
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRadical pathways for the formation of non-canonical nucleobases in prebiotic environments†
Sarabjeet Kaur and
Purshotam Sharma*
and
Purshotam Sharma*
Computational Biochemistry Laboratory, Department of Chemistry and Centre for Advanced Studies in Chemistry, Panjab University, Chandigarh, 160014, India. E-mail: psharma@pu.ac.in
First published on 11th November 2019
Abstract
Due to the inability of canonical nucleobases (adenine, uracil, guanine and cytosine) to spontaneously form ribonucleosides and base pairs in free form in solution, RNA is believed to be preceded by a primitive information polymer (preRNA). The preRNA is proposed to contain non-canonical, heterocyclic bases that possess the above-mentioned capabilities. An extensive search for such candidate heterocycles has recently revealed that barbituric acid (BA), melamine (MM) and 2,4,6-triaminopyrimidine (TAP) have the capability to spontaneously form ribonucleosides and supramolecular assemblies that are held by Watson–Crick type hydrogen-bonded base pairs involving BA, MM, TAP and cyanuric acid (CA) heterocycles. However, despite this evidence, the prebiotic formation pathways of these heterocycles have not been fully explored. Further, for these heterocycles to interact and assemble into informational polymers under prebiotic conditions, it is expected that they should have formed in the proximity of each other. In this context, the present work employs density functional theory to propose the associated radical based formation pathways starting from cyanamide. Our pathways suggest that cyanamide, its derivatives (malonic acid and urea) and malononitrile can form BA, MM, CA and TAP in the presence of ammonia and hydroxyl radicals. In addition to originating from a common precursor, similarities in the highest reaction barriers (13 to 20 kcal mol−1) obtained for these pathways suggest that these heterocycles may likely form under similar conditions. Specifically, these pathways are relevant to high energy events such as meteoritic impact during the late heavy bombardment period on the early earth, which would have created conditions where radicals might have formed in reasonable concentrations. Overall, the present study emphasizes the importance of cyanamide in prebiotic heterocycle formation.
Introduction
Owing to their ability to spontaneously form ribonucleosides and hydrogen-bonded base pairs (similar to canonical base pairs in RNA helices) in free form, it has been suggested that alternate heterocycles (i.e. urazole and cytazole) could have preceded canonical nucleobases.1 This idea subsequently developed into a theory, which posits that the extant RNA was preceded by a primitive RNA (i.e. pre-RNA) that involved alternate heterocycles (prebiotic nucleobases) rather than canonical nucleobases. Subsequent investigations on a large (around 81) number of candidate heterocycles revealed that barbituric acid (BA), melamine (MM), cyanuric acid (CA) and 2,4,6-triaminopyrimidine (TAP) are four such promising prebiotic nucleobase candidates2 (Scheme 1). Specifically, BA, MM and TAP spontaneously react with ribose or ribose 5′-monophosphate (rMP) to form corresponding nucleosides or nucleotides.3,4 and further form BA:MM3 and BA:TAP5 base pairs analogous to Watson–Crick (WC)-type pairs (A:U and G:C) in water. Furthermore, BA, MM and TAP have the ability to form supramolecular assemblies that include BA:MM,3 BA:TAP,5 CA:MM6,7 and CA:TAP4,8 pairs as constituent motifs. | ||
| Scheme 1 Outline of the formation pathways of TAP, MM, CA and BA heterocycles. | ||
To understand the origin of first informational polymer (i.e. preRNA) on earth, it is important to analyze the feasibility of formation of its primary constituent components, i.e. prebiotic nucleobases. In this context, incessant input of UV and cosmic radiation on the early earth,9 coupled with the high-energy impact of extraterrestrial bodies during the late heavy bombardment period,10 points towards prebiotic conditions where free radicals would have played an important role in facilitating reactions leading to the formation of such heterocycles. Indeed, simulations of the late heavy bombardment period of early earth involving a laser-induced dielectric breakdown of formamide produced radicals such as ˙CN and ˙NH, which may have played an important role in the prebiotic formation of canonical nucleobases.11 Further, canonical nucleobases were synthesized in high-energy (4500 K) electric discharge and laser-driven plasma impact simulations of a reducing atmosphere containing NH3 and CO, where the involvement of ˙CN was suggested.12 Additional studies have suggested the role of radical pathways in bringing about prebiotic transformations, in the presence of urea ice13 or radiation,14,15 particularly during the late heavy bombardment.11,16 Furthermore, based upon the hypothesis that organic compounds in meteorites were formed in the solar nebula through Fischer–Tropsch-type reactions, MM and CA were synthesized from CO, NH3 and H2 in the presence of powdered meteorite.17,18 In a subsequent study, urea was subjected to freeze–thaw cycles, thereby creating microenvironments in the ice that provided appropriate conditions for the prebiotic formation of CA, MM and BA.19 Subsequent theoretical investigations suggest the involvement of radicals in such prebiotic reactions.20,21
Given the relevance of BA, MM, CA and TAP in context of the formation of prebiotic supramolecular assemblies,3–8 it is reasonable to hypothesize that these heterocycles might have formed and then coexisted in a common prebiotic environment, which would have ultimately led to their assembly presumably by non-covalent interactions. Indeed, such a scenario can be considered as a preliminary step for the formation of subsequent more-evolved, covalently-linked prebiotic polymers similar to extant nucleic acids. To evaluate the hypothesis of coexistence of these prebiotic heterocycles through their formation from a common precursor under similar conditions, the present work attempts to explore unified pathways for their formation in an early earth scenario using density functional theory (DFT) based quantum chemical calculations. Specifically, we propose and evaluate radical mechanisms for the plausible formation of BA, CA, MM and TAP using cyanamide (Scheme 1), which is a prebiotic condensing agent,22,23 and a suggested precursor for natural nucleotides.24 The proposed pathways include prebiotic heterocycle formation through reaction of cyanamide, urea (prebiotically derived from cyanamide25) or malonic acid (that can, in turn, be obtained from urea14) with each other or with other prebiotically-relevant species such as malononitrile,26,27 ˙NH2 (ref. 11, 12 and 28) ˙OH,13,20,29,30 and NH3 (ref. 30 and 31) through the involvement of the catalytic ˙NH2/NH3 pair.20,29,30 We believe that our proposed pathways will provide fundamental insights about underlying mechanisms of formation of non-canonical nucleobases (components of preRNA) and hopefully inspire future experimental studies in this direction.
Computational details
Gas-phase geometry optimizations and zero-point vibrational energy (ZPVE)-corrected electronic energy calculations of the stationery points (i.e. reactants, transition states, intermediates or products) along the reaction coordinates associated with the formation of BA, CA, MM or TAP were carried out using B3LYP/6-311G(d,p). This method was chosen in synchrony with recent computational studies on radical pathways of prebiotically-important reactions.13,20,29–31 Stationary points on the reaction surfaces were characterized as minima (i.e. reactants, intermediates or products) or first-order saddle points (i.e. transition states) using vibrational frequency calculations. We note that relative energies of some of the transition states were negative, which may occur due to numerical noise or inclusion of zero-point energy corrections. Similar barriers have been obtained in previous analogous reaction pathways.20 Such pathways have been considered as barrierless (i.e. the relative energies of the associated transitions states have been considered as 0 kcal mol−1) in the present work. Further, intrinsic reaction coordinate scans were carried out to confirm the pathways connecting the transition states to reactants and products of each step. Despite careful evaluation of the pathways through gas-phase quantum chemical calculations, we did not perform solvent phase calculations, mainly because the concentration of radical initiators such as ˙OH and ˙NH2 may not be sufficient in the aqueous phase, due to predominance of radical quenching with water molecules or recombination reactions leading to the formation of neutral species.To test the method dependence of the derived results and confirm the feasibility of the proposed pathways, optimizations were re-performed using three additional functionals (i.e. ωB97XD,32 M06–2X,33 and B2PLYP34). These calculations reveal deviations of 0–7 kcal mol−1 compared to all calculated energy barriers, and up to 5.0 kcal mol−1 reduction in the highest barrier calculated using B3LYP (Table S1†). Further, the mean and maximum absolute deviation of B3LYP energies with respect to the M06-2X energies is 2.6 kcal mol−1 and 7.2 kcal mol−1, respectively (Table S2†). Regardless, recalculation at other DFT functionals does not significantly affect the conclusions derived from the present work. As a result, B3LYP/6-311G(d,p) calculations have been discussed in this work, in order to allow meaningful comparisons with previous studies.21,24,29,31 All quantum chemical calculations were carried out using the Gaussian 09 suite of programs.35
Results
Formation of CA
The initial step for CA formation involves the barrierless attack of ˙OH on the cyano group of cyanamide (1) to give 2 (Table 1, Scheme 2, Fig. 1 and S1†). This step is followed by hydrogen abstraction from NH3 by 2 through a barrier of 7.8 kcal mol−1. The resulting neutral intermediate 3 crosses a barrier of 4.1 kcal mol−1 through the attack of ˙NH2 to form species 4 with the radical centre at the terminal amino group. A subsequent attack of 4 on the imine group of another molecule of 3 across a barrier of 11.6 kcal mol−1 generates the radical intermediate 5.| Relative energies for CA formation (in kcal mol−1) | Relative energies for MM formation (in kcal mol−1) | ||||
|---|---|---|---|---|---|
| Conversion | TS | P | Conversion | TS | P |
| a Each reactant is considered in its free optimized state while calculating the electronic energies. | |||||
| 1 + ˙OH → 2 | 0 | −31.8 | 1 + ˙NH2 → 18 | 1.2 | −23.4 |
| 2 → 3 | 7.8 | 7.5 | 18 → 19 | 9.5 | 9.3 |
| 3 → 4 | 4.1 | −2.4 | 19 → 20a | 3.4 | −2.4 |
| 4 → 5 | 11.6 | −0.5 | 20a + 19 → 21 | 12.0 | 1.0 |
| 5 → 6 | 7.6 | 3.7 | 21 → 22 | 10.4 | −1.5 |
| 6 + 4 → 7 | 10.8 | 6.4 | 22 → 23 | 2.8 | 1.2 |
| 7 → 8 | 6.7 | 3.8 | 23 → 24 | 2.1 | −13.8 |
| 8 → 9 | 2.0 | −5.2 | 24 → 25 | 8.5 | 8.2 |
| 9 → 10 | 17.4 | 5.9 | 25 → 26 | 1.0 | −4.1 |
| 10 → 11 | 0 | 3.6 | 26 → 27 | 14.7 | −6.0 |
| 11 → 12 | 0 | −8.5 | 27 → 28 | 12.8 | 1.2 |
| 12 → 13 | 19.9 | −14.2 | |||
| 13 → 14 | 0 | −9.8 | |||
| 14 → 15 | 16.6 | −10.2 | |||
| 15 → 16 | 0 | −10.7 | |||
| 16 → 17 | 3.1 | −11.8 | |||
 | ||
| Scheme 2 Proposed radical mechanism along with the corresponding transition states (TSs) for the formation of cyanuric acid (17) from cyanamide (1). | ||
 | ||
| Fig. 1 Potential energy curves depicting the B3LYP/6-311G(d,p) (blue), ωB97XD/6-311G(d,p) (black), M06-2X/6-311G(d,p) (red), and B2PLYP/6-311G(d,p) (green) level relative electronic energies (kcal mol−1) of the various species associated with proposed mechanism of (a) CA and (b) MM. Energies of transition states and products are reported relative to the energies of the reactants of each step using free reactant state as the reference state. | ||
The next step involves the formation of 6, through the abstraction of hydrogen from NH3 by 5 across a barrier of 7.6 kcal mol−1. Subsequent formation of species 8 is accomplished by 10.8 kcal mol−1 attack of 4 on 6 to form 7, followed by hydrogen abstraction from NH3 by 7 through 6.7 kcal mol−1 barrier. Further, a low (2.0 kcal mol−1) barrier formation of radical centre at the terminal amino group of 8 gives 9, which then undergoes high (17.4 kcal mol−1) barrier cyclization. The resulting cyclized intermediate 10 converts into neutral intermediate 11 by the barrierless attack of NH3. A subsequent barrierless abstraction of hydrogen from one of the –OH groups of 11 by ˙NH2, followed by high (19.9 kcal mol−1) barrier release of ˙NH2, results in the introduction of a carbonyl group in 13. In a similar manner, the other two carbonyl groups of CA (17) are formed by hydrogen abstraction from the remaining two –OH groups of 13, followed by the release of two additional ˙NH2. Overall, the formation of CA is accomplished in 16 steps and consumes three molecules of cyanamide, three ˙OH and one NH3.
Formation of MM
MM formation proceeds through the attack of ˙NH2 on cyanamide 1 to generate 18, which possesses the radical centre at nitrogen (Table 1, Scheme 3, Fig. 1 and S2†). 18 is then attacked by NH3 through 9.5 kcal mol−1 barrier to give 19, which is converted into its radical form (20) by passing a low (3.4 kcal mol−1) barrier. 20 can adopt two conformations (i.e. 20a and 20b, Fig. S5†) depending upon the location of the attack of ˙NH2 on 19. Nevertheless, due to high energy associated with the impact events, rapid rotation across the bond renders a mixture of two conformers (20a and 20b). However, only 20a can proceed towards MM formation. Specifically, the attack of 20a on another molecule of 19 across a barrier of 12 kcal mol−1 forms the radical dimer 21, which releases ˙NH2 by crossing 10.4 kcal mol−1 barrier to form 22. ˙NH2 then attacks the terminal amino group of 22 through 2.8 kcal mol−1 barrier to form 23, which is then attacked at its cyano position by a second cyanamide molecule to produce 24 with the radical centre at nitrogen. The formation of 25 (trimer of 19), is then accomplished by the abstraction of hydrogen from NH3 by the radical centre of 24 across a barrier of 8.5 kcal mol−1. Subsequently, 25 undergoes a low (1 kcal mol−1) barrier attack of ˙NH2 at the terminal amino group and generates 26, which cyclizes into 27 by the high (14.7 kcal mol−1) barrier attack of terminal ˙NH– group on the sp2 carbon. Finally, the release of ˙NH2 from 27 across a barrier of 12.8 kcal mol−1 forms MM (28). Overall, the formation of MM is accomplished in 11 steps and consumes three cyanamide molecules and one ˙NH2. Further, one NH3 is released and two ˙NH2/NH3 pairs play catalytic roles. | ||
| Scheme 3 Proposed radical mechanism along with the corresponding transition states (TSs) for the formation of melamine (28) from cyanamide (1). | ||
Formation of BA
The first step for BA formation involves the production of urea radical (30) from urea 29 by the attack of ˙NH2 radical (Table 2, Scheme 4, Fig. 2 and S3†). 30 then attacks the carbonyl carbon of malonic acid (31) across 8.9 kcal mol−1 barrier to form the radical intermediate 32. Release of ˙OH from 32 across a barrier of 12.1 kcal mol−1 retrieves the third carbonyl group of BA, thereby forming 33. A low (5.5 kcal mol−1) barrier attack of ˙NH2 on the terminal amino group of 33 generates intermediate 34 having the radical center at nitrogen. 34 involves 13.1 kcal mol−1 intramolecular attack on its electron-deficient carbonyl carbon to form cyclic intermediate 35. Release of ˙OH from 35 across a barrier of 11.1 kcal mol−1 gives BA (36) as the end product. Overall, the conversion of urea and malonic acid into BA is achieved in 6 steps and involves the consumption of two ˙NH2 and the generation of two NH3 molecules and two ˙OH.| Relative energies for BA formation (in kcal mol−1) | Relative energies for TAP formation (in kcal mol−1) | ||||
|---|---|---|---|---|---|
| Conversion | TS | P | Conversion | TS | P |
| a Each reactant is considered in its free optimized state while calculating the electronic energies. | |||||
| 29 → 30 | 4.8 | 2.4 | 37 + 20b → 38 | 10.0 | −9.6 |
| 30 + 31 → 32 | 8.9 | 5.8 | 38 → 39 | 0 | 10.8 |
| 32 → 33 | 12.1 | 7.7 | 39 → 40 | 3.3 | −8.3 |
| 33 → 34 | 5.5 | 2.1 | 40 → 41 | 8.2 | −13.3 |
| 34 → 35 | 13.1 | 1.5 | 41 → 42 | 10.3 | 9.5 |
| 35 → 36 | 11.1 | 5.2 | 42 → 43 | 3.4 | −2.7 |
| 43 → 44 | 2.4 | −6.2 | |||
| 44 → 45 | 13.6 | −20.2 | |||
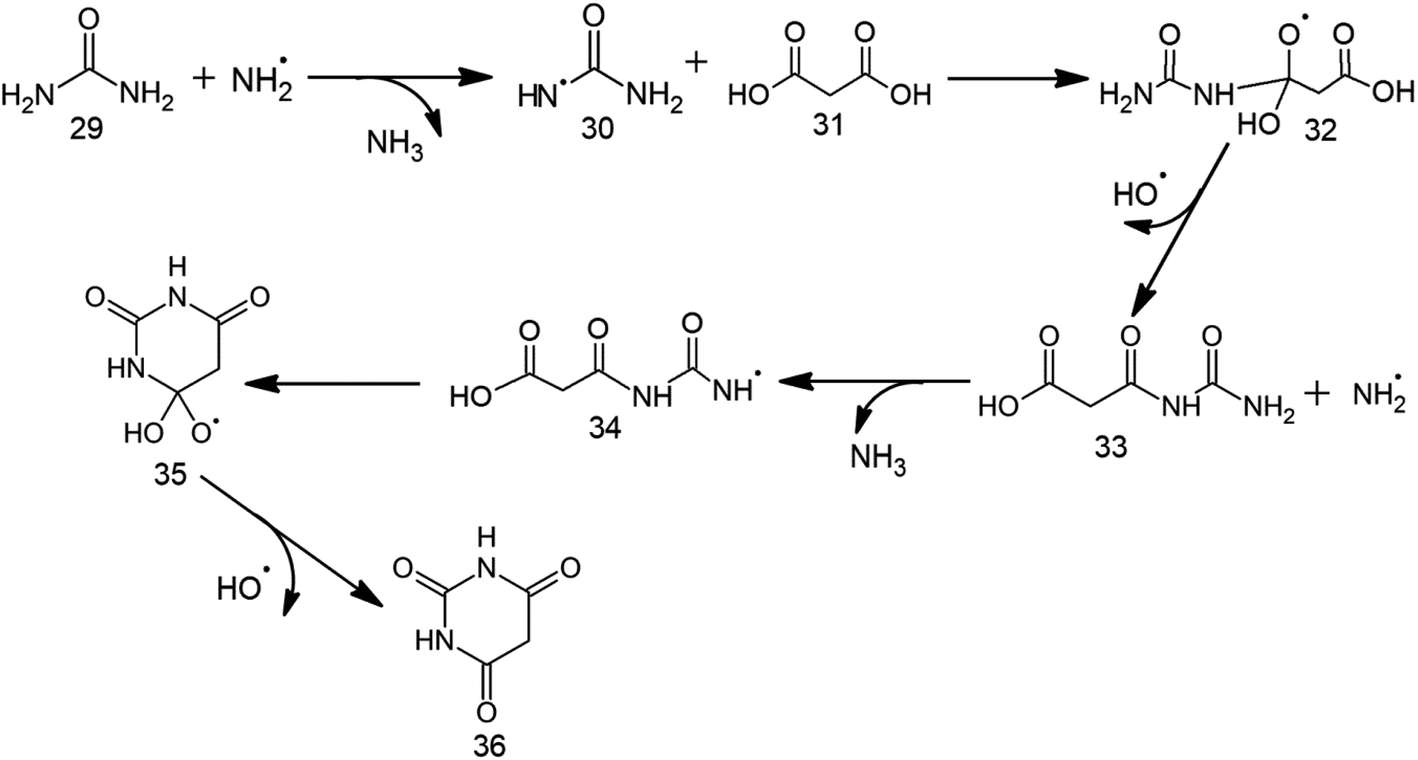 | ||
| Scheme 4 Proposed radical mechanism along with corresponding transition states (TSs) for the formation of barbituric acid (30) from urea (23) and malonic acid (25). | ||
 | ||
| Fig. 2 Potential energy curves depicting the B3LYP/6-311G(d,p) (blue), ωB97XD/6-311G(d,p) (black), M06-2X/6-311G(d,p) (red), and B2PLYP/6-311G(d,p) (green) level relative electronic energies (kcal mol−1) of the various species associated with proposed mechanism of (a) BA and (b) TAP. Energies of transition states and products are reported relative to the energies of the reactants of each step using free reactant state as the reference state. | ||
Formation of TAP
Although conformation 20a (Fig. S5†) of intermediate 20 proceeds toward MM formation (vide supra), the alternate conformation (20b) is involved in TAP formation. However, at high temperatures (characteristic of impact events), the two rotamers (20a and 20b) can switch into each other. Therefore, a mixture of both melamine and 2,4,6-triaminopyrimidine will be formed under these conditions. Specifically, 20b attacks the cyano carbon of malononitrile (37) through 10.0 kcal mol−1 barrier to form intermediate 38 (Table 2, Scheme 5, Fig. 2 and S4†), which barrierlessly abstracts a hydrogen from NH3 and converts into neutral intermediate 39. The terminal amino group of 39 then undergoes low (3.3 kcal mol−1) barrier attack by ˙NH2. The resulting radical intermediate 40 further cyclizes by crossing a modest (8.2 kcal mol−1) barrier to form 41, which bears radical at nitrogen. Intermediate 41 abstracts a hydrogen from NH3 through a 10.3 kcal mol−1 barrier to form a neutral, imine-containing analogue of 2,4,6-triaminopyrimidine (42). Three subsequent ammonia-assisted imine-enamine tautomerizations of 42 through 3.4, 2.4 and 13.6 kcal mol−1 barriers give TAP (45). Overall, starting from cyanamide, TAP formation is achieved in a total of 11 steps. Further, the formation of TAP from malononitrile involves consumption of one NH3. However, three NH3 molecules (that assist in imine-enamine tautomerism) and two ˙NH2/NH3 pairs act as catalysts.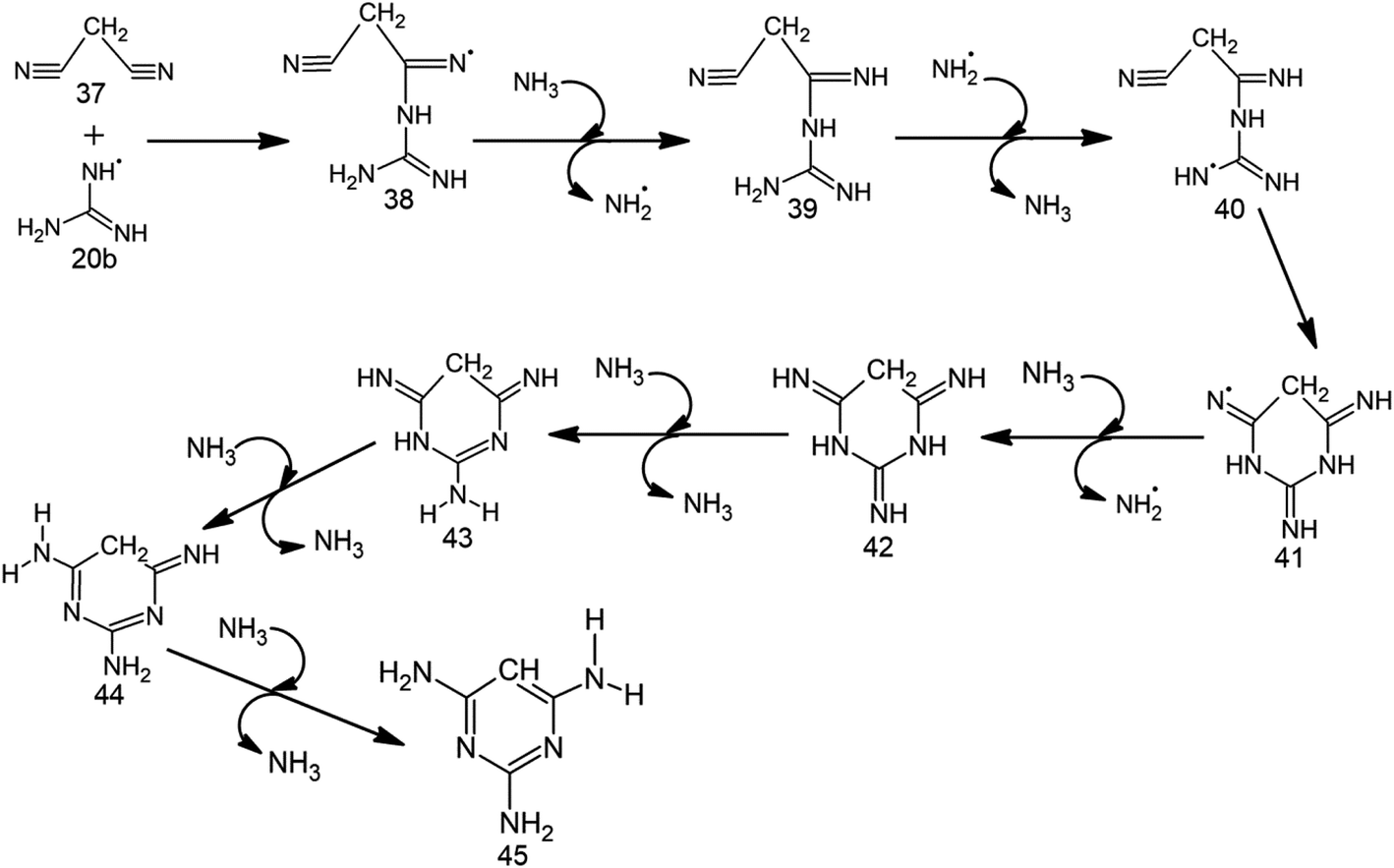 | ||
| Scheme 5 Proposed radical mechanism along with corresponding transition states (TSs) for the formation of 2,4,6-triaminopyrimidine (45) from malononitrile (37) and 20b. | ||
Discussion
The present work focuses on the formation pathways for four prebiotic heterocycles, i.e. BA, MM, CA and TAP in an early earth environment from a common precursor, i.e. cyanamide (Scheme 1). Cyanamide, is a suggested precursor to natural nucleotides,24 and plays an important role in prebiotic reactions.22,23,36,37 Although cyanamide can directly convert to CA or MM through the involvement of ˙OH (CA) and ˙NH2/NH3 pair (CA and MM), BA formation requires urea,38 which can be prebiotically derived from hydrolysis of cyanamide25,39 or can be directly derived from other prebiotic sources such as irradiation of prebiotic ammonium cyanide.40 In addition, BA formation requires malonic acid, which can be prebiotically formed by the γ irradiation of urea involving water radiolysis.14 In contrast, the formation of TAP requires the reaction of cyanamide with malononitrile and involves the ˙NH2/NH3 pair. Malononitrile used in this pathway is a prebiotically important molecule26 and its derivative (aminomalononitrile) is a trimer of HCN,41 a likely photochemical product of primitive atmosphere.42 NH3, a component of early earth atmosphere,43 ˙NH2/NH3 pair (derived from NH3), and ˙OH (photochemically formed from the dissociation of water vapors44) used in these pathways have been previously suggested to play important role in prebiotic reactions.13,20,29–31 For example, ˙OH is observed in triazine synthesis where urea:water ice mixtures act as its source.13 Similarly, ˙NH2 is a photolysis product of early earth NH3.45The sixteen-step pathway for CA formation requires three molecules of cyanamide, three ˙OH, one NH3 and three catalytic ˙NH2/NH3 pairs (Scheme 2 and Fig. S1†). Despite barrierless formation of five intermediates, the remaining eleven steps involve 2–20 kcal mol−1 reaction barriers. Specifically, the highest barrier (19.9 kcal mol−1) step pertains to the formation of first carbonyl group, which is comparable to the highest barrier (20.6 kcal mol−1) obtained for previously proposed pathways involving urea and ˙NH2.21 Further, our proposed pathway involves trimerization of intermediate 3, which is analogous to the experimentally-proposed trimerization of cyanic acid to CA.46 In contrast, MM formation consumes three molecules of cyanamide (required for the formation of three molecules of intermediate 19 that forms MM, Scheme 3 and Fig. S2†) and one ˙NH2, and involves two catalytic ˙NH2/NH3 pairs. This route involves eleven steps with barrier heights ranging from 1–15 kcal mol−1. Further, the highest (i.e. cyclization) barrier of 15 kcal mol−1 is significantly smaller than the ˙NH2 assisted MM formation from urea (26 kcal mol−1).21
On the other hand, BA formation involves one molecule each of urea and malonic acid, two ˙NH2 radicals and release of two ˙OH and two NH3 (Scheme 4 and Fig. S3†). This six-step pathway is characterized by the highest (i.e. cyclization) barrier of 13.1 kcal mol−1, which is significantly lower than the highest barrier (40.0 kcal mol−1) involved in previously-proposed radical pathway for BA formation from formamide and acetylene.29 However, the mechanism for the prebiotic TAP formation involves cyanamide (the precursor of intermediate 19), malononitrile, one NH3 and two catalytic ˙NH2/NH3 pairs (Scheme 5 and Fig. S4†). This eleven-step pathway involves the highest barrier of 13.6 kcal mol−1. Although various substituted 2,4,6-triaminopyrimidines have been obtained in literature by refluxing malonic nitriles with guanidine in alcohol,47,48 TAP formation under prebiotic (radical) conditions is not reported in the literature. The present work thus tries to fill this gap by suggesting a route appropriate to the early-earth scenario.
The overall highest barrier observed in the synthesis of four heterocycles (BA, MM, CA and TAP) is 20 kcal mol−1 (Fig. 1 and 2), which is significantly smaller than the barriers to similar prebiotic reactions, such as formation of pyrimidines (40 kcal mol−1)29 and purines (43 kcal mol−1)31 from formamide and formation of purines (36 kcal mol−1) from cyanamide.24 This suggests that the formation of prebiotic heterocycles (thereby, preRNA) might have been more feasible than canonical nucleobases on the early earth. Further, out of all four heterocycles, BA formation is most favored, since it takes place in relatively smaller (six) number of steps and involves modest energy barriers (5–13 kcal mol−1) compared to CA, MM or TAP formation having higher (eleven or sixteen) number of steps and higher energy barriers (0 to 20 kcal mol−1, Fig. 1 and 2). Regardless, the barrier associated with the energetically most demanding formation step of all four prebiotic heterocycles is very similar (13 to 20 kcal mol−1, Fig. 1 and 2). This, in addition to the common origin of their starting precursors, indicates that BA, MM, CA and TAP might have formed under similar prebiotic conditions through the suggested pathways.
The structural similarities of the considered prebiotic heterocycles with their precursors further lend support to the plausibility of our suggested formation pathways. Specifically, urea and malonic acid contain all the skeletal components required for BA formation. For instance, the –NH2–CO–NH2– segment of urea and –CO–CH2–CO– segment of malonic acid can be directly incorporated into the BA skeleton. Similarly, the –(NH)2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH– segment of MM is obtained from guanidine (intermediate 19), which is in turn formed from cyanamide. Further, the constituent components of CA (i.e. –NH–CO–NH– segment) can be obtained from cyanamide (N
NH– segment of MM is obtained from guanidine (intermediate 19), which is in turn formed from cyanamide. Further, the constituent components of CA (i.e. –NH–CO–NH– segment) can be obtained from cyanamide (N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–NH2) and ˙OH, whereas the –(NH2)CCH–C(NH2) and –(NH)2CNH– segments of TAP can be obtained from malononitrile and guanidine, respectively. Thus, the proposed pathways automatically optimize the atom economy, since all the components of the heterocycles can be obtained from one or two precursors and radicals (˙OH and/or ˙NH2).
C–NH2) and ˙OH, whereas the –(NH2)CCH–C(NH2) and –(NH)2CNH– segments of TAP can be obtained from malononitrile and guanidine, respectively. Thus, the proposed pathways automatically optimize the atom economy, since all the components of the heterocycles can be obtained from one or two precursors and radicals (˙OH and/or ˙NH2).
Overall, our results suggest that the common precursor to the formation of prebiotic heterocycles (BA, MM, CA and TAP) under similar environmental conditions will bring them close to form prebiotic supramolecular assemblies held by non-covalent interactions,3–8 which would have ultimately paved way to the formation of more complex prebiotic information polymers. Nevertheless, side reactions associated with radical pathways cannot be neglected and by-products formation is ensued. However, any sufficiently high energy event such as impacts of extraterrestrial bodies can produce enough radicals (initiators and radical intermediates) for such pathways to give product heterocycles and compensate side reactions such as radical quenching by recombination. Regardless, elucidation of the routes that led to the formation of prebiotic heterocycles in a similar environment can strengthen our understanding of primitive genetics.
Conclusions
In the present study, we propose that the precursor cyanamide and its derivatives, urea and malonic acid can react with other relevant precursors such as malononitrile, ˙NH2 and ˙OH radicals (products of reactions in early earth atmosphere) to give MM, TAP, CA and BA under prebiotic conditions. The proposed pathways are characterized by smaller (upto 20 kcal mol−1) barriers to the energetically most demanding reaction step compared to other similar prebiotic reactions (26–43 kcal mol−1).21,24,29,31 Further, in the suggested pathways, one or two types of precursors and radicals (˙OH and/or ˙NH2) can form all the skeletal components of the heterocycles, thereby optimizing the atom economy.Most importantly, we posit that for the prebiotic heterocycles to form supramolecular assemblies that later evolved into the first information polymer, their proximity in a prebiotic environment is crucial. In this context, our proposed pathways, which are characterized by a common, starting precursor (cyanamide) and similar, low barriers, might help these heterocycles to form at the same place and under similar environmental conditions. This will, in turn, ensure their proximity to form prebiotic supramolecular assemblies held by non-covalent interactions. Altogether, such a scenario would have ultimately paved way to their evolution into more complex information-bearing polymers.
The present work will thus hopefully contribute to some extent in getting a clearer idea of the origin of the first informational polymer. However, future experimental studies based on the idea of the common origin of heterocycles and the involvement of suggested precursors (cyanamide and urea) might give more insights.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
PS thanks the Department of Science and Technology (DST) and University Grants Commission (UGC), New Delhi for financial support through the DST INSPIRE (IFA14-CH162) and the UGC FRP (F.4-5(176-FRP/2015(BSR))) programs, respectively. SK thanks the Department of Science and Technology (DST) for financial support through WOS-A (SR/WOS-A/CS-99/2018) program. We thank Ms Himani Ahuja for preliminary contribution to this project.References
- V. Kolb, J. Dworkin and S. Miller, J. Mol. Evol., 1994, 38, 549–557 CrossRef CAS.
- B. J. Cafferty and N. V. Hud, Isr. J. Chem., 2015, 55, 891–905 CrossRef CAS.
- B. J. Cafferty, D. M. Fialho, J. Khanam, R. Krishnamurthy and N. V. Hud, Nat. Commun., 2016, 7, 11328 CrossRef CAS.
- M. C. Chen, B. J. Cafferty, I. Mamajanov, I. Gállego, J. Khanam, R. Krishnamurthy and N. V. Hud, J. Am. Chem. Soc., 2014, 136, 5640–5646 CrossRef CAS PubMed.
- T. M. Bohanon, S. Denzinger, R. Fink, W. Paulus, H. Ringsdorf and M. Weck, Angew. Chem., Int. Ed. Engl., 1995, 34, 58–60 CrossRef CAS.
- M. Ma and D. Bong, Langmuir, 2011, 27, 8841–8853 CrossRef CAS.
- C. T. Seto and G. M. Whitesides, J. Am. Chem. Soc., 1990, 112, 6409–6411 CrossRef CAS.
- B. J. Cafferty, I. Gállego, M. C. Chen, K. I. Farley, R. Eritja and N. V. Hud, J. Am. Chem. Soc., 2013, 135, 2447–2450 CrossRef CAS.
- C. S. Cockell and G. Horneck, Photochem. Photobiol., 2001, 73, 447–451 CrossRef CAS.
- C. F. Chyba, Nature, 1987, 330, 632–635 CrossRef.
- M. Ferus, D. Nesvorný, J. Šponer, P. Kubelík, R. Michalčíková, V. Shestivská, J. E. Šponer and S. Civiš, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 657–662 CrossRef CAS.
- M. Ferus, F. Pietrucci, A. M. Saitta, A. Knížek, P. Kubelík, O. Ivanek, V. Shestivska and S. Civiš, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 4306–4311 CrossRef CAS.
- C. Menor-Salván and M. R. Marín-Yaseli, Chem.–Eur. J., 2013, 19, 6488–6497 CrossRef.
- R. Navarro-Gonzalez, A. Negron-Mendoza and E. Chacon, Origins Life Evol. Biospheres, 1989, 19, 109–118 CrossRef CAS.
- M. Nuevo, Y. J. Chen, W. J. Hu, J. M. Qiu, S.-R. Wu, H. S. Fung, C. C. Chu, T. S. Yih, W. H. Ip and C. Y. R. Wu, Astrobiology, 2014, 14, 119–131 CrossRef CAS.
- S. Miyakawa, K. Murasawa, K. Kobayashi and A. B. Sawaoka, J. Am. Chem. Soc., 1999, 121, 8144–8145 CrossRef CAS.
- R. Hayatsu, M. H. Studier, A. Oda, K. Fuse and E. Anders, Geochim. Cosmochim. Acta, 1968, 32, 175–190 CrossRef CAS.
- M. H. Studier, R. Hayatsu and E. Anders, Geochim. Cosmochim. Acta, 1972, 36, 189–215 CrossRef CAS.
- C. Menor-Salván, D. M. Ruiz-Bermejo, M. I. Guzmán, S. Osuna-Esteban and S. Veintemillas-Verdaguer, Chem.–Eur. J., 2009, 15, 4411–4418 CrossRef.
- Y. A. Jeilani, H. T. Nguyen, B. H. Cardelino and M. T. Nguyen, Chem. Phys. Lett., 2014, 598, 58–64 CrossRef CAS.
- Y. A. Jeilani, T. M. Orlando, A. Pope, C. Pirim and M. T. Nguyen, RSC Adv., 2014, 4, 32375–32382 RSC.
- R. Lohrmann and L. Orgel, Science, 1968, 161, 64–66 CrossRef CAS.
- R. A. Sanchez and L. E. Orgel, J. Mol. Biol., 1970, 47, 531–543 CrossRef CAS.
- A. A. Ingar, R. W. Luke, B. R. Hayter and J. D. Sutherland, ChemBioChem, 2003, 4, 504–507 CrossRef CAS.
- J. Hulshof and C. Ponnamperuma, Orig. Life, 1976, 7, 197–224 CrossRef CAS.
- S. Becker, I. Thoma, A. Deutsch, T. Gehrke, P. Mayer, H. Zipse and T. Carell, Science, 2016, 352, 833–836 CrossRef CAS.
- W. Traube, Der aufbau der xanthinbasen aus der cyanessigsäure. Synthese des hypoxanthins und adenins, Springer, 1904, pp. 40–56 Search PubMed.
- M. Ferus, R. Michalcikova, V. Shestivská, J. Sponer, J. E. Sponer and S. Civis, J. Phys. Chem. A, 2014, 118, 719–736 CrossRef CAS.
- H. T. Nguyen, Y. A. Jeilani, H. M. Hung and M. T. Nguyen, J. Phys. Chem. A, 2015, 119, 8871–8883 CrossRef CAS.
- Y. A. Jeilani, P. N. Williams, S. Walton and M. T. Nguyen, Phys. Chem. Chem. Phys., 2016, 18, 20177–20188 RSC.
- Y. A. Jeilani, H. T. Nguyen, D. Newallo, J. M. D. Dimandja and M. T. Nguyen, Phys. Chem. Chem. Phys., 2013, 15, 21084–21093 RSC.
- Q. Wu and W. Yang, J. Chem. Phys., 2003, 118, 2498–2509 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- S. Grimme and F. Neese, J. Chem. Phys., 2007, 127, 154116 CrossRef PubMed.
- M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. Petersson, Gaussian Inc., Wallingford, CT, USA, 2009.
- C. M. Tapiero and J. Nagyvary, Nature, 1971, 231, 42–43 CrossRef CAS.
- A. A. Ingar, R. W. Luke, B. R. Hayter and J. D. Sutherland, ChemBioChem, 2003, 4, 504–507 CrossRef CAS.
- E. Grimaux, Bull. Soc. Chim. Fr., 1879, 31, 146 Search PubMed.
- H. Hetherington and J. Braham, J. Am. Chem. Soc., 1923, 45, 824–829 CrossRef CAS.
- R. Lohrmann, J. Mol. Evol., 1972, 1, 263–269 CrossRef CAS.
- R. A. Sanchez, J. P. Ferbis and L. E. Orgel, J. Mol. Biol., 1967, 30, 223–253 CAS.
- K. J. Zahnle, J. Geophys. Res.: Atmos., 1986, 91, 2819–2834 CrossRef CAS.
- J. F. Kasting, Science, 1993, 259, 920–926 CrossRef CAS.
- T. B. Vander Wood and M. H. Thiemens, J. Geophys. Res.: Oceans, 1980, 85, 1605–1610 CrossRef CAS.
- W. R. Kuhn and S. K. Atreya, Icarus, 1979, 37, 207–213 CrossRef CAS.
- A. Senier and T. Walsh, J. Chem. Soc., 1902, 81, 290–291 RSC.
- P. B. Russell and G. H. Hitchings, J. Am. Chem. Soc., 1952, 74, 3443–3444 CrossRef CAS.
- W. Traube, Ber. Dtsch. Chem. Ges., 1904, 37, 4544–4547 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra08001e |
| This journal is © The Royal Society of Chemistry 2019 |