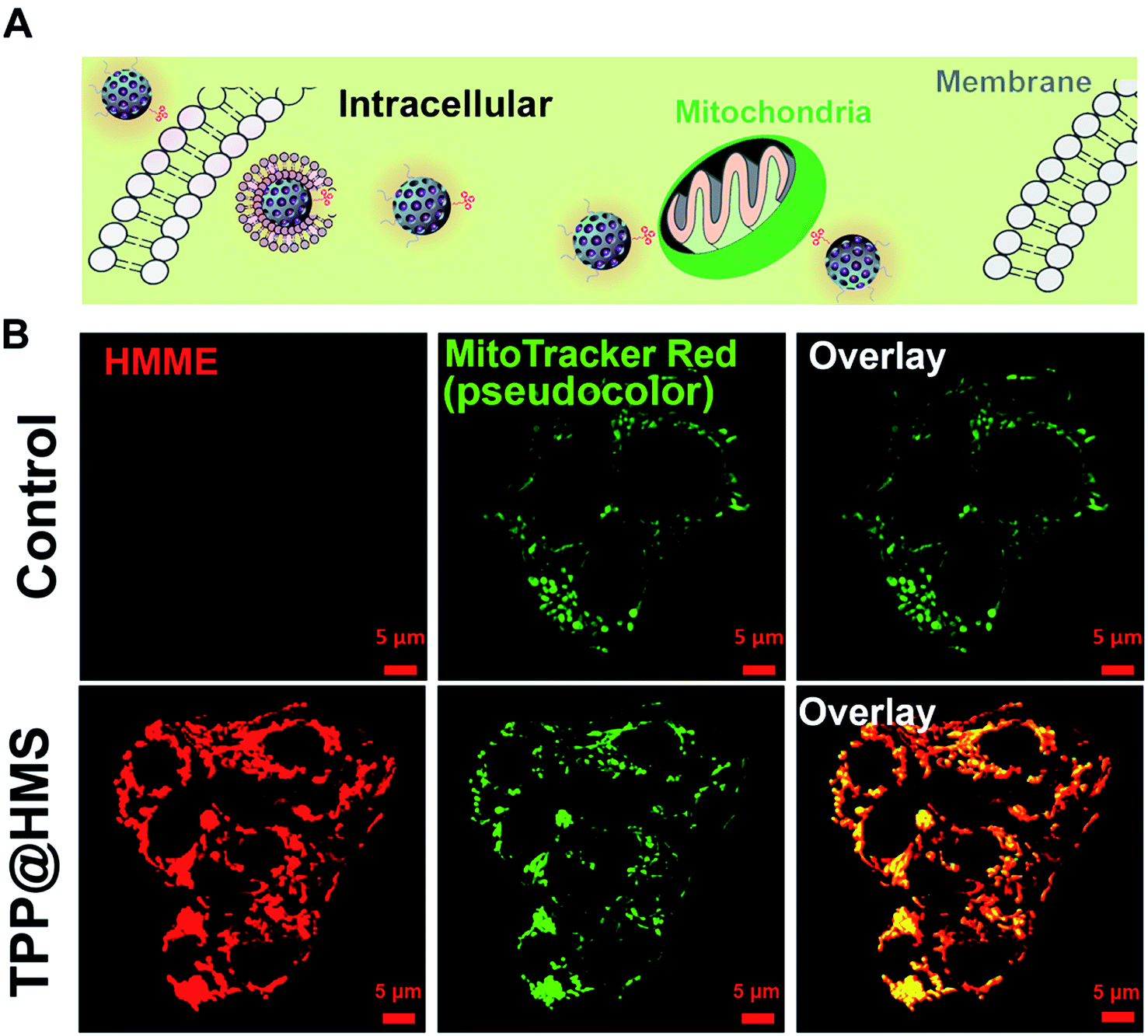
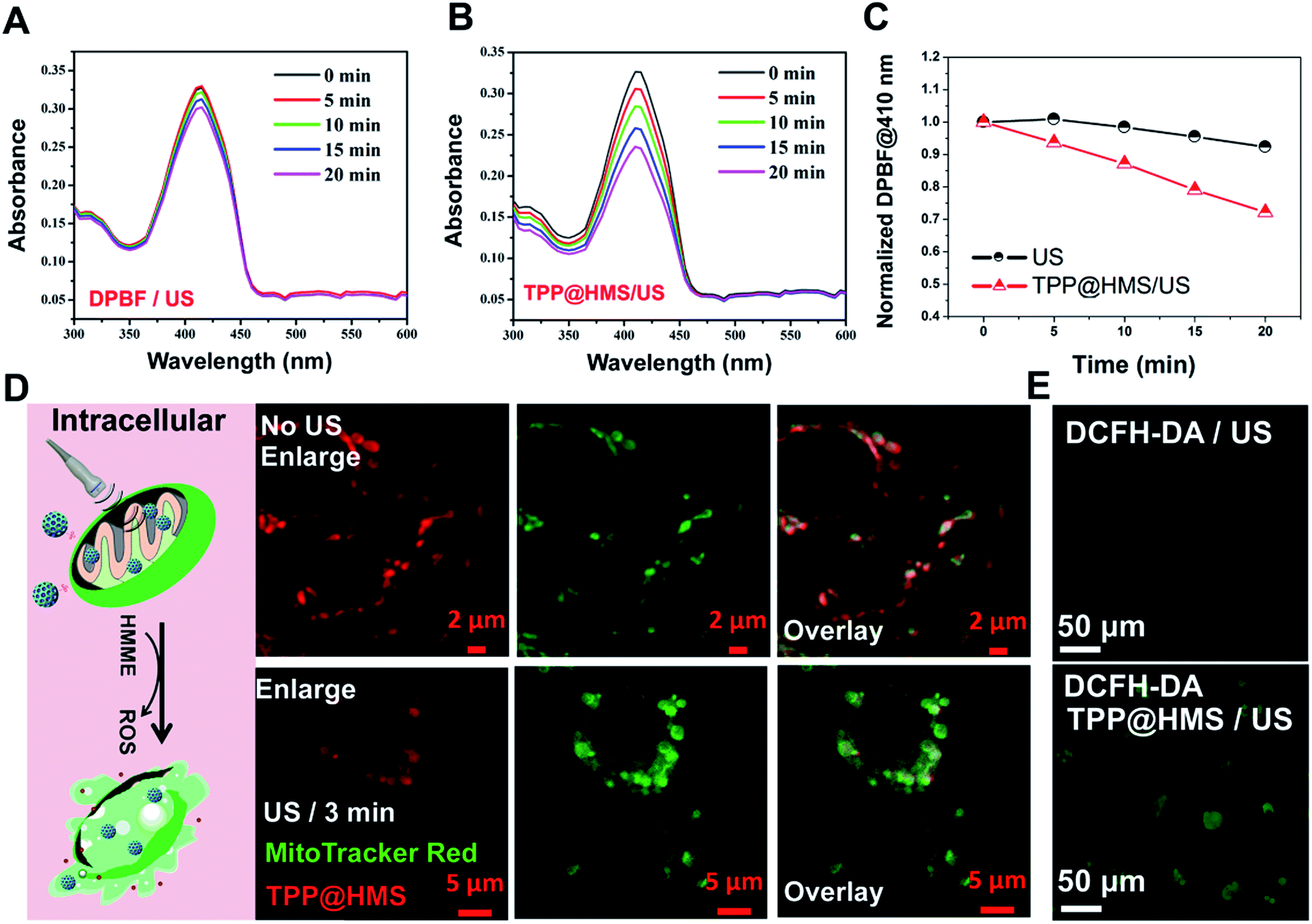
Mitochondria-specific damage of cancer cells is an effective strategy with remarkable therapeutic potency in cancer therapy.1–4 To investigate the mitochondrial targeting ability of HMS with decoration of TPP-COOH, HepG2 cells as a typical cancer cell model were chosen. As shown in Fig. 2A and B, the confocal laser scanning microscopy (CLSM) images showed good overlap between TPP@HMS with red fluorescence and MitoTracker Red with green fluorescence (pseudo-color) after co-incubation for 6 h, and Pearson's correlation coefficient (PCCs) was calculated to be 0.86 using the ImageJ software. PBS-treated HepG2 cells stained with MitoTracker Red were as a control. To achieve excellent therapeutic efficiency for the treatment of cancer, in situ mitochondria ROS generation is a very promising approach.5,6 Inspired by this, we checked the 1O2 generation ability of our prepared TPP@HMS. The 1O2 indicator 1,3-diphenylisobenzofuran (DPBF) was used to measure 1O2 upon 1 WHz US (1 W cm−2).24 The change in optical density was investigated by monitoring the US-vis spectrum. As shown in Fig. 3A and B, the absorbance of the DPBF solution containing TPP@HMS ranging from 350 to 500 nm significantly decreased after US, and the decrease was time dependent, suggesting the excellent 1O2 generation ability of TPP@HMS. In contrast, the DPBF solution without TPP@HMS did not obviously change under the same US power condition (Fig. 3B and C). Moreover, the mitochondrial morphology of the cancer cells was also examined by CLSM after TPP@HMS treatment upon US (1 WHz, 0.75 W cm−2) for 3 min. As shown in Fig. 3D, obvious mitochondrial swelling/disintegration was detected in the TPP@HMS-treated HepG2 cells after US treatment compared to the control. Furthermore, the intracellular ROS were investigated using the green fluorescence indicator 2,7-dichlorodihydrofluorescein diacetate (DCFH-DA) (Fig. 3E).20–22 Green fluorescence with a much higher fluorescence intensity was obviously detected in TPP@HMS-treated HepG2 cells after US treatment compared to the PBS-treated cells. These results suggested that our prepared TPP@HMS could act as an active mitochondria-specific SDT nanoagent for further treatment of cancer cells.
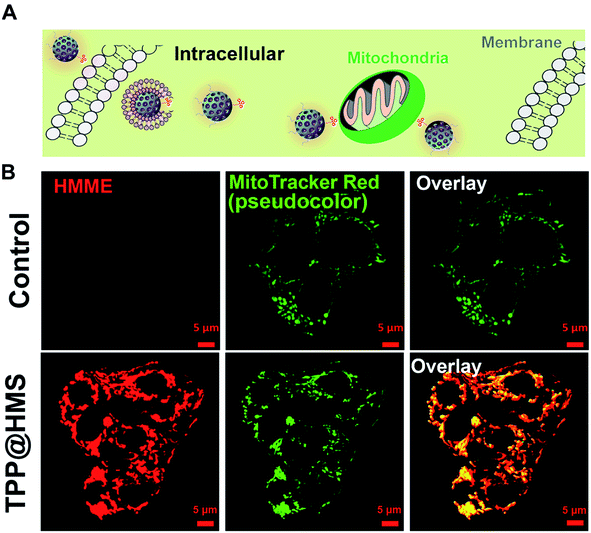 |
| | Fig. 2 (A) Schematic illustration of HepG2 cells with TPP@HMS. (B) Intracellular distribution of TPP@HMS in HepG2 cells after co-incubation for 6 h, and imaged by CLSM. The intracellular mitochondria were stained by MitoTracker Red (green, pseudocolor). | |
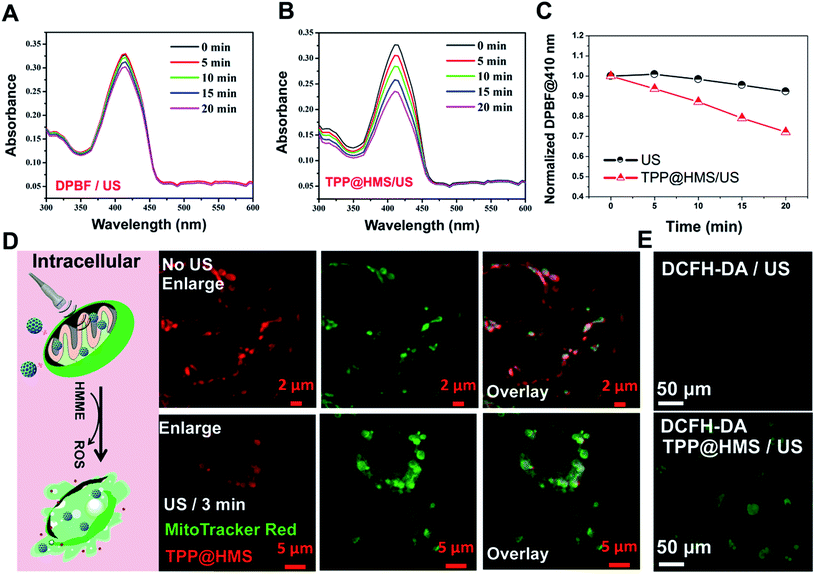 |
| | Fig. 3 Absorbance spectra of (A) DPBF with US (100 μM) and (B) DPBF (100 μM) in TPP@HMS with ROS generation upon US (1 MHz, 0.75 W cm−2) for different intervals, where DPBF with US is the positive control. (C) Normalized absorption coefficient of DPBF at 410 nm based on (A and B). (D) CLSM images of mitochondria morphology after HepG2 cells were treated with TPP@HMS for 6 h with or without US (0.75 W cm−2, 3 min) treatment. (E) CLSM image of intracellular ROS generation by DCFH-DA detector after PBS or TPP@HMS-treated cells were exposed to US (0.75 W cm−2, 3 min) treatment. | |
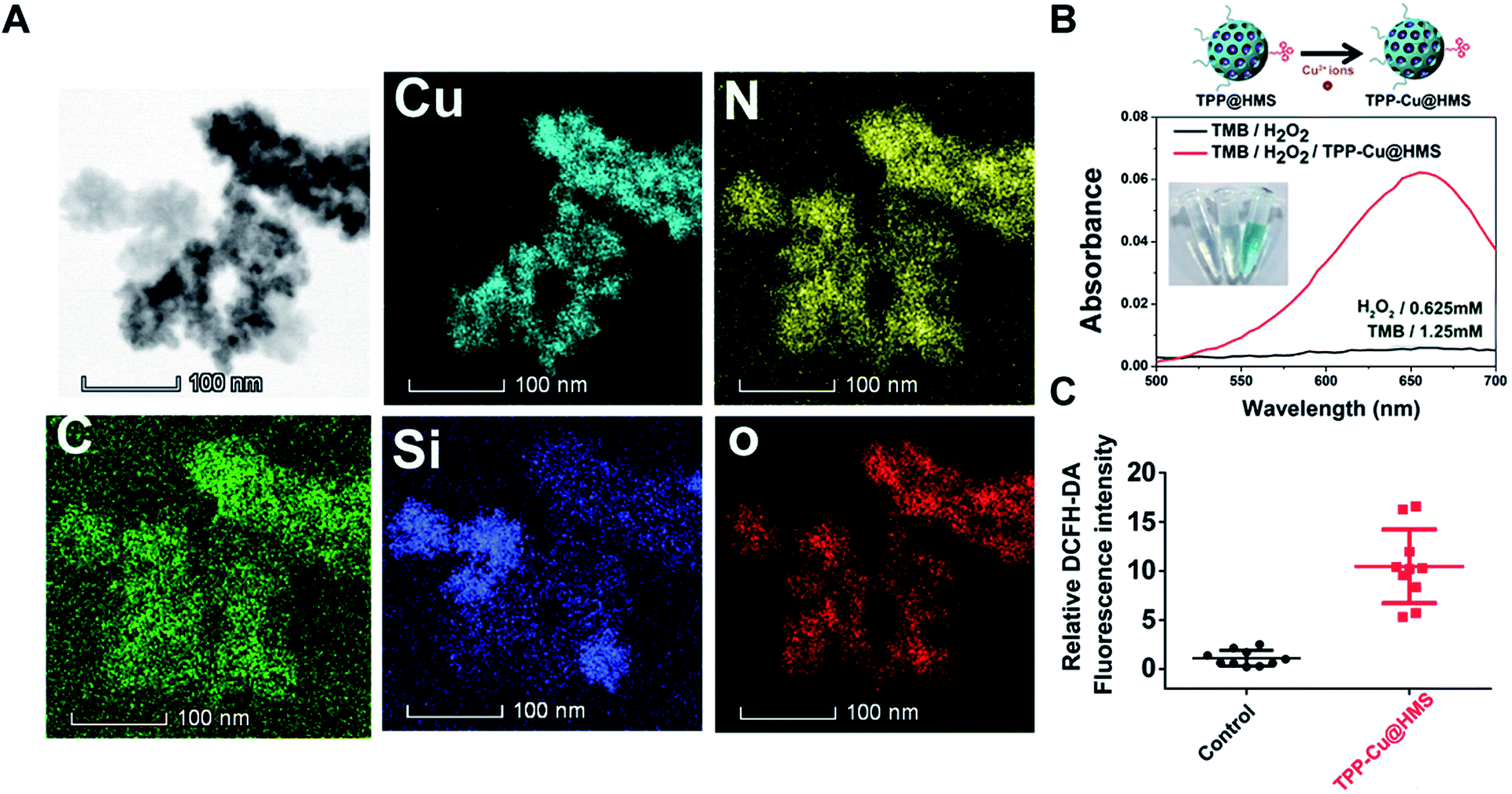
In the TME, hypoxia and acidic pH play an important role in the heterogeneity of tumors.24 The limited oxygen in the TME can diminish the SDT effect, which may cause tumor relapse after SDT treatment. In this study, we combined a Fenton-like agent as an oxygen-independent CDT approach to convert the H2O2 in the TME into ·OH to induce tumor cells apoptosis/necrosis and SDT, overcoming the above limitation for enhanced SDT efficiency. Cu2+ ions as a Fenton-like agent have been widely used in CDT therapy.19,20,25 To enhance the Cu2+ ion accumulation in tumors, the oral administration of Cu2+-containing compounds is a currently available approach that is commonly utilized in copper deficiency in clinic. Unfortunately, the systemic administration of Cu2+ compounds may induce heavy-metal poisoning.26,27 Herein, Cu2+ was coordinated with TPP@HMS via the NH2-medium coordination effect to form TPP-Cu@HMS at neutral pH according to our previously reported work.28 STEM-HAADF was performed to confirm the existence of Cu in TPP-Cu@HMS. Fig. 4A shows the presence of Cu, N, Si, O, and C elements in TPP-Cu@HMS, and Cu was homogenously distributed throughout TPP-Cu@HMS, indicating the existence of Cu2+. Next, the bio-stability of TPP-Cu@HMS in physiological conditions (at pH 7.4) was investigated. The results showed 13.5% of Cu2+ was released from TPP-Cu@HMS after 24 h incubation, indicating its relative stability in physiological conditions (Fig. S1†). Afterwards, we further checked the catalytic performance of TPP-Cu@HMS in TMB (250 μM) aqueous solution in the presence of H2O2. After co-incubation, the absorbance of TMB obviously increased in TPP-Cu@HMS with the presence of H2O2 (0.625 mM), and the color of solution changed to wathet blue due to the Cu2+ ions from TPP-Cu@HMS converting H2O2 to ·OH. In contrast, the TMB solution in the presence of H2O2 without TPP-Cu@HMS did not show any change. Moreover, we checked the ROS generation of TPP-Cu@HMS in cancer cells using the DCFH-DA indicator after co-incubation for 24 h, and the results showed relatively high ROS generation in TPP-Cu@HMS-treated HepG2 cells compared with the control groups. These results are similar to the intracellular ·OH generation from TPP-Cu@HMS without US (Fig. S2†). Thus, the results indicate that TPP-Cu@HMS can act as an activatable SDT-CDT nanoagent for further cancer therapy.
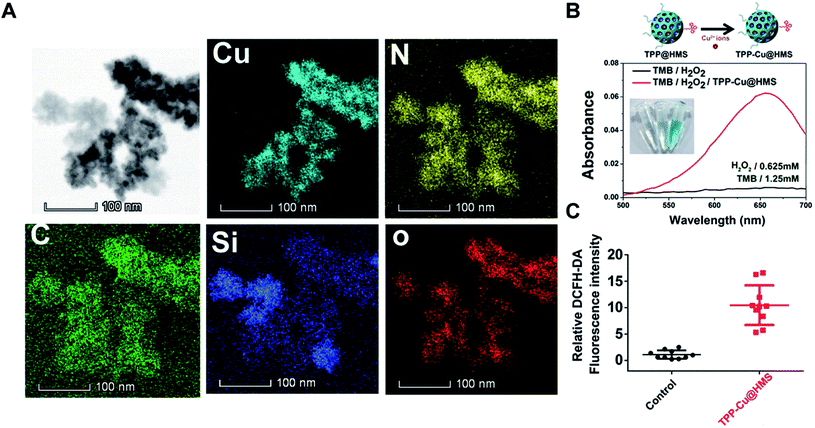 |
| | Fig. 4 (A) TEM elemental mapping of C, N, O, Cu and Si edge signals of TPP-Cu@HMS. (B) UV-vis spectrum and photographs (inset) of TMB aqueous solution treated with H2O2 with or without TPP-Cu@HMS. (C) Relative DCFH-DA fluorescence intensity of HepG2 cells after TPP-Cu@HMS treatment without US. | |
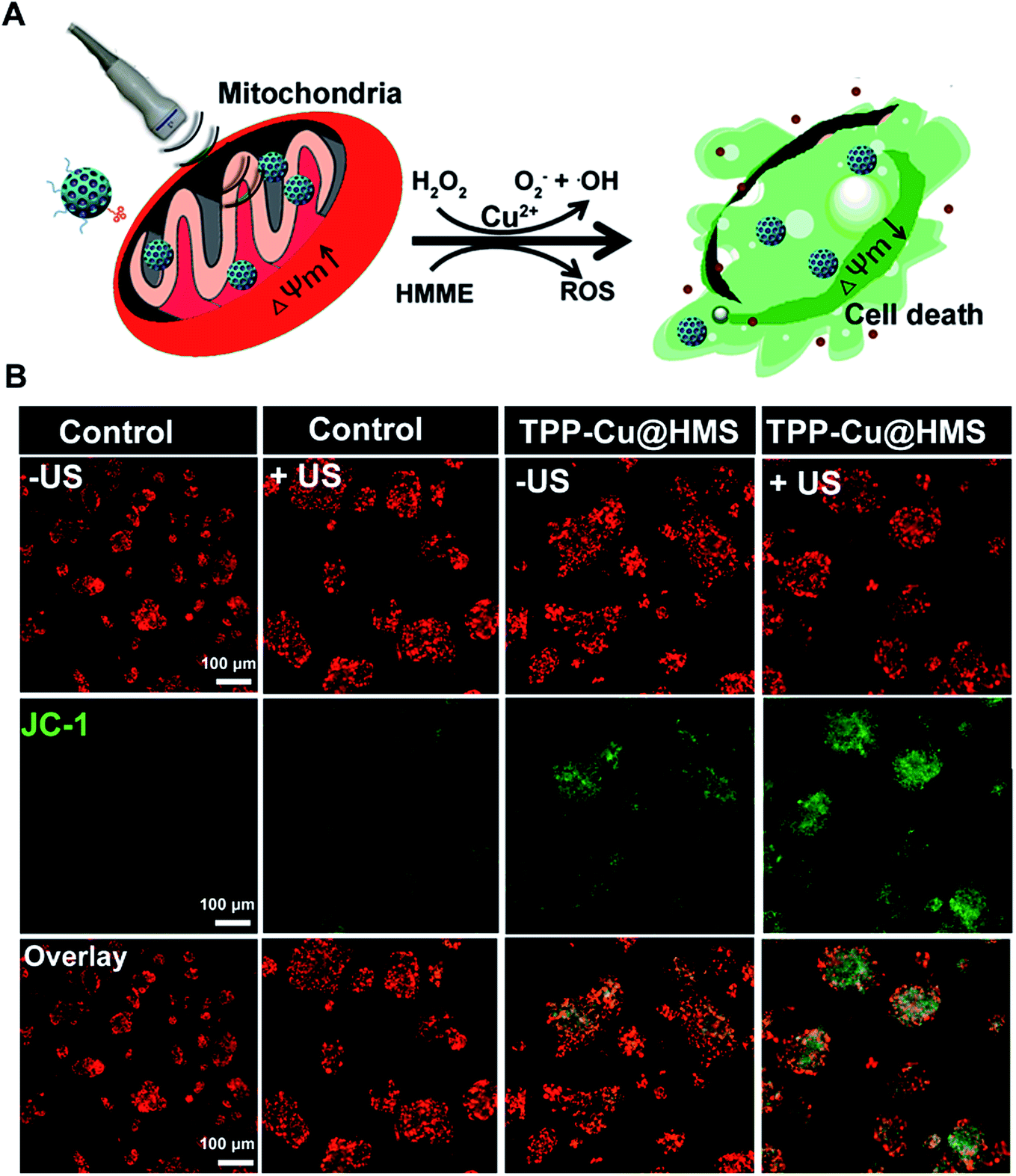
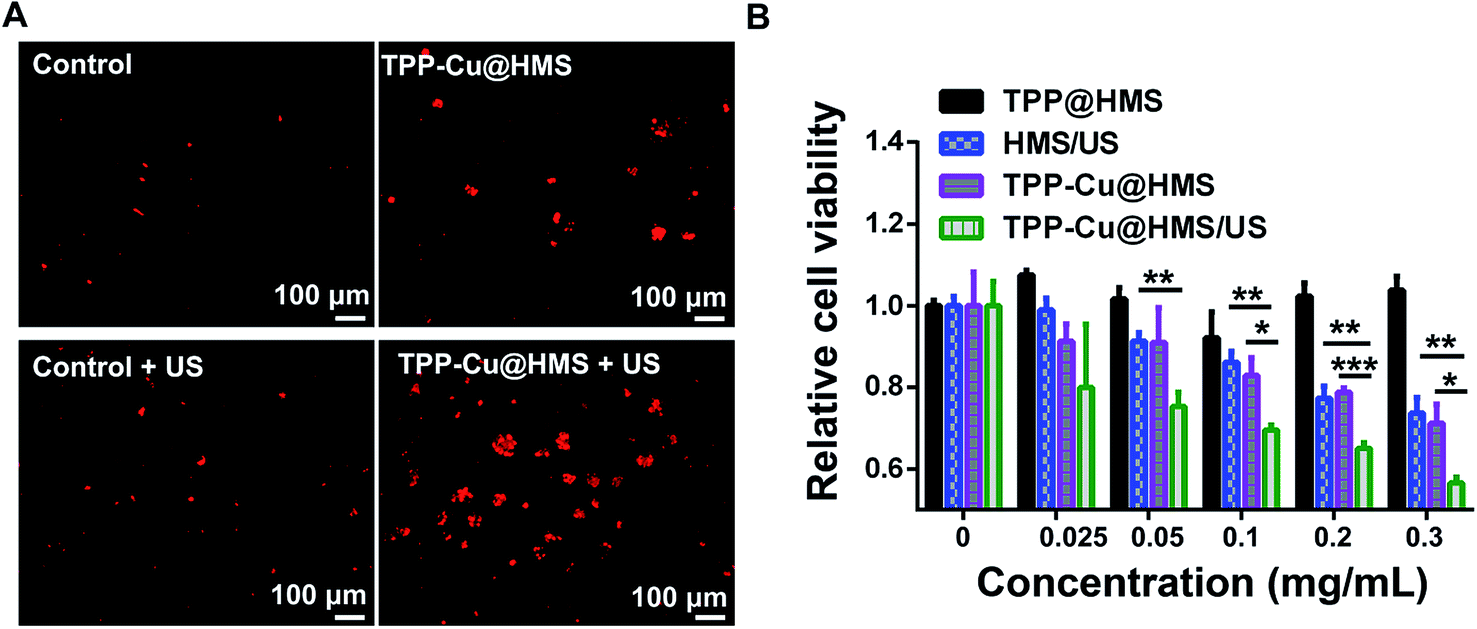
The mitochondrial membrane permeability (MMP) is essential in mitochondria apoptosis and cell death. Encouraged by the above results, we investigated the mitochondrial membrane (Δψm) using the JC-1 dye detection kit (the potential sensitive indicator) and imaging by CLSM. As illustrated in Fig. 5, the JC-1 monomers with green fluorescence were more obviously and increased in the TPP-Cu@HMS-treated HepG2 cells after 1 WHz US (0.75 W cm−2) for 3 min. The ratio of JC-1 aggregates with red fluorescence/JC-1 monomers with green fluorescence was reduced, indicating the high in situ ROS (1O2 and ·OH) generation and accurate photodamage on mitochondria. However, the JC-1 monomer fluorescence intensity was low in the TPP-Cu@HMS-treated HepG2 cells alone. In contrast, the HepG2 cells without any treatment showed overwhelming red fluorescence. Noteworthily, green fluorescence was observed in the US-treated HepG2 cells in the absence of TPP-Cu@HMS mainly due to the cavitation effect of ultrasound.22,23 Next, the synergistic therapeutic efficiency of TPP-Cu@HMS was investigated by PI dead staining and analyzed through fluorescence microscopy. A significant amount of dead cells with red fluorescence was obviously detected in the TPP-Cu@HMS-treated HepG2 cells after received 1 WHz US (0.75 W cm−2) for 3 min compared to the TPP-Cu@HMS-treated cells alone and US alone (Fig. 6A). Meanwhile, to quantify the TPP-Cu@HMS cytotoxicity after US treatment, Cell Counting Kit-8 assay (CCK-8) was performed. As illustrated in Fig. 6B, the cell viability of the TPP@HMS-treated cancer cells alone showed no obvious cytotoxicity after 24 h incubation, and the mortality rate was very low even with the concentration of TPP@HMS up to 0.3 mg mL−1, indicating that our prepared TPP@HMS has good biocompatibility. In contrast, the viable cells of TPP-Cu@HMS-treated HepG2 cells upon 1 WHz US (0.75 W cm−2) for 3 min significantly decreased to 56.6%, suggesting the synergistic treatment of TPP-Cu@HMS compared to TPP-Cu@HMS-treated HepG2 cells alone without US treatment and HMS-treated HepG2 cells with US treatment. Overall, the results highlight the potential of our prepared TPP-Cu@HMS as a powerful therapeutic agent for enhanced mitochondria-specific cancer therapy.
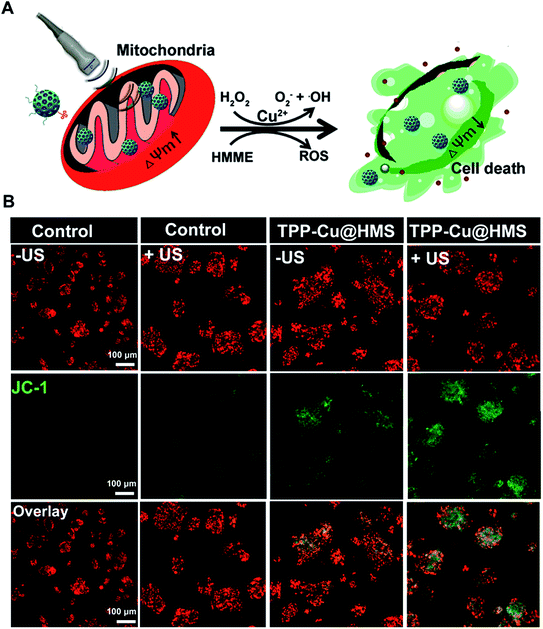 |
| | Fig. 5 (A) Schematic illustration of the mitochondrial membrane potential change (ΔΨm) after US. (B) CLSM images of JC-1-stained (green channel), and TPP-Cu@HMS (red channel)-treated HepG2 cells under US treatment. | |
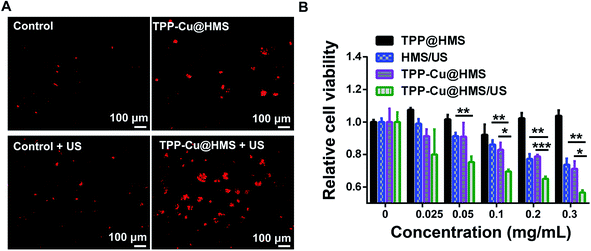 |
| | Fig. 6 (A) Localized photo-killing effects of TPP@HMS or TPP-Cu@HMS with or without US (1 MHz, 0.75 W cm−2) on HepG2 cells. (B) Cell viability of the HMS, TPP@HMS or TPP-Cu@HMS treated HepG2 cells with or without US (1 MHz, 0.75 W cm−2) for 3 min, where the cell viability was determined using the CCK-8 assay. The statistical analysis was performed with the two-tailed paired Student's t-test, where *p < 0.05, **p < 0.01, ***p < 0.001 (n = 4). | |
Conclusion
In summary, we introduced a promising therapeutic agent and integration of SDT and CDT to in situ generate ROS (1O2 and ·OH) for mitochondria-specific cancer therapy. TPP-Cu@HMS was easily obtained and endowed with mitochondria-specific targeting ability by decoration with TPP-COOH ligands. After US treatment, TPP-Cu@HMS generated a high amount of 1O2 and ·OH in the mitochondria and effectively induced mitochondrial disintegration and damage. According to the in vitro therapeutic data, TPP-Cu@HMS effectively induced apoptosis in cancer cells compared to the individual treatments. Therefore, we highlight the current strategy as a promising approach for enhanced cancer therapy.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
Kecan Lin and Ziguo Lin contributed equally to this work. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. This work was supported by the Natural Science Foundation of China (Grant No. 61775037, 61805041). Bethune Aishikun Surgical Excellence Fund and Joint Funds for the Innovation of Science and Technology, Fujian province (Grant No. 2018Y9119).
Notes and references
- M. L. Boland, A. H. Chourasia and K. F. Macleod, Front. Oncol., 2013, 3, 292 CrossRef PubMed.
- M. Jeena, L. Palanikumar, E. M. Go, I. Kim, M. G. Kang, S. Lee, S. Park, H. Choi, C. Kim and S.-M. Jin, Nat. Commun., 2017, 8, 26 CrossRef CAS PubMed.
- D. A. Bader, S. M. Hartig, V. Putluri, C. Foley, M. P. Hamilton, E. A. Smith, P. K. Saha, A. Panigrahi, C. Walker and L. Zong, Nature Metabolism, 2019, 1, 70 CrossRef PubMed.
- M. Ott, V. Gogvadze, S. Orrenius and B. Zhivotovsky, Apoptosis, 2007, 12, 913–922 CrossRef CAS PubMed.
- O. M. Koval, E. K. Nguyen, V. Santhana, T. P. Fidler, S. C. Sebag, T. P. Rasmussen, D. J. Mittauer, S. Strack, P. C. Goswami and E. D. Abel, Sci. Signaling, 2019, 12, 1439 CrossRef PubMed.
- Y. Chen, W. Ai, X. Guo, Y. Li, Y. Ma, L. Chen, H. Zhang, T. Wang, X. Zhang and Z. Wang, Small, 2019, 1902352 CrossRef PubMed.
- A. Vadivelmurugan, R. Anbazhagan, V. Arunagiri, J. Y. Lai and H. C. Tsai, RSC Adv., 2019, 9, 25592–25601 RSC.
- Y. Zheng, X. Ji, B. Yu, K. Ji, D. Gallo, E. Csizmadia, M. Zhu, M. R. Choudhury, L. K. C. De La Cruz and V. Chittavong, Nat. Chem., 2018, 10, 787 CrossRef CAS PubMed.
- W. Zhang, X. Hu, Q. Shen and D. Xing, Nat. Commun., 2019, 10, 1704 CrossRef PubMed.
- L. Zhang, H. Yi, J. Song, J. Huang, K. Yang, B. Tan, D. Wang, N. Yang, Z. Wang and X. Li, ACS Appl. Mater. Interfaces, 2019, 11, 9355–9366 CrossRef CAS PubMed.
- F. Zhou, S. Wu, B. Wu, W. R. Chen and D. Xing, Small, 2011, 7, 2727–2735 CrossRef CAS PubMed.
- P. Yuan, X. Mao, X. Wu, S. S. Liew, L. Li and S. Q. Yao, Angew. Chem., 2019, 131, 7739–7743 CrossRef.
- X.-Q. Wang, M. Peng, C.-X. Li, Y. Zhang, M. Zhang, Y. Tang, M.-D. Liu, B.-R. Xie and X.-Z. Zhang, Nano Lett., 2018, 18, 6804–6811 CrossRef CAS PubMed.
- X. Lin, J. Song, X. Chen and H. Yang, Angew. Chem., 2019 DOI:10.1002/ange.201906823.
- S. G. Rhee, Science, 2006, 312, 1882–1883 CrossRef PubMed.
- X. Lin, Y. Qiu, L. Song, S. Chen, X. Chen, G. Huang, J. Song, X. Chen and H. H. Yang, Nanoscale Horiz., 2019, 4, 747–756 RSC.
- K. L. Zhang, Y. J. Wang, J. Sun, J. Zhou, C. Xing, G. Huang, J. Li and H. H. Yang, Chem. Sci., 2019, 10, 1555–1561 RSC.
- D. Zhang, Z. Cai, N. Liao, S. Lan, M. Wu, H. Sun, Z. Wei, J. Li and X. Liu, Chem. Sci., 2018, 9, 7390–7399 RSC.
- D. Zhang, Z. Lin, S. Lan, H. Sun, Y. Zeng and X. Liu, Mater. Chem. Front., 2019, 3, 656–663 RSC.
- L. S. Lin, J. Song, L. Song, K. Ke, Y. Liu, Z. Zhou, Z. Shen, J. Li, Z. Yang, W. Tang, G. Niu, H. H. Yang and X. Chen, Angew. Chem., 2018, 57, 4902–4906 CrossRef CAS PubMed.
- D. Zhang, Y. Zheng, Z. Lin, S. Lan, X. Zhang, A. Zheng, J. Li, G. Liu, H. H. Yang, X. Liu and J. Liu, Small, 2019, 1902636 CrossRef CAS PubMed.
- T. C. Lei, G. F. Glazner, M. Duffy, L. Scherrer, S. Pendyala, B. Li, X. Wang, H. Wang and Z. Huang, Photodiagn. Photodyn. Ther., 2012, 9, 232–242 CrossRef CAS PubMed.
- J. Shi, R. Ma, L. Wang, J. Zhang, R. Liu, L. Li, Y. Liu, L. Hou, X. Yu and J. Gao, Int. J. Nanomed., 2013, 8, 2361 CrossRef PubMed.
- S. Lan, Z. Lin, D. Zhang, Y. Zeng and X. Liu, ACS Appl. Mater. Interfaces, 2019, 11, 9804–9813 CrossRef CAS PubMed.
- L. Lin, T. Huang, J. Song, X. Y. Ou, Z. Wang, H. Deng, R. Tian, Y. Liu, J. F. Wang and Y. Liu, J. Am. Chem. Soc., 2019, 141, 9937–9945 CrossRef CAS PubMed.
- W. Wu, L. Yu, Q. Jiang, M. Huo, H. Lin, L. Wang, Y. Chen and J. Shi, J. Am. Chem. Soc., 2019, 141, 11531–11539 CrossRef CAS PubMed.
- Z. Chen, H. Meng, G. Xing, C. Chen, Y. Zhao, G. Jia, T. Wang, H. Yuan, C. Ye and F. Zhao, Toxicol. Lett., 2006, 163, 109–120 CrossRef CAS PubMed.
- D. Zhang, A. Zheng, J. Li, M. Wu, L. Wu, Z. Wei, N. Liao, X. Zhang, Z. Cai, H. H. Yang, G. Liu, X. Liu and J. Liu, Theranostics, 2017, 7, 164–179 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra08142a |
| ‡ K. Lin and Z. Lin contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence