
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparative separation of seven phenolic acids from Xanthii Fructus using pH-zone-refining counter-current chromatography combined with semi-preparative high performance liquid chromatography†
Tianyu Maab,
Lihua Xuab,
Xinming Wangab,
Jia Lia,
Lanping Guo*c and
Xiao Wang *b
*b
aCollege of Pharmacy, Shandong University of Traditional Chinese Medicine, Jinan, 250355, China
bKey Laboratory of TCM Quality Control Technology, Shandong Analysis and Test Center, Qilu University of Technology (Shandong Academy of Sciences), Jinan, 250014, China. E-mail: wangx@sdas.org
cState Key Laboratory Breeding Base of Dao-di Herbs, National Resource Center for Chinese Materia Medica, China Academy of Chinese Medical Sciences, Beijing, 100700, China. E-mail: glp01@126.com; Fax: +86-531-82964889; Tel: +86-531-82605319
First published on 11th November 2019
Abstract
Phenolic acids represented by caffeoylquinic acids in Xanthii Fructus have various pharmacological activities such as anti-inflammatory, anti-nociceptive, anti-oxidative and anti-allergic effects. In this study, pH-zone-refining counter-current chromatography was successfully applied in the segmentation of crude samples and further separation of phenolic acids from Xanthii Fructus. We initially segmented 1.6 g of the crude sample to yield three sample fractions using a two-phase solvent system composed of EtOAc–ACN–H2O (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:5, v/v/v) with 10 mM TFA added to the organic phase as the stationary phase and 10 mM NH3·H2O added to the aqueous phase as the mobile phase. The first fraction was separated using EtOAc–H2O (1:1, v/v) (10 mM TFA was added in the upper phase and 20 mM NH3·H2O was added in the lower phase) solvent system, the second fraction containing low-content compounds was separated using semi-preparative high performance liquid chromatography, and the third fraction contained one pure compound. As a result, seven phenolic acids including six caffeoylquinic acid isomers (3-caffeoylquinic acid, 4-caffeoylquinic acid, 5-caffeoylquinic acid, 1,5-O-dicaffeoylquinic acid, 3,5-O-dicaffeoylquinic acid, and 4,5-O-dicaffeoylquinic acid) and caffeic acid were successfully isolated from Xanthii Fructus with purities above 90%. This study demonstrated that pH-ZRCCC is an efficient preparative separation method for phenolic acids, especially isomeric caffeoylquinic acids, from natural products.
:1:5, v/v/v) with 10 mM TFA added to the organic phase as the stationary phase and 10 mM NH3·H2O added to the aqueous phase as the mobile phase. The first fraction was separated using EtOAc–H2O (1:1, v/v) (10 mM TFA was added in the upper phase and 20 mM NH3·H2O was added in the lower phase) solvent system, the second fraction containing low-content compounds was separated using semi-preparative high performance liquid chromatography, and the third fraction contained one pure compound. As a result, seven phenolic acids including six caffeoylquinic acid isomers (3-caffeoylquinic acid, 4-caffeoylquinic acid, 5-caffeoylquinic acid, 1,5-O-dicaffeoylquinic acid, 3,5-O-dicaffeoylquinic acid, and 4,5-O-dicaffeoylquinic acid) and caffeic acid were successfully isolated from Xanthii Fructus with purities above 90%. This study demonstrated that pH-ZRCCC is an efficient preparative separation method for phenolic acids, especially isomeric caffeoylquinic acids, from natural products.
1 Introduction
Xanthii Fructus is the dry ripe fruit of Xanthium sibiricum Patr. with involucre which is also called “Cang Er-Zi” in Chinese.1 As a commonly used traditional Chinese medicine, Xanthii Fructus is often used to treat cold headaches, allergic rhinitis, urticaria, pruritis and rheumatoid arthritis based on its abundant chemical constituents such as phenolic acids, flavones, saponins and sesquiterpene lactones.2,3 Among them, phenolic acids, as the main components in traditional Chinese medicine, have received wide attention because of their clear anti-inflammatory, anti-nociceptive, anti-oxidative and anti-allergic effects.4,5 However, most of the phenolic acids in Xanthii Fructus are caffeoylquinic acid (CQA) isomers such as 3-CQA, 4-CQA, 5-CQA, 1,5-di-CQA, 3,5-di-CQA and 4,5-di-CQA which are difficult to separate because of their similar chemical structures.6,7At present, most of the CQAs separations are still achieved by silica gel column chromatography combined with semi-preparative high performance liquid chromatography (HPLC).8,9 It has also been reported to use high-speed counter-current chromatography (HSCCC) combined with semi-preparative HPLC to obtain pure CQA isomers.10,11 However, the conventional column chromatography has many disadvantages such as long separation time and large solvent consumption. As for HSCCC, though it significantly improves the separation efficiency, its weak sample loading capacity is also unavoidable. Therefore, it is necessary to develop an efficient and environmentally friendly method for the CQA isomers separation.
pH-zone-refining counter-current chromatography (pH-ZRCCC) is a displacement chromatography technology developed by HSCCC. As a liquid–liquid chromatography, pH-ZRCCC not only retains the advantages of HSCCC that elimination of the irreversible adsorption of solid stationary phase and high separation efficiency, but also increases the sample loading amount to g-level.12 It was reported that pH-ZRCCC had been successfully applied to the separation of di-CQA isomers from Lonicerae japonicae Flos.13,14 Therefore, it was considered to separate CQA isomers from Xanthii Fructus using pH-ZRCCC.
In this study, we developed a segmentation strategy using pH-ZRCCC to segment the crude sample of Xanthii Fructus with various phenolic acids into three fractions, which were then separated using pH-ZRCCC and semi-preparative HPLC. As a result, seven phenolic acids including six CQA isomers (Fig. 1) were separated from Xanthii Fructus successfully.
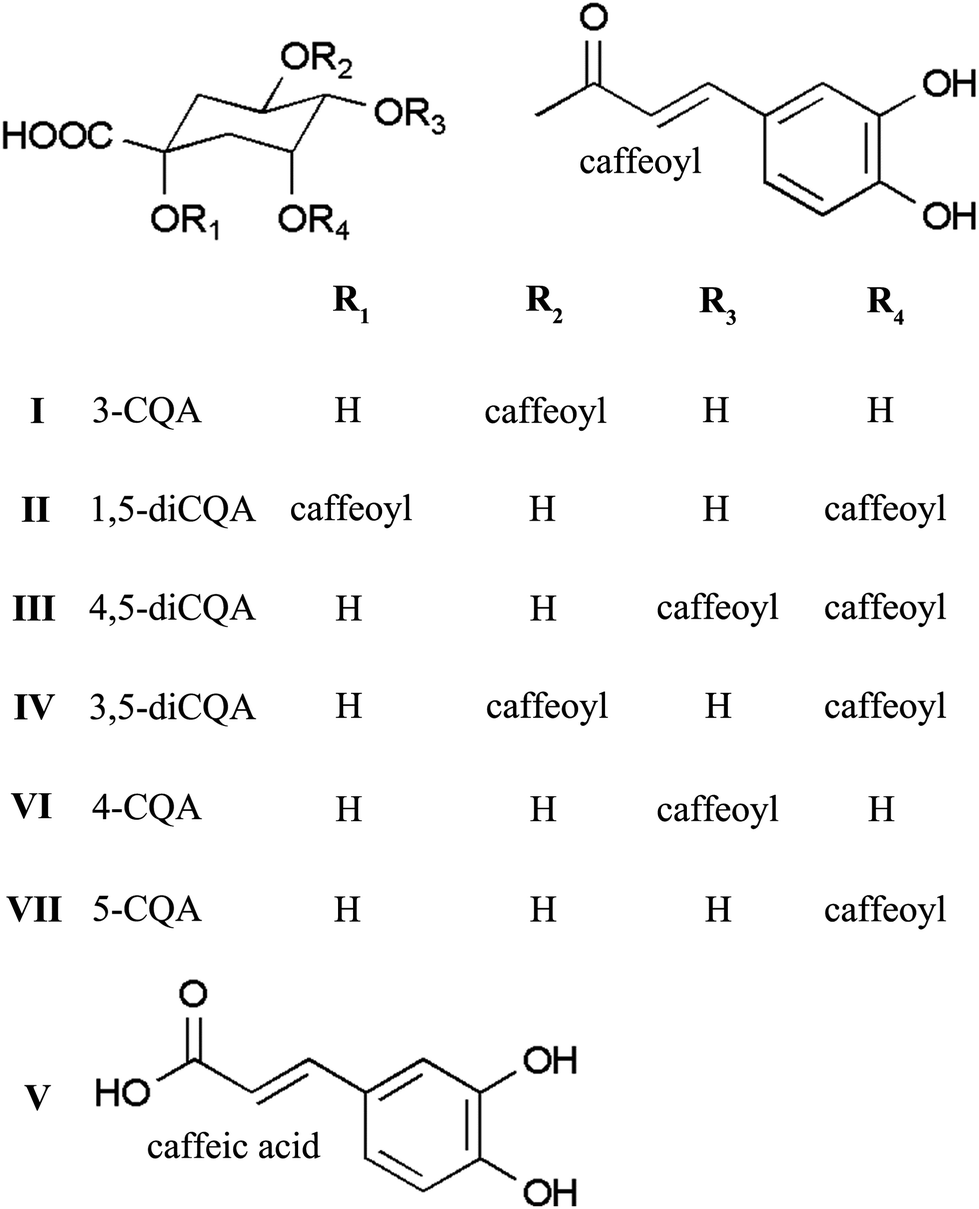 | ||
| Fig. 1 Chemical structures of seven phenolic acids from Xanthii Fructus. | ||
2 Materials and methods
2.1 Apparatus
A TBE-300C instrument (Tauto Biotech, Shanghai, China) was used for the phenolic acids separation which consists of a series of three preparative coil columns (total volume of 300 mL) and a 20 mL sample loop. The rotate speed adjustment range is 0–1000 rpm. A TBP5002 constant flow pump (Tauto Biotech) was used to pump the solvent into the separation columns. The absorbance of effluent was detected by an UV detector (8823B, Beijing BINTA Instrument Technology Co., Ltd., Beijing, China) at 254 nm and the separation chromatograms were recorded by a 3057-11 portable recorder (Yokogawa Sichuan Instrument Factory, Sichuan, China). The pH value of effluent was measured using a pH/mV meter (UB-7, Denver Instrument Co., Ltd., Beijing, China). The column temperature during the separation process was controlled by a DC-0506 constant temperature circulator (Tauto Biotech) at 25 °C.All samples were analyzed using an Acchrom S6000 system (Acchrom-Tech Co., Ltd, Beijing, China) consisting of an auto sampler, a column oven, a photodiode array detector and four binary gradient pumps. The chromatographic column was a Compass C18 column (250 × 4.6 mm, 5 μm; Rigol Technologies Co., Ltd, Beijing, China).
The identification and the purity testing of the purified compounds were performed on the Acquity UPLC (Waters, Milford, MA) coupled with Impact II UHR-QqTOF (Ultra-High Resolution Qq-Time-Of-Flight) mass spectrometers (Bruker, Billerica, MA). NMR spectra of purified compounds were measured by an ADVANCE DPX 400 spectrometer (Bruker, Billerica, MA).
2.2 Materials and reagents
Analytical grade 95% ethanol, petroleum ether (Pet, 60–90 °C), ethyl acetate (EtOAc), hydrochloric acid (HCl), n-butanol (n-BuOH), acetonitrile (ACN), methanol (MeOH), trifluoroacetic acid (TFA), and ammonia water (NH3·H2O) used for sample extraction and pH-ZRCCC separation were products of Sinopharm Chemical Reagent Limited Company (Shanghai, China). Chromatography grade ACN (Concord Technology (Tianjin) Co. Ltd., Tianjin, China) and formic acid (Kermel Chemical Reagent Co. Ltd., Tianjin, China) were used for HPLC. The deionized water used in this study was prepared by a Direct-Q 8 UV-R water purification system (Millipore, Bedford, MA).Xanthii Fructus was purchased from Bo Zhou herbal medicine market and was identified by Prof. Xu Lingchuan.
2.3 Preparation of crude sample and Fr. 1 sample recovery
Xanthii Fructus (1.5 kg) was weighed and extracted three times with 50% ethanol for 2 h each time, the solid–liquid ratio was 1:14 (w/v). The obtained 21 L of extract was filtered and concentrated at 50 °C. The residue was redissolved to 800 mL and extracted three times with an equal volume of Pet. The lower phase was acidified to pH 2.0 with HCl, then extracted five times with an equal volume of EtOAc. After the concentration of EtOAc phase, the crude sample had a mass of 13.2 g.
For Fr. 1 sample recovery, the effluent of Fr. 1 was collected, concentrated and diluted with 100 mL of water, then acidified to pH 2 with HCl and extracted with equal volume of EtOAc five times. The organic phase was concentrated for further pH-ZRCCC separation.
2.4 Preparation of two-phase solvent system and sample solution for pH-ZRCCC
The two-phase solvent system used for pH-ZRCCC separation was prepared by adding the selected solvents to the separatory funnel in proportion. After equilibrium, upper phase was acidified with TFA as the stationary phase, and lower phase was basified with NH3·H2O as the mobile phase. Both phases were placed into ultrasonic cleaner for degassing 2 min before used.The sample was weighted and dissolved using 8 mL of the acidified upper phase and the same volume of the lower phases without NH3·H2O for pH-ZRCCC separation.
2.5 Separation procedure of pH-ZRCCC
Before separating the sample, the upper phase was pumped in at the flow rate of 25 mL min−1 until the separation columns were filled. Then the sample solution was injected through the sample loop while the rotation speed was adjusted to 800 rpm and the lower phase was pumped in at a flow rate of 2 mL min−1. The effluent was monitored at 254 nm using a UV detector and collected at 6 min intervals. The pH of each fraction was measured using a pH/mV meter. After the separation finished, the residual solvents in columns were pushed out using an air compressor, and the retention rate was calculated as the volume of residual upper phase divided by the volume of residual solvent.2.6 Purification of Fr. 2 sample using semi-preparative HPLC
Before purification, the Fr. 2 sample was dissolved with 5 mL of MeOH and filtrated through a 0.45 μm membrane. The mobile phase was composed of ACN–0.1% aqueous formic acid solution (11:89, v/v). The injection volume was 250 μL, the solvent flow rate was 3 mL min−1 and the UV detection wavelength was at 254 nm.
2.7 HPLC analysis of samples and identifications of purified compounds
The mobile phase for HPLC analysis of samples was composed of ACN (A) – 0.1% formic acid aqueous solution (B) with the gradient of 0–6 min, 5–13% A; 6–15 min, 13% A; 15–25 min, 13–21% A; 25–40 min, 21% A. The solvent flow rate was 1 mL min−1 and the UV detection wavelength was at 330 nm.All purified compounds were identified by HPLC-ESI-QTOF-MS, 1H-NMR and 13C-NMR with tetramethylsilane (TMS) as the internal standard.
3 Results and discussion
3.1 HPLC analysis of samples
The HPLC chromatograms of crude sample and purified compounds are shown in Fig. 2. Based on the peak area normalization at 330 nm, the percentage of compound I in the crude sample is 50.2%, the percentages of compounds II–V in Fr. 1 sample are 30.1% (peak II), 4.2% (peak III), 9.8% (peak IV) and 28.4% (peak V), while compounds VI–VII in Fr. 2 sample account for 69.1% (peak VI) and 21.0% (peak VII), respectively.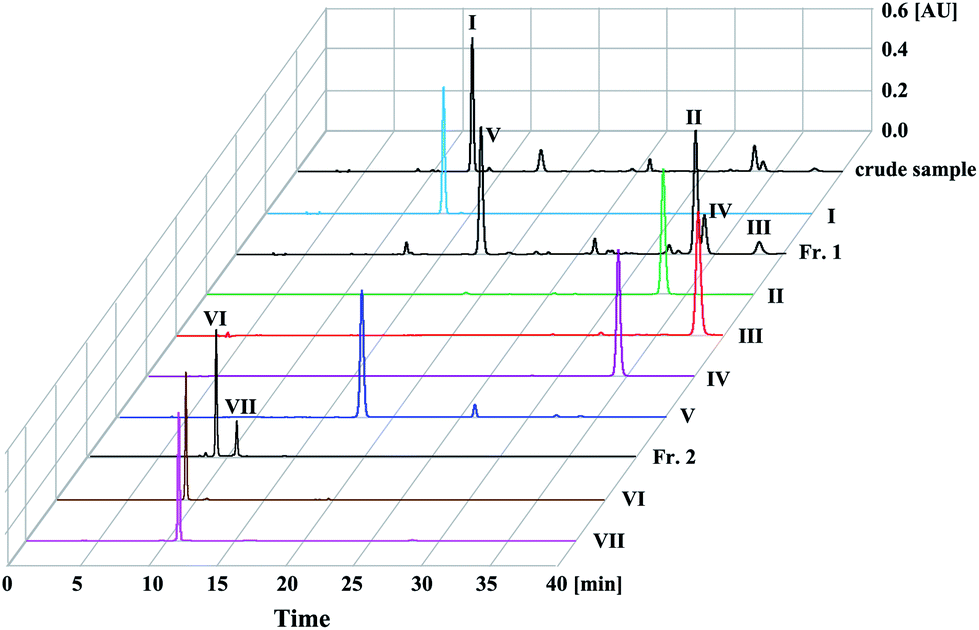 | ||
| Fig. 2 HPLC analysis of the crude sample, Fr. 1, Fr. 2 samples and purified compounds from Xanthii Fructus. Experimental conditions: Compass C18 column (250 mm × 4.6 mm i.d., 5 μm); mobile phase, acetonitrile (A) and 0.1% aqueous formic acid solution (B), gradient: 0–6 min, 5–13% A; 6–15 min, 13% A; 15–25 min, 13–21% A; 25–40 min, 21% A. Column temperature, 25 °C; flow rate, 1.0 mL min; UV detection wavelength, 330 nm. | ||
3.2 Optimization of the pH-ZRCCC conditions
According to our experience of phenolic acids separation,13 we first selected a two-phase solvent system composed of EtOAc–ACN–H2O (4:1:5, v/v/v) with short phenolic acids retention time to separate 1.6 g of the crude sample. 10 mM TFA and 10 mM NH3·H2O were added into upper phase and lower phase, separately. From the pH-ZRCCC chromatogram (Fig. 3(a)) we found that only two compounds (I–II) were separated in 8 h and the separation interval was up to 4 h. However, there were still three target compounds that were not eluted, and the two compounds which appeared in the HPLC chromatogram of crude sample at about ten minutes (Fig. 2) were eluted as a mixture without forming a platform at about 2 hours due to their low content. From this separation we found it was difficult to elute all compounds with one solvent system for the variety of compounds in Xanthii Fructus. Therefore, a segmentation strategy was proposed. We pushed out the remaining solvent in columns after compound I was eluted and recovered them as described in Section 2.3 to obtain 531 mg sample, recorded as Fr. 1, for further pH-ZRCCC separation. The effluent of the mixture eluted at 2 h was collected and concentrated to obtain 11.6 mg sample as Fr. 2, which was further purified using semi-preparative HPLC. The HPLC chromatograms of Fr. 1 and Fr. 2 were shown in Fig. 2. In addition, the effluent of compound I was collected and dried in freeze drier to obtain 178 mg sample with the purity of 98.6%.
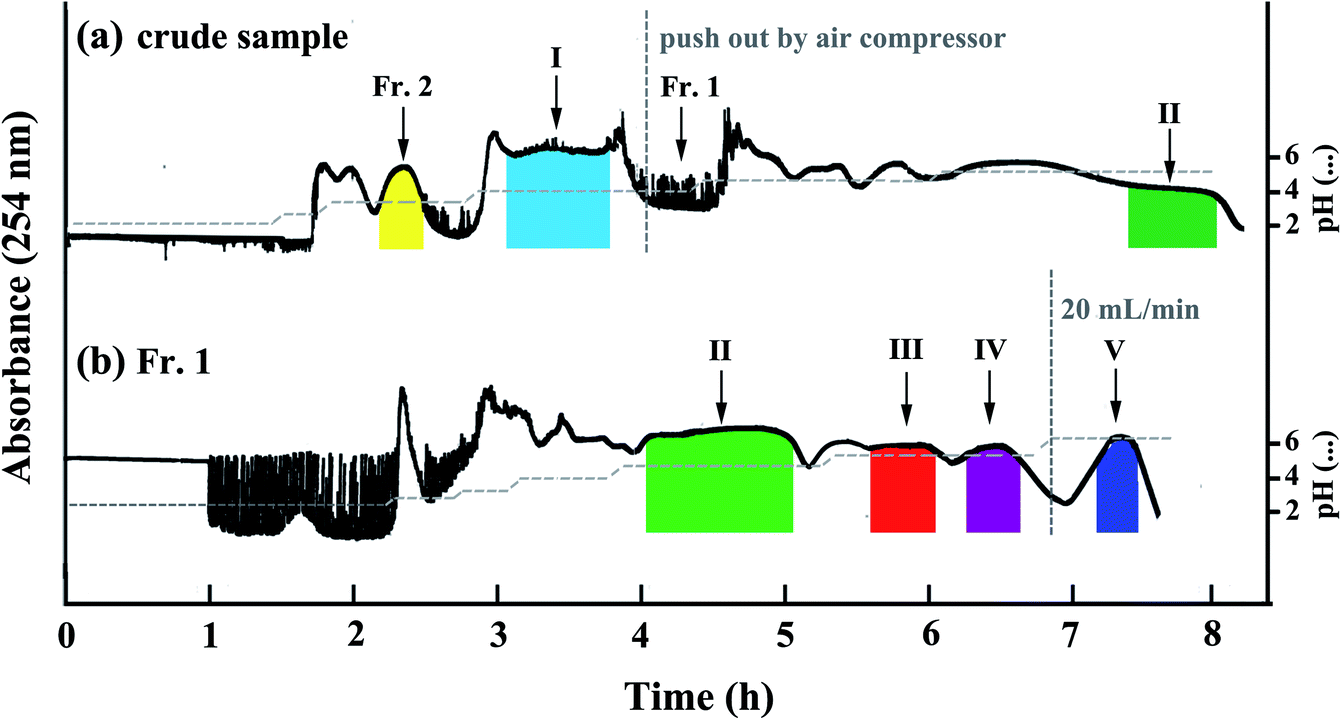 | ||
| Fig. 3 pH-ZRCCC chromatograms of crude sample and Fr. 1 sample from Xanthii Fructus. (a) EtOAc–n-BuOH–H2O (4:1:5, v/v/v) (10 mM TFA as the retainer, 10 mM NH3·H2O as the eluter), crude sample size, 1.6 g, retention rate, 50.0%; (b) EtOAc–H2O (1:1, v/v) (10 mM TFA as the retainer, 20 mM NH3·H2O as the eluter), Fr. 1 sample size, 531 mg, retention rate, 55.4%; experimental conditions: flow rate, 2.0 mL min; revolution speed, 800 rpm; ultraviolet detection wavelength, 254 nm. | ||
For pH-ZRCCC separation of Fr. 1, because the separation of the target compounds could last for a long time using EtOAc–ACN–H2O (4:1:5, v/v/v) (10 mM TFA was added as the retainer and 10 mM NH3·H2O was added as the eluter), a two-phase solvent system which is capable of rapid elution and good resolution of the target compounds is required. It was reported that increasing the concentration of eluter could increase the concentration of compounds on the rectangular platform and shorten retention time, so we increased the concentration of NH3·H2O to 20 mM.15 And according to our previous study,13 addition of ACN in solvent system could result in the reduced resolution of CQAs, so we omitted the ACN and turned the two-phase solvent system into EtOAc–H2O (1:1, v/v) with 10 mM TFA and 20 mM NH3·H2O to purify 531 mg of Fr. 1 sample. After 7 h of separation, the flow rate of mobile phase was increased to 20 mL min−1 to elute the last compound, which had a long retention time. Finally, four irregular rectangular platforms with four pure phenolic acids were observed as shown in Fig. 3(b). After these four parts were dried in a freeze drier, 30.6 mg of compound II, 4.3 mg of compound III, 12.7 mg of compound IV and 34.1 mg of compound V were obtained with the purities of 97.4%, 96.8%, 97.6% and 93.6%, respectively.
Fr. 2 sample (11.6 mg) was separated using semi-preparative HPLC as described in Section 2.6. After dried in freeze drier, 7.5 mg of compound VI and 2.0 mg of compound VII were obtained with the purities of 97.9% and 98.8%. We also determined the purities of these seven compounds using HPLC-ESI-QTOF-MS and the purities of compound I–VII were all above 90%. Purity values of seven compounds are shown in Table 1.
| Compound | Purity | [M + H]+ |
|---|---|---|
| I | 90.2% | 355.08 |
| II | 96.7% | 517.10 |
| III | 97.7% | 517.10 |
| IV | 94.8% | 517.10 |
| V | 91.6% | 181.04 |
| VI | 91.9% | 355.08 |
| VII | 92.9% | 355.08 |
3.3 Identification of compounds
The purified compounds I–VII were identified by comparison of their MS, 1H-NMR and 13C-NMR data (Tables 1–3) with the literature.16–18 These seven compounds were identified as 3-CQA (compound I), 1,5-di-CQA (compound II), 4,5-di-CQA (compound III), 3,5-di-CQA (compound IV), caffeic acid (compound V), 4-CQA (compound VI) and 5-CQA (compound VII), respectively.| Position | Compound | ||||||
|---|---|---|---|---|---|---|---|
| I | II | III | IV | V | VI | VII | |
| 2 | 1.76–2.01 (2H, m) | 2.15–2.45 (2H, m) | 1.99–2.20 (2H, m) | 1.90–2.19 (2H, m) | 2.03–2.25 (2H, m) | 1.84–1.99 (2H, m) | |
| 3 | 5.08 (1H, d, J = 4.0) | 4.03 (1H, s) | 4.13 (1H, s) | 5.18 (1H, m) | 4.27 (1H, s) | 3.84 (1H, d, J = 4.0) | |
| 4 | 3.57–3.58 (1H, m) | 3.54 (1H, m) | 4.91 (1H, dd, J = 16.0, 8.0) | 3.74 (1H, d, J = 8.0) | 3.96 (1H, s) | 3.59 (1H, s) | |
| 5 | 3.93–3.94 (1H, m) | 5.22–5.28 (1H, m) | 5.46–6.52 (1H, m) | 5.27–5.33 (1H, m) | 4.60 (1H, s) | 5.17–5.19 (1H, m) | |
| 6 | 1.76–2.01 (2H, m) | 2.15–2.45 (2H, m) | 1.99–2.09 (2H, m) | 1.90–2.00 (2H, m) | 2.23–2.33 (2H, m) | 1.84–1.99 (2H, m) | |
| 2′ | 7.05 (1H, s) | 7.08 (1H, s) | 7.01 (1H, s) | 7.06 (1H, d, J = 12.0) | 7.02 (1H, s) | 7.02 (1H, s) | 7.02 (1H, s) |
| 5′ | 6.78 (1H, d, J = 8.0) | 6.75 (1H, d, J = 4.0) | 6.73 (1H, d, J = 4.0) | 6.77 (1H, d, J = 8.0) | 6.76 (1H, d, J = 8.0) | 6.76 (1H, d, J = 8.0) | 6.76 (1H, d, J = 8.0) |
| 6′ | 6.99 (1H, d, J = 8.0) | 6.96 (1H, dd, J = 16.0, 8.0) | 6.95 (1H, dd, J = 16.0, 8.0) | 6.98 (1H, s) | 6.94 (1H, d, J = 8.0) | 6.98 (1H, d, J = 8.0) | 6.97 (1H, d, J = 8.0) |
| 7′ | 7.43 (1H, d, J = 16.0) | 7.47 (1H, d, J = 16.0) | 7.46 (1H, d, J = 12.0) | 7.50 (1H, d, J = 8.0) | 7.38 (1H, d, J = 16.0) | 7.41 (1H, d, J = 16.0) | 7.46 (1H, d, J = 16.0) |
| 8′ | 6.16 (1H, d, J = 16.0) | 6.24 (1H, d, J = 16.0) | 6.21 (1H, d, J = 16.0) | 6.25 (1H, s) | 6.17 (1H, d, J = 16.0) | 6.18 (1H, d, J = 16.0) | 6.20 (1H, d, J = 16.0) |
| 2′′ | 7.03 (1H, s) | 7.01 (1H, s) | 7.06 (1H, d, J = 12.0) | ||||
| 5′′ | 6.74 (1H, d, J = 4.0) | 6.71 (1H, d, J = 4.0) | 6.77 (1H, d, J = 8.0) | ||||
| 6′′ | 6.96 (1H, m) | 6.95 (1H, dd, J = 16.0, 8.0) | 6.98 (1H, s) | ||||
| 7′′ | 7.41 (1H, d, J = 16.0) | 7.42 (1H, d, J = 12.0) | 7.46 (1H, d, J = 8.0) | ||||
| 8′′ | 6.18 (1H, d, J = 16.0) | 6.16 (1H, d, J = 16.0) | 6.21 (1H, s) | ||||
| Position | Compound | ||||||
|---|---|---|---|---|---|---|---|
| I | II | III | IV | V | VI | VII | |
| 1 | 74.0 | 81.8 | 76.0 | 73.1 | 80.4 | 73.4 | |
| 2 | 37.7 | 35.2 | 34.2 | 36.8 | 34.8 | 35.7 | |
| 3 | 68.6 | 69.0 | 68.9 | 70.8 | 66.8 | 71.4 | |
| 4 | 70.9 | 72.5 | 71.3 | 69.6 | 67.7 | 71.7 | |
| 5 | 71.4 | 70.8 | 68.6 | 71.7 | 74.7 | 67.9 | |
| 6 | 36.8 | 37.6 | 38.4 | 36.5 | 35.8 | 38.6 | |
| 7 | 175.5 | 173.9 | 176.0 | 177.8 | 173.5 | 176.7 | |
| 1′ | 126.1 | 126.2 | 125.9 | 126.1 | 126.3 | 126.0 | 126.2 |
| 2′ | 115.2 | 115.4 | 115.4 | 115.2 | 115.1 | 115.1 | 115.1 |
| 3′ | 146.0 | 145.4 | 145.9 | 146.1 | 146.1 | 146.0 | 144.9 |
| 4′ | 148.8 | 149.1 | 149.0 | 148.7 | 148.6 | 148.8 | 148.6 |
| 5′ | 116.2 | 116.4 | 116.3 | 116.3 | 116.3 | 116.3 | 116.3 |
| 6′ | 121.8 | 121.7 | 121.8 | 121.7 | 121.4 | 121.7 | 121.5 |
| 7′ | 145.4 | 144.5 | 146.1 | 145.2 | 144.4 | 145.5 | 146.0 |
| 8′ | 114.8 | 114.9 | 114.3 | 115.6 | 116.4 | 115.3 | 115.5 |
| 9′ | 166.2 | 166.7 | 166.6 | 166.9 | 168.8 | 165.8 | 166.5 |
| 1′′ | 126.0 | 125.9 | 126.0 | ||||
| 2′′ | 115.4 | 115.4 | 115.0 | ||||
| 3′′ | 145.4 | 145.9 | 146.1 | ||||
| 4′′ | 148.7 | 149.0 | 148.9 | ||||
| 5′′ | 115.5 | 116.3 | 116.3 | ||||
| 6′′ | 121.1 | 121.8 | 121.5 | ||||
| 7′′ | 144.5 | 146.1 | 145.0 | ||||
| 8′′ | 114.9 | 114.3 | 115.3 | ||||
| 9′′ | 165.5 | 166.6 | 166.6 | ||||
4 Conclusions
In this study, a segmentation strategy using pH-ZRCCC was developed to separate the crude sample of Xanthii Fructus, which contained multiple compounds and therefore cannot be separated in one step. We first used pH-ZRCCC to segment the crude sample into three fractions. Then pH-ZRCCC and semi-preparative HPLC were used for the phenolic acids purifications according to the content of phenolic acids in each fraction of the sample. As a result, seven phenolic acids including 3-CQA, 4-CQA, 5-CQA, 1,5-di-CQA, 4,5-di-CQA, 3,5-di-CQA and caffeic acid were successfully isolated from Xanthii Fructus. Compared with the previously reports of HSCCC separations of phenolic acids,10,11 pH-ZRCCC has greatly improved the separation efficiency due to its high sample loading capacity. Moreover, the purities of phenolic acids isolated by pH-ZRCCC were much higher, so there was no need to use semi-preparative HPLC for further purification. All the results demonstrated that pH-ZRCCC is an efficient method for the preparative separation of phenolic acids, especially isomeric caffeoylquinic acids, from natural products.Funding
This work was supported by the Major Special Projects of National Natural Science Foundation of China (81891014); China Agriculture Research System (CARS-21); and Priority Research Program of the Shandong Academy of Sciences (Lanping Guo).Conflicts of interest
There are no conflicts to declare.References
- Pharmacopoeia Commission of China, The Pharmacopoeia of People's Republic of China, China Medical Science Press, China, Vol. 1, 2015, p. 162 Search PubMed
.
- A. Kamboj and A. K. Saluja, Int. J. Green Pharm., 2010, 4, 129–139 CrossRef
- W. Peng, Q. L. Ming, P. Han, Q. Y. Zhang, Y. P. Jiang, C. J. Zheng, T. Han and L. P. Qin, Phytomedicine, 2014, 21, 824–829 CrossRef CAS
- M. H. Huang, B. S. Wang, C. S. Chiu, S. Amagaya, W. T. Hsieh, S. S. Huang, P. H. Shie and G. J. Huang, J. Ethnopharmacol., 2011, 135, 545–552 CrossRef PubMed
- W. Peng, P. Han, L. Y. Yu, Y. Chen, B. Z. Ye, L. P. Qin, H. L. Xin and T. Han, Rev. Bras. Farmacogn., 2019, 29, 46–53 CrossRef CAS
- T. Han, Q. Y. Zhang, H. Zhang, J. Wen, Y. Wang, B. K. Huang, K. Rahman, H. C. Zheng and L. P. Qin, Anal. Chim. Acta, 2009, 634, 272–278 CrossRef CAS
- H. Jiang, L. Yang, X. D. Xing, M. L. Yan, X. Y. Guo, B. Y. Yang, Q. H. Wang and H. X. Kuang, J. Pharm. Biomed. Anal., 2018, 153, 117–125 CrossRef CAS
- J. Chen, S. Mangelinckx, L. Ma, Z. T. Wang, W. L. Li and D. N. Kimpe, Fitoterapia, 2014, 99, 1–6 CrossRef CAS PubMed
- X. W. Jiang, J. P. Bai, Q. Zhang, X. L. Hu, X. Tian, J. Zhu, J. Liu, W. H. Meng and Q. C. Zhao, Phytochem. Lett., 2016, 15, 159–163 CrossRef CAS
- Z. J. Zheng, X. Wang, P. L. Liu, M. Li, H. J. Dong and X. G. Qiao, Molecules, 2018, 23, 429 CrossRef
- W. Guo, L. Wang, Y. Gao, B. N. Zhao, D. J. Wang, W. J. Duan and Z. Y. Yu, J. Chromatogr. B, 2015, 981–982, 27–32 CrossRef CAS
- Y. Ito, J. Chromatogr. A, 2005, 1065, 145–168 CrossRef CAS
- T. Y. Ma, H. J. Dong, H. Lu, H. Q. Zhao, L. P. Guo and X. Wang, J. Chromatogr. A, 2018, 1578, 61–66 CrossRef CAS
- S. Q. Tong, J. Z. Yan and Y. X. Guan, J. Chromatogr. A, 2008, 1212, 48–53 CrossRef CAS
- X. Wang, Y. L. Geng, F. W. Li, X. G. Shi and J. H. Liu, J. Chromatogr. A, 2006, 1115, 267–270 CrossRef CAS
- J. Q. Yu, Z. P. Wang, H. Zhu, G. Li and X. Wang, Acta Pharmacol. Sin., 2016, 51, 1110–1116 Search PubMed
- D. D. Huang, J. L. Li, F. Y. Yao, G. H. Huang, D. Xue and L. N. Sun, Chin. J. Exp. Tradit. Med. Formulae, 2014, 20, 100–104 CAS
- F. Y. Ni, L. Liu, Y. L. Song, X. J. Wang, Y. W. Zhao, W. Z. Huang, Z. Z. Wang and W. Xiao, China J. Chin. Mater. Med., 2015, 40, 269–274 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra06969k |
| This journal is © The Royal Society of Chemistry 2019 |