
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biofunctional few-layer metal dichalcogenides and related heterostructures produced by direct aqueous exfoliation using phospholipids†
Aled T. Williamsa,
Roberto Donno b,
Nicola Tirellib and
Robert A. W. Dryfe*ac
b,
Nicola Tirellib and
Robert A. W. Dryfe*ac
aSchool of Chemistry, University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: robert.dryfe@manchester.ac.uk
bLaboratory of Polymers and Biomaterials, Fondazione Istituto Italiano di Tecnologia, Via Morego, 30, Genoa, Italy
cNational Graphene Institute, University of Manchester, Oxford Road, Manchester, M13 9PL, UK
First published on 13th November 2019
Abstract
We report a novel, inexpensive and green method for preparing aqueous dispersions of various biofunctional transition-metal dichalcogenides (MoS2, WS2, TiS2 and MoSe2) and their related heterostructures directly via ultrasonic exfoliation mediated by the presence of phospholipids. The dispersions predominantly consist of few-layer flakes coated with 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), as confirmed by Raman, photoluminescence and X-ray photoelectron spectroscopies. The phospholipid coating renders the flakes biofunctional, which coupled with the unique properties of transition-metal dichalcogenides and their heterostructures, suggests this method will have great potential in biological applications.
Transition-metal dichalcogenides (TMDCs) and other species of layered inorganic compounds are currently attracting much interest as the next generation of two-dimensional (2D) materials beyond graphene.1 These materials, notably MoS2, WS2 and hexagonal boron nitride (hBN), have already found applications in fields such as optoelectronics and energy storage due to their unique physical properties.2–6 Structurally, TMDCs are 2D layers of transition metal atoms, M, each covalently bound to two chalcogen atoms, X, and held together by van der Waals forces. The combination of metal and chalcogen influences the electronic properties of the material, for example TiS2 is a semimetal whilst MoS2 is a semiconductor in its naturally occurring, 2H, phase.7 In addition to MX2 compounds, similar structures are also embodied by hBN, Bi2Te3, transition-metal oxides, silicene, germanene and phosphorene (exfoliated black phosphorus) providing a range of new 2D materials for exploitation based on their individual attributes.1,8 There is also growing interest in hetero-structures formed from these materials, i.e. stacked combinations of different 2D species, offering further diversity in the properties that can be harvested from these layered materials.2,9–11
Solution processing of 2D materials is essential for industrial applications12 where the time-intensive and costly approaches of micromechanical cleavage and chemical vapour deposition hinder the commercial viability of some technologies. Several liquid-phase exfoliation methods have been reported over recent years, the majority of which have been based on earlier methods applied to the exfoliation of graphite.13,14 Notably, direct sonication of bulk powders in high boiling-point (HBP) solvents such as N-methyl-2-pyrrolidone (NMP) has been shown to produce monolayer to few-layer (layer number, n ≤ 3) dispersions of TMDCs and other layered inorganic materials with reasonable stabilities and concentrations (up to 40 mg mL−1).15,16 However, direct sonication using HBP solvents raises obvious economic, environmental and safety concerns and furthermore, HBP solvent residues in thin films fabricated from such dispersions can be difficult to remove, which can be detrimental to subsequent applications in electronics.17 Alternative preparation techniques, such as chemical and electrochemical Li-intercalation18–20 and various surface functionalization strategies using organic molecules,19,21 often involve multiple steps and undesirable reaction conditions.
Aqueous exfoliation methods can address many of these issues. However, successful liquid-phase exfoliation requires that the enthalpy of mixing per unit volume associated with the dispersal of the 2D flakes is close to zero, and it has been shown that for TMDCs22 optimal solvents are characterized by surface tensions in the region of 40 mJ m−2, with water falling outside this range at 72.75 mJ m−2 (at 20 °C).23 However, aqueous dispersions of hydrophobic flakes can be stabilized electrostatically or sterically, and a number of recent reports detail the use of surfactants or polymers in facilitating the aqueous exfoliation of layered inorganic materials via the application of ultrasonic energy.24–28 Herein, we report on a new exfoliation method, where phospholipids are used as electrostatic stabilizing agents in preparing aqueous dispersions of TMDCs directly via sonication of the bulk materials. Phospholipids have the additional advantage of imparting bio-compatibility to the 2D materials. We also demonstrate that the mixing and subsequent sonication of these dispersions result in the formation of hetero-structured materials, whose optoelectronic characteristics can be modified by the respective ratios of the parent materials.
Briefly, our method involves sonicating MX2 powders in ultra-pure water containing phospholipid for 12 h, followed by a centrifugation to purify the dispersions from large aggregates (full details can be found in the ESI†). We have previously described how this procedure can be applied to graphene exfoliation,29 where we have shown that the amount of 2D-material dispersed was dependent on the phospholipid concentration and the fluidity of the phospholipid aliphatic chains. Herein, we report that the same method can be used to produce meta-stable aqueous dispersions for each TMDC tested, namely MoS2, WS2, TiS2 and MoSe2, as well as hBN. For the dispersions characterised in this study, MX2 and hBN powders were sonicated in ultra-pure water with 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) at a concentration of 0.2 mg mL−1, resulting in dispersed concentrations in the range of 0.01–0.1 mg mL−1, depending on the 2D material used. DOPC was selected as the lipid of choice on the basis of our graphene dispersion work,29 as the fluidity of this phospholipid chain was shown to impart a good level of dispersion stability compared to other phospholipids. Thermogravimetric analysis (TGA) performed on freeze-dried dispersions of MoS2, WS2 and TiS2 suggest that the method typically produces fresh dispersions containing 10–20% weight of dispersed 2D-material (refer to Fig. S1 in the ESI†), comparable to that observed for graphene dispersions produced by the same method.29
Ultraviolet-visible (UV-vis) spectroscopy confirmed the presence of dispersed few-layer MX2 species, with the following characteristic major absorption peaks: 393, 450, 608 and 665 nm for MoS2;3,30,31 412, 697 and 805 nm for MoSe2;3,32 593 nm for TiS2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 33 and 624 nm for WS2.3,30 No absorption peaks could be discerned from the Rayleigh scattering in the spectrum for hBN. The spectra are presented in Fig. 1A together with photographs of the dispersions.
33 and 624 nm for WS2.3,30 No absorption peaks could be discerned from the Rayleigh scattering in the spectrum for hBN. The spectra are presented in Fig. 1A together with photographs of the dispersions.
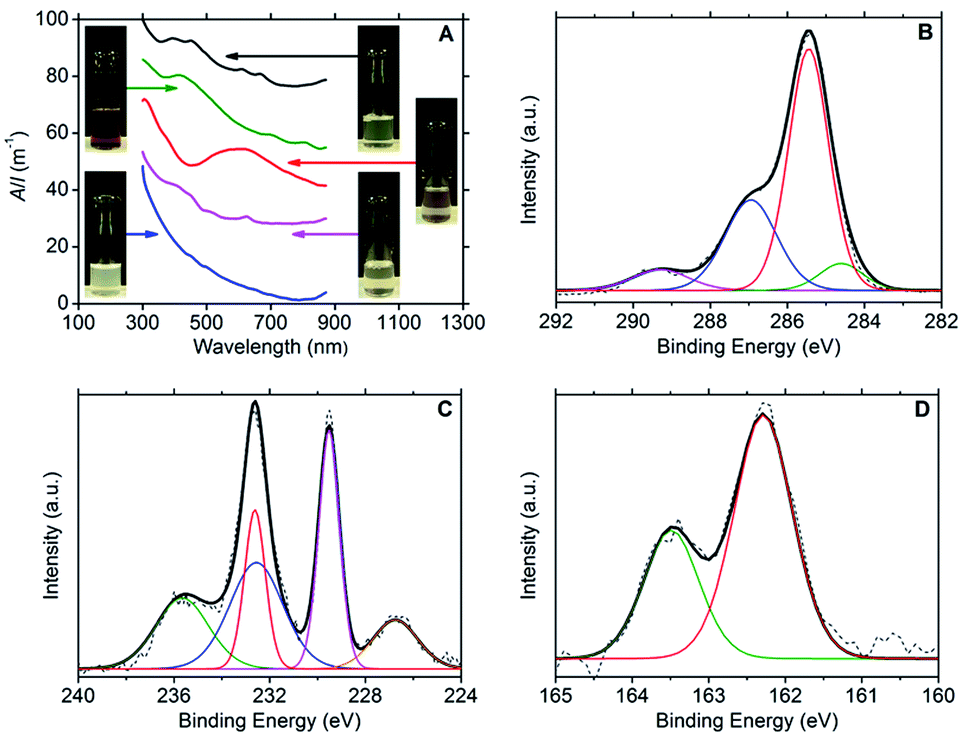 | ||
Fig. 1 (A) UV-vis absorbance spectra of the aqueous DOPC/2D-material dispersions (from top to bottom): MoS2 (black line), MoSe2 (green line), TiS2 (red line), WS2 (purple line) and hBN (blue line). The stacked spectra are in proportion to each other. (B) C1s XPS spectrum of a DOPC/MoS2 dispersion drop cast onto Si/SiO2 substrate. In the fitting, the green line corresponds to the C–C sp2 peak, the red line to the C–C sp3 peak, the blue line to the C–O/C–N peak and the purple line to the O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O peak. (C) Mo3d XPS spectrum of a DOPC/MoS2 dispersion drop cast onto Si/SiO2 substrate. In the fitting, the orange line corresponds to the S2s peak, the purple line to the Mo4+3d5/2 peak, the red line to the Mo4+3d3/2 peak, the blue line to the Mo6+3d5/2 peak and the green line to the Mo6+3d3/2 peak. (D) S2p XPS spectrum of a DOPC/MoS2 dispersion drop cast onto Si/SiO2 substrate. The fitting is for the S2p doublet, with the red line depicting the S2p3/2 peak and the green line the S2p1/2 peak. O peak. (C) Mo3d XPS spectrum of a DOPC/MoS2 dispersion drop cast onto Si/SiO2 substrate. In the fitting, the orange line corresponds to the S2s peak, the purple line to the Mo4+3d5/2 peak, the red line to the Mo4+3d3/2 peak, the blue line to the Mo6+3d5/2 peak and the green line to the Mo6+3d3/2 peak. (D) S2p XPS spectrum of a DOPC/MoS2 dispersion drop cast onto Si/SiO2 substrate. The fitting is for the S2p doublet, with the red line depicting the S2p3/2 peak and the green line the S2p1/2 peak. | ||
Hydrodynamic diameter values obtained via dynamic light scattering (DLS) measurements are presented in Table 1, providing rough estimates (between tens and hundreds of nm, due to the approximations in the technique) of colloid size which were found to be comparable to those observed for graphene dispersions produced by the same method.29 The dispersed TMDCs were characterized by negative ζ potential values at neutral pH (see Table 1), although smaller in magnitude to those observed for the graphene dispersions (−34 mV). Shorter-term stability is expected as a result of this observation and this is confirmed by comparison of the time-dependent reductions in optical density presented in Table 1. It is worth noting that the TiS2 dispersions, approximately 12 h after preparation, produce a strong sulphurous odour: it is known that this material is prone to oxidation in aqueous solution, most likely via the following reaction:34
| TiS2 + xH2O → TiS2−xOx + xH2S |
| Material | ζ (mV) | D (nm) | ODR day 10a | ODR day 20a | ODR day 30a | ODR day 50a |
|---|---|---|---|---|---|---|
| a Optical density measured at A608 (MoS2), A805 (MoSe2), A565 (TiS2), A624 (WS2), A300 (hBN) and A660 (graphene), where subscripts are wavelengths in nanometres. Graphene data at A660 taken from ref. 29.b Optical density data for graphene measured at A660 at time points: 22, 41 and 56 days. | ||||||
| MoS2 | −20 | 142 | 8% | 16% | 32% | 71% |
| MoSe2 | −28 | 164 | 14% | 23% | 34% | 87% |
| TiS2 | −19 | 495 | 56% | 69% | 81% | 92% |
| WS2 | −15 | 255 | 14% | 29% | 51% | 85% |
| hBN | −23 | 295 | 70% | 74% | 80% | 85% |
| Graphene | −34 | 190 | — | 11%b | 25%b | 43%b |
As the hBN dispersions were found to precipitate at a significantly faster rate to the other 2D materials, no further characterisation work was conducted on hBN dispersions as part of this study.
X-ray photoelectron spectroscopy (XPS) was used to confirm the presence of DOPC and MX2 in the dispersions; refer to Fig. 1B–D (and Fig. S2–S4 in the ESI†). For DOPC, the C1s spectrum for all samples (dispersions of MoS2, MoSe2, WS2 and TiS2) showed peaks representatives of DOPC at average binding energies of 284.5 eV (C–C sp2), 285.3 eV (C–C sp3), 286.9 eV (combined signal for C–O and C–N) and 289.2 eV (O–CO).29 Furthermore, the phosphorous and nitrogen peaks were found to be present at similar percentage atomic concentrations appearing at average binding energies of 134.0 eV and 134.9 eV (P2p3/2 and P2p1/2) and 402.9 eV respectively (N1s). For MoS2, the Mo3d3/2 and Mo3d5/2 doublet peaks were located at 232.6 eV and 229.5 eV respectively, with the S2s peak at 226.7 eV and the S2p doublet at 163.5 eV and 162.3 eV (S2p1/2 and S2p3/2). These binding energies are indicative of the expected Mo4+ and S2− chemical states.35–38 However, the presence of a peak at 235.6 eV is indicative of the Mo3d3/2 doublet peak for Mo6+, and fitting reveals the complementary Mo3d5/2 peak at 232.5 eV, which strongly suggests that MoO3 is present at approximately the same concentration as MoS2 (percentage atomic concentrations of 47% Mo4+ and 53% Mo6+).37–40 The Mo6+ state was more heavily detected for the MoSe2 dispersions (percentage atomic concentrations of 11% Mo4+ and 89% Mo6+). The Mo3d3/2 and Mo3d5/2 doublet peaks for Mo6+ appear at 236.1 eV and 232.9 eV respectively, the same doublet peaks for Mo4+ being found at lower intensities at 232.5 eV and 229.4 eV, with the Se3d3/2 and Se3d5/2 doublet peaks appearing at 56.6 eV and 55.8 eV.36 For WS2, two chemical states were also detected for tungsten, W4+ and W6+. The W4f5/2 and W4f7/2 doublet peaks expected for WS2 are observed at 34.6 eV and 32.7 eV with the S2p doublet peaks appearing at 163.4 eV and 162.2 eV (S2p1/2 and S2p3/2). The W4f5/2 and W4f7/2 doublet peaks characteristic of the W6+ chemical state are seen at 38.1 eV and 36.0 eV, and represent the dominant binding energies of the W4f signal (percentage atomic concentrations of 12% W4+ and 88% W6+),35,36,41 which coupled with the weak S2p signal suggests that the samples contained significant amounts of WO3. For TiS2, the Ti2p1/2 and Ti2p3/2 doublet peaks appear at 464.8 eV and 459.1 eV respectively, with the S2p doublet peaks appearing at 165.2 eV and 163.9 eV (S2p1/2 and S2p3/2). These binding energies are indicative of the expected Ti4+ and S2− chemical states.42,43 However, as with that observed for the WS2 sample, a weak S2p signal intensity was observed when compared to the Ti2p signal intensity. This observation, in conjunction with the dispersion stability data, suggests that oxidised titanium is also present in significant quantity. The partial oxidation of the TMDCs, detected by the XPS, could explain the lower long-term stability of the DOPC-stabilised dispersions relative to the graphene sample (see Table 1). Consistent with this, it is noted that the TiS2 sample has the lowest level of dispersion stability.
Raman spectroscopy was used to further characterise the DOPC/MX2 dispersions and to confirm the presence of few-layer flakes. Analysis was performed on drop-cast samples using Si/SiO2 substrates. For MoS2, spectra diagnostic of few-layer flakes predominated with the A1g and E12g typically appearing at 406 cm−1 and 382 cm−1 respectively, separated by 24 cm−1, yielding maximum photoluminescent (PL) emission at 675 nm (1.84 eV), which is consistent with literature values for few-layer MoS2.44–49 Similarly, the spectra measured for the WS2 samples were found to be representative of few-layer material,47–52 with A1g and E12g appearing at 419 cm−1 and 351 cm−1 respectively, separated by 68 cm−1 and yielding maximum photoluminescent (PL) emission at 633 nm (1.96 eV). Few-layer flakes were also detected for MoSe2 and TiS2. For MoSe2, this was confirmed by the following representative peaks: 351 cm−1 (B12g), 285 cm−1 (E12g) and 241 cm−1 (A1g)48,49,53 and for TiS2: 377 cm−1 (A2u), 333 cm−1 (A1g) and 233 cm−1 (Eg),33,49,54 with the A1g:A2u peak intensity ratio at approximately 2.0 (compare to a value of 1.6 measured for the bulk powder starting-material).33 Weaker PL emissions were observed for the samples derived from the MoSe2 and TiS2 dispersions, as compared to those from MoS2 and WS2 dispersions. Fig. 2A and S5–S8 in the ESI† give typical examples of the Raman and PL spectra measured.
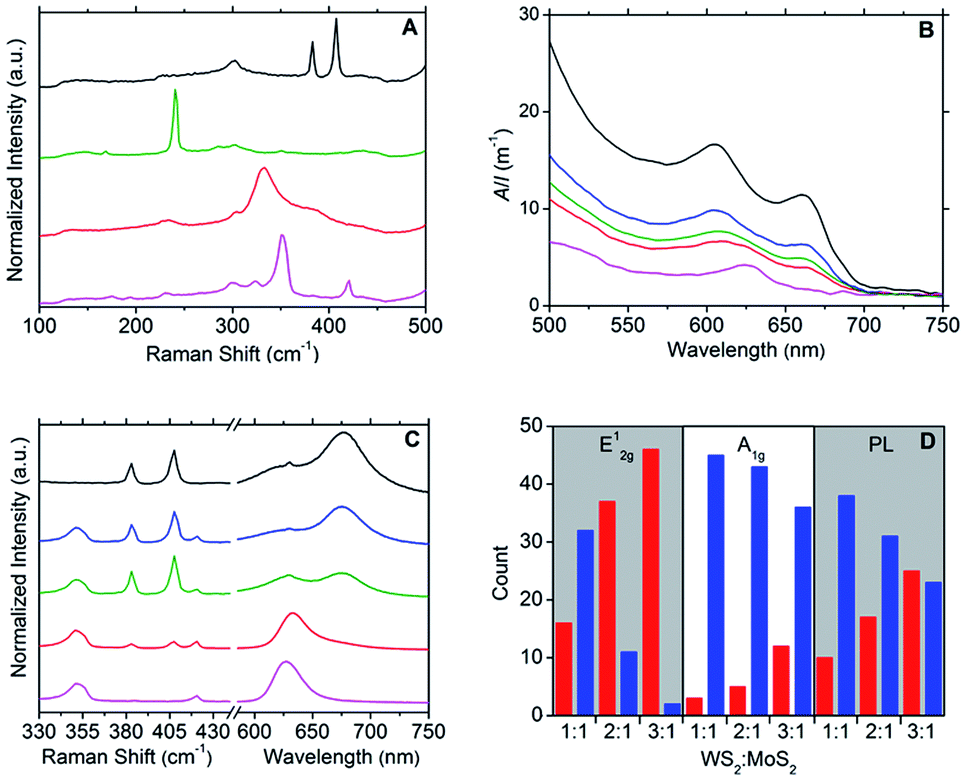 | ||
| Fig. 2 (A) Raman spectra (532 nm excitation) of the DOPC/MX2 dispersions drop cast onto Si/SiO2 substrates, corresponding (from top to bottom) to MoS2 (black line), MoSe2 (green line), TiS2 (red line) and WS2 (purple line). (B) UV-vis spectra of the following aqueous DOPC/2D-material dispersions (from top to bottom): MoS2 (black line), 1:1 WS2:MoS2 heterostructures (blue line), 2:1 WS2:MoS2 heterostructures (green line), 3:1 WS2:MoS2 heterostructures (red line) and WS2 (purple line). (C) Typical Raman and PL spectra (532 nm excitation) measured for the following DOPC/2D-material dispersions drop cast onto Si/SiO2 substrates (from top to bottom): MoS2 (black line), 1:1 WS2:MoS2 heterostructures (blue line), 2:1 WS2:MoS2 heterostructures (green line), 3:1 WS2:MoS2 heterostructures (red line) and WS2 (purple line). (D) Histograms showing the relative proportions of dominant peak intensities in the Raman (E12g and A1g) and PL spectra (λmax) of aqueous DOPC-mediated dispersions of 1:1, 2:1 and 3:1 WS2:MoS2 heterostructures. Instances where the peak intensities arising from WS2 are greater than those from MoS2 are binned on the left-hand side (red columns) with the converse case binned on the right-hand side (blue columns); a bin size of 48 measurements (at different sample spots) was used in all instances. | ||
Finally, atomic force microscopy (AFM) performed on drop-cast samples of DOPC/MoS2 dispersions (clean mica substrates were used) showed structures similar to those observed for graphene dispersions produced by the same method,29 with an average height of 40 nm. The AFM height images showed objects characterized by “steps” with a height of 5 nm (or multiple of 5 nm), which is a thickness comparable to that of a DOPC bilayer;55–58 refer to Fig. S9 in the ESI† for further details.
Supplemental to the novel properties offered by true 2D materials and their few-layer counterparts, mixed van der Waals stacking of different 2D materials to form heterostructures, endow few-layer materials with scope to impart bespoke properties.2,9–11,38,59 In addition to our method detailed above for the production of aqueous DOPC/MX2 dispersions, we also present a fast and single-step method to prepare aqueous DOPC/heterostructure dispersions. Briefly, the method involves mixing the desired parent DOPC/MX2 dispersions at specific volume ratios, after which the mixtures are sonicated for 10 minutes to facilitate the de-stacking and subsequent re-stacking of the parent layers into hetero-layered stacks. The method was used to prepare aqueous DOPC/WS2:MoS2 dispersions, where the resulting optoelectronic properties were found to be tuneable to the relative quantities of the parent dispersions used. UV-vis spectroscopy (Fig. 2B) was used as a simple screening tool for the selection of appropriate volume ratios of DOPC/WS2 (x) and DOPC/MoS2 (y) dispersions to produce heterostructure dispersions with desired optoelectronic properties. For example, the UV-vis spectrum of the DOPC/WS2:MoS2 dispersion prepared using the x:y ratio at 2:1 was found to be intermediate in character between those of the parent dispersions, with flakes (deposited on Si/SiO2 substrates) also displaying intermediate character with regard to Raman and PL response; refer to Fig. 2C. Further illustration of this optoelectronic tuning is shown via the histograms presented in Fig. 2D, which are based on Raman and PL data measured from a total of 48 flakes per DOPC/WS2:MoS2 dispersion (x:y ratio at 1:1, 2:1 and 3:1). The measured flakes were categorised into two conditions: one where the Raman (A1g and E12g) and PL (λmax) intensities were greater for peaks emanating from WS2 (approximately 419 cm−1 for A1g, 350 cm−1 for E12g and 633 nm for PL) and the other where the intensities were greater for peaks emanating from MoS2 (approximately 407 cm−1 for A1g, 383 cm−1 for E12g and 675 nm for PL). As expected, the intensities of the peaks arising from WS2 become greater as x increases and as such, the findings show that manipulation of the x:y volume ratio allows for the tuning of the predominant optoelectronic properties of heterostructure flakes.
Conclusions
In summary, a simple method has been presented to produce aqueous dispersions of phospholipid-coated few-layer transition-metal dichalcogenides (MoS2, WS2, TiS2 and MoSe2) via single-step exfoliation of the bulk starting materials in water. Furthermore, the resulting dispersions can be subsequently mixed at pre-defined volume ratios to produce heterostructured dispersions thereby allowing for the tuning of the optoelectronic properties. The phospholipid coating renders the flakes biofunctional, which coupled with the unique properties of transition-metal dichalcogenides, indicates the great potential of this method for use in biological applications, for example in vivo sensing or drug transport.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We thank Dr Andrew Thomas for XPS measurements. The authors gratefully acknowledge the Faculty of Medical and Human Sciences of The University of Manchester, and the EPSRC Programme Grant “2DHealth” (grant reference EP/P00119X/1) for funding, and the Bio-AFM facility for technical support.Notes and references
- S. Z. Butler, S. M. Hollen, L. Y. Cao, Y. Cui, J. A. Gupta, H. R. Gutierrez, T. F. Heinz, S. S. Hong, J. X. Huang, A. F. Ismach, E. Johnston-Halperin, M. Kuno, V. V. Plashnitsa, R. D. Robinson, R. S. Ruoff, S. Salahuddin, J. Shan, L. Shi, M. G. Spencer, M. Terrones, W. Windl and J. E. Goldberger, ACS Nano, 2013, 7, 2898–2926 CrossRef CAS PubMed.
- X. P. Hong, J. Kim, S. F. Shi, Y. Zhang, C. H. Jin, Y. H. Sun, S. Tongay, J. Q. Wu, Y. F. Zhang and F. Wang, Nat. Nanotechnol., 2014, 9, 682–686 CrossRef CAS PubMed.
- Q. H. Wang, K. Kalantar-Zadeh, A. Kis, J. N. Coleman and M. S. Strano, Nat. Nanotechnol., 2012, 7, 699–712 CrossRef CAS.
- M. L. Tsai, S. H. Su, J. K. Chang, D. S. Tsai, C. H. Chen, C. I. Wu, L. J. Li, L. J. Chen and J. H. He, ACS Nano, 2014, 8, 8317–8322 CrossRef CAS.
- E. G. D. Firmiano, A. C. Rabelo, C. J. Dalmaschio, A. N. Pinheiro, E. C. Pereira, W. H. Schreiner and E. R. Leite, Adv. Energy Mater., 2014, 4, 1301380 CrossRef.
- G. H. Lee, Y. J. Yu, X. Cui, N. Petrone, C. H. Lee, M. S. Choi, D. Y. Lee, C. Lee, W. J. Yoo, K. Watanabe, T. Taniguchi, C. Nuckolls, P. Kim and J. Hone, ACS Nano, 2013, 7, 7931–7936 CrossRef CAS PubMed.
- M. Chhowalla, H. S. Shin, G. Eda, L. J. Li, K. P. Loh and H. Zhang, Nat. Chem., 2013, 5, 263–275 CrossRef.
- R. Mas-Balleste, C. Gomez-Navarro, J. Gomez-Herrero and F. Zamora, Nanoscale, 2011, 3, 20–30 RSC.
- C. M. Huang, S. F. Wu, A. M. Sanchez, J. J. P. Peters, R. Beanland, J. S. Ross, P. Rivera, W. Yao, D. H. Cobden and X. D. Xu, Nat. Mater., 2014, 13, 1096–1101 CrossRef CAS.
- H. Fang, C. Battaglia, C. Carraro, S. Nemsak, B. Ozdol, J. S. Kang, H. A. Bechtel, S. B. Desai, F. Kronast, A. A. Unal, G. Conti, C. Conlon, G. K. Palsson, M. C. Martin, A. M. Minor, C. S. Fadley, E. Yablonovitch, R. Maboudian and A. Javey, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 6198–6202 CrossRef CAS PubMed.
- H. Wang, F. C. Liu, W. Fu, Z. Y. Fang, W. Zhou and Z. Liu, Nanoscale, 2014, 6, 12250–12272 RSC.
- D. J. Finn, M. Lotya, G. Cunningham, R. J. Smith, D. McCloskey, J. F. Donegan and J. N. Coleman, J. Mater. Chem. C, 2014, 2, 925–932 RSC.
- Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Y. Sun, S. De, I. T. McGovern, B. Holland, M. Byrne, Y. K. Gun'ko, J. J. Boland, P. Niraj, G. Duesberg, S. Krishnamurthy, R. Goodhue, J. Hutchison, V. Scardaci, A. C. Ferrari and J. N. Coleman, Nat. Nanotechnol., 2008, 3, 563–568 CrossRef CAS PubMed.
- U. Khan, A. O'Neill, M. Lotya, S. De and J. N. Coleman, Small, 2010, 6, 864–871 CrossRef CAS.
- J. N. Coleman, M. Lotya, A. O'Neill, S. D. Bergin, P. J. King, U. Khan, K. Young, A. Gaucher, S. De, R. J. Smith, I. V. Shvets, S. K. Arora, G. Stanton, H. Y. Kim, K. Lee, G. T. Kim, G. S. Duesberg, T. Hallam, J. J. Boland, J. J. Wang, J. F. Donegan, J. C. Grunlan, G. Moriarty, A. Shmeliov, R. J. Nicholls, J. M. Perkins, E. M. Grieveson, K. Theuwissen, D. W. McComb, P. D. Nellist and V. Nicolosi, Science, 2011, 331, 568–571 CrossRef CAS.
- A. O'Neill, U. Khan and J. N. Coleman, Chem. Mater., 2012, 24, 2414–2421 CrossRef.
- T. C. Hernandez, A. C. F. Blanco, A. T. Williams, M. Velicky, H. V. Patten, A. Colina and R. A. W. Dryfe, Electroanalysis, 2015, 27, 1026–1034 CrossRef.
- J. Zheng, H. Zhang, S. H. Dong, Y. P. Liu, C. T. Nai, H. S. Shin, H. Y. Jeong, B. Liu and K. P. Loh, Nat. Commun., 2014, 5, 2995 CrossRef.
- X. Huang, Z. Y. Zeng and H. Zhang, Chem. Soc. Rev., 2013, 42, 1934–1946 RSC.
- R. A. Gordon, D. Yang, E. D. Crozier, D. T. Jiang and R. F. Frindt, Phys. Rev. B, 2002, 65, 125407 CrossRef.
- L. Zhou, B. Z. He, Y. Yang and Y. G. He, RSC Adv., 2014, 4, 32570–32578 RSC.
- G. Cunningham, M. Lotya, C. S. Cucinotta, S. Sanvito, S. D. Bergin, R. Menzel, M. S. P. Shaffer and J. N. Coleman, ACS Nano, 2012, 6, 3468–3480 CrossRef CAS PubMed.
- N. B. Vargaftik, B. N. Volkov and L. D. Voljak, J. Phys. Chem. Ref. Data, 1983, 12, 817–820 CrossRef CAS.
- R. J. Smith, P. J. King, M. Lotya, C. Wirtz, U. Khan, S. De, A. O'Neill, G. S. Duesberg, J. C. Grunlan, G. Moriarty, J. Chen, J. Z. Wang, A. I. Minett, V. Nicolosi and J. N. Coleman, Adv. Mater., 2011, 23, 3944–3948 CrossRef CAS.
- Y. G. Yao, L. Tolentino, Z. Z. Yang, X. J. Song, W. Zhang, Y. S. Chen and C. P. Wong, Adv. Funct. Mater., 2013, 23, 3577–3583 CrossRef CAS.
- M. D. J. Quinn, N. H. Ho and S. M. Notley, ACS Appl. Mater. Interfaces, 2013, 5, 12751–12756 CrossRef CAS PubMed.
- L. Guardia, J. I. Paredes, R. Rozada, S. Villar-Rodil, A. Martinez-Alonso and J. M. D. Tascon, RSC Adv., 2014, 4, 14115–14127 RSC.
- A. Gupta, V. Arunachalam and S. Vasudevan, J. Phys. Chem. Lett., 2015, 6, 739–744 CrossRef CAS.
- A. T. Williams, R. Donno, N. Tirelli and R. A. W. Dryfe, RSC Adv., 2018, 8, 19220–19225 RSC.
- T. P. Nguyen, W. Sohn, J. H. Oh, H. W. Jang and S. Y. Kim, J. Phys. Chem. C, 2016, 120, 10078–10085 CrossRef CAS.
- I. Bilgin, F. Liu, A. Vargas, A. Winchester, M. K. L. Man, M. Upmanyu, K. M. Dani, G. Gupta, S. Talapatra, A. D. Mohite and S. Kar, ACS Nano, 2015, 9, 8822–8832 CrossRef CAS PubMed.
- N. Berahim, I. S. Amiri, T. Anwar, S. R. Azzuhri, M. N. S. Mohd Nasir, R. Zakaria, W. Y. Chong, C. K. Lai, S. H. Lee, H. Ahmad, M. A. Ismail and P. Yupapin, Results Phys., 2019, 12, 7–11 CrossRef.
- P. C. Sherrell, K. Sharda, C. Grotta, J. Ranalli, M. S. Sokolikova, F. M. Pesci, P. Palczynski, V. L. Bemmer and C. Mattevi, ACS Omega, 2018, 3, 8655–8662 CrossRef CAS PubMed.
- E. Long, S. O'Brien, E. A. Lewis, E. Prestat, C. Downing, C. S. Cucinotta, S. Sanvito, S. J. Haigh and V. Nicolosi, npj 2D Mater. Appl., 2017, 1, 22 CrossRef.
- M.-H. Chiu, C. Zhang, H.-W. Shiu, C.-P. Chuu, C.-H. Chen, C.-Y. S. Chang, C.-H. Chen, M.-Y. Chou, C.-K. Shih and L.-J. Li, Nat. Commun., 2015, 6, 7666 CrossRef CAS.
- J. Guo, Y. Shi, X. Bai, X. Wang and T. Ma, J. Mater. Chem. A, 2015, 3, 24397–24404 RSC.
- G. Tai, T. Zeng, J. Yu, J. Zhou, Y. You, X. Wang, H. Wu, X. Sun, T. Hu and W. Guo, Nanoscale, 2016, 8, 2234–2241 RSC.
- Z. Lin, B. R. Carvalho, E. Kahn, R. Lv, R. Rao, H. Terrones, M. A. Pimenta and M. Terrones, 2D Materials, 2016, 3, 022002 CrossRef.
- H. Nan, Z. Wang, W. Wang, Z. Liang, Y. Lu, Q. Chen, D. He, P. Tan, F. Miao, X. Wang, J. Wang and Z. Ni, ACS Nano, 2014, 8, 5738–5745 CrossRef CAS.
- V. Madhavi, P. Kondaiah, S. S. Rayudu, O. M. Hussain and S. Uthanna, Mater. Express, 2013, 3, 135–143 CrossRef CAS.
- K. M. McCreary, A. T. Hanbicki, G. G. Jernigan, J. C. Culbertson and B. T. Jonker, Sci. Rep., 2016, 6, 19159 CrossRef CAS.
- C. Lin, X. Zhu, J. Feng, C. Wu, S. Hu, J. Peng, Y. Guo, L. Peng, J. Zhao, J. Huang, J. Yang and Y. Xie, J. Am. Chem. Soc., 2013, 135, 5144–5151 CrossRef CAS.
- D. Tonti, C. Pettenkofer and W. Jaegermann, Electrochem. Solid-State Lett., 2000, 3(5), 220–223 CrossRef CAS.
- R. Ganatra and Q. Zhang, ACS Nano, 2014, 8, 4074–4099 CrossRef CAS.
- K. F. Mak, C. Lee, J. Hone, J. Shan and T. F. Heinz, Phys. Rev. Lett., 2010, 105, 136805 CrossRef.
- A. Splendiani, L. Sun, Y. Zhang, T. Li, J. Kim, C.-Y. Chim, G. Galli and F. Wang, Nano Lett., 2010, 10, 1271–1275 CrossRef CAS.
- M. A. Bissett, A. G. Hattle, A. J. Marsden, I. A. Kinloch and R. A. W. Dryfe, ACS Omega, 2017, 2, 738–745 CrossRef CAS.
- Y. Niu, S. Gonzalez-Abad, R. Frisenda, P. Marauhn, M. Drüppel, P. Gant, R. Schmidt, S. N. Taghavi, D. Barcons, J. A. Molina-Mendoza, M. S. De Vasconcellos, R. Bratschitsch, D. Perez De Lara, M. Rohlfing and A. Castellanos-Gomez, Nanomaterials, 2018, 8, 725 CrossRef PubMed.
- M. Bissett, S. Worrall, I. Kinloch and R. A. W. Dryfe, Electrochim. Acta, 2016, 201, 30–37 CrossRef CAS.
- W. Zhao, Z. Ghorannevis, L. Chu, M. Toh, C. Kloc, P.-H. Tan and G. Eda, ACS Nano, 2013, 7, 791–797 CrossRef CAS PubMed.
- A. Berkdemir, H. R. Gutiérrez, A. R. Botello-Méndez, N. Perea-López, A. L. Elías, C.-I. Chia, B. Wang, V. H. Crespi, F. López-Urías, J.-C. Charlier, H. Terrones and M. Terrones, Sci. Rep., 2013, 3, 1755 CrossRef.
- G. V. Bianco, M. Losurdo, M. M. Giangregorio, A. Sacchetti, P. Prete, N. Lovergine, P. Capezzuto and G. Bruno, RSC Adv., 2015, 5, 98700–98708 RSC.
- P. Tonndorf, R. Schmidt, P. Böttger, X. Zhang, J. Börner, A. Liebig, M. Albrecht, C. Kloc, O. Gordan, D. R. T. Zahn, S. Michaelis de Vasconcellos and R. Bratschitsch, Opt. Express, 2013, 21, 4908–4916 CrossRef CAS.
- K. Dolui and S. Sanvito, Europhys. Lett., 2016, 115, 47001 CrossRef.
- M. Hirtz, A. Oikonomou, T. Georgiou, H. Fuchs and A. Vijayaraghavan, Nat. Commun., 2013, 4, 2591 CrossRef PubMed.
- S. T. Wang, M. Fukuto and L. Yang, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2008, 77, 031909 CrossRef CAS PubMed.
- A. Dols-Perez, L. Fumagalli and G. Gomila, Colloids Surf., B, 2014, 116, 295–302 CrossRef CAS PubMed.
- Z. V. Leonenko, E. Finot, H. Ma, T. E. S. Dahms and D. T. Cramb, Biophys. J., 2004, 86, 3783–3793 CrossRef CAS PubMed.
- L. Liang and V. Meunier, Nanoscale, 2014, 6, 5394–5401 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra07764b |
| This journal is © The Royal Society of Chemistry 2019 |