DOI:
10.1039/C9RA07096F
(Paper)
RSC Adv., 2019,
9, 37067-37078
A mitochondria-targeted delivery system of doxorubicin and evodiamine for the treatment of metastatic breast cancer†
Received
4th September 2019
, Accepted 28th October 2019
First published on 13th November 2019
Abstract
For mitochondria-targeted nano-drug delivery systems against cancer, effectively targeting and releasing the drug into mitochondria are the keys to improve the therapeutic effect. In this study, mitochondria-targeted and reduction-sensitive micelles were developed to co-deliver doxorubicin (DOX) and evodiamine (EVO) for the treatment of metastatic breast cancer. After entering cancer cells, the micelles first targeted mitochondria through triphenylphosphonium cations. Then, the disulfide bonds of the micelles were cleaved by GSH, and both DOX and EVO were released near the mitochondria. The released EVO subsequently destroyed the mitochondrial membrane, resulting in a large amount of DOX entering the mitochondria and improving the anti-tumor effect of DOX. These mitochondria-targeted and reduction-sensitive micelles loaded with doxorubicin and evodiamine showed significant inhibition of the tumor cell growth both in vitro and in vivo.
1. Introduction
According to the American Cancer Society's estimate of the numbers of new cancer cases and deaths that will occur in the United States in 2019, breast cancer still ranks first among women in the United States and alone accounts for 30% of all new cancer diagnoses in women.1 At present, chemotherapy is still the main treatment for metastatic breast cancer.2,3 Small molecule chemotherapeutic drugs do not have tumor targeting function, leading to serious toxicity and side effects on normal tissues, and their effect on tumor treatment is not ideal.4–6 The accurate delivery of drugs to the targeted tumor site is key to improving traditional chemotherapy, which can be achieved by nanocarrier-based drug delivery systems. The nanocarrier-based drug delivery system has unique advantages, including improving the solubility of hydrophobic drugs,7–9 prolonging blood circulation of drugs, and increasing drug accumulation in the tumor tissue in solid tumors through the enhanced permeation and retention (EPR) effect, which can greatly help to reduce the side effects of chemotherapeutic drugs and enhance their pharmacological effects.10–13
However, the current nanocarrier-based drug delivery systems still face great challenges in the treatment of metastatic breast cancer. On the one hand, increasing drug accumulation in tumor tissues through EPR is limited.14–16 Since the final target site of chemotherapy drugs is the organelles in the tumor cells, if the final accumulation of drugs in the organelles is insufficient, the killing effect of drugs on the tumor cells may be very weak.17–19 On the other hand, long-term chemotherapy stimulation can increase the metastasis and invasion of cancer cells.20 For these reasons, drug delivery systems targeting cell organs are promoted. They can effectively increase the pharmacological effect of drugs while reducing the drug resistance and metastasis of cancer cells.21–23 Since most chemotherapeutic drugs are generally DNA-toxic reagents, the strategy of targeting cell organelles mainly focuses on organelles containing DNA, including nuclei24,25 and mitochondria.26–28 Drug delivery systems targeting the nuclei have been intensively studied. By grafting nuclear-targeted peptides on the surface of nanocarriers or controlling the size of nanocarriers in the nuclear pore size range, they can target the nucleus well. Nuclear DNA can self-repair, which is not good for cancer therapy. However, mitochondrial DNA lacks repair pathways.29 Moreover, mitochondria are the “energy factories” of cells and can directly affect the survival of cancer cells.30,31 Mitochondrial DNA also lacks introns and contains only the necessary genes for the expression of mitochondrial proteins, tRNA and rRNA. Thus, the mitochondrial DNA-targeted therapy may have better therapeutic effects than the nuclear DNA-targeted therapy.32
Although the mitochondrial DNA-targeted therapy has many advantages, it is not easy for drugs to enter the mitochondria smoothly, because the mitochondrial membrane is a dense double layer as a biological protection to prevent harmful substances from entering mitochondria.33 For this reason, it is difficult for common drugs to enter the mitochondrion. Therefore, after entering the cancer cells, targeting the mitochondria and overcoming mitochondrial membrane barriers need to be considered. Evodiamine (EVO), a plant alkaloid,34,35 is reported to increase mitochondrial membrane depolarization and disrupt mitochondrial structure,36 resulting in increased cytochrome-c release from mitochondria into the cytosol as well as nucleus.37 For this reason, EVO can be used as a reagent to destroy the mitochondrial membrane and further increase the amount of chemotherapeutic drugs entering the mitochondria, improving the anti-tumor effect of chemotherapeutic drugs. Simultaneously, EVO is also able to inhibit the migration of cancer cells, thereby reducing the metastasis of tumors.38
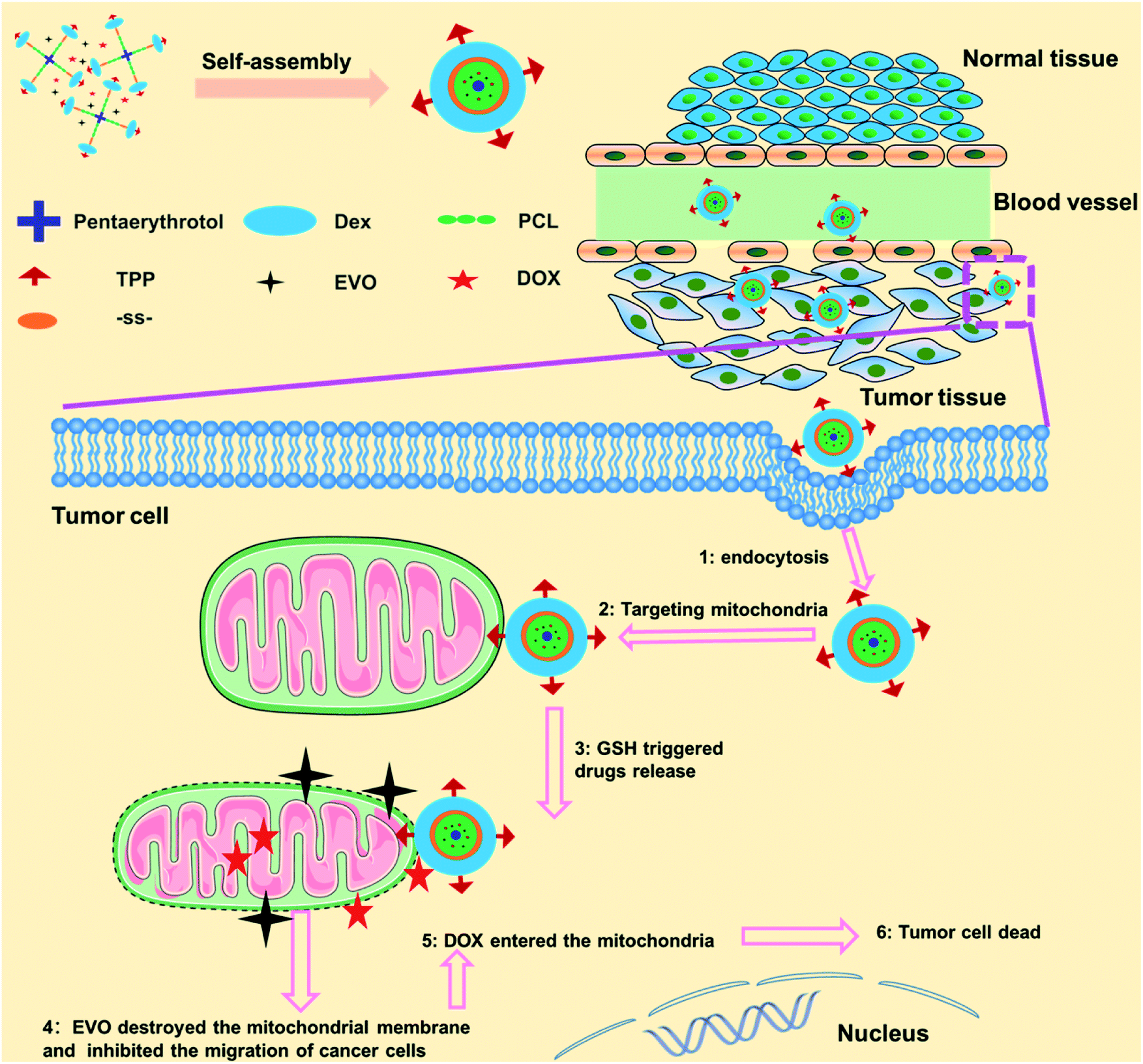
Herein, we developed a mitochondria-targeted stimulus-responsive micelle co-encapsulating DOX and EVO. This micelle was formed by the polymer (TPP-Dex-ss-SPCL), composed of dextran (Dex) as the hydrophilic segment, (4-ethoxy-4-oxobutyl)triphenylphosphonium bromide (TPP) as the targeting group grafted on Dex, and the star-shaped polycaprolactone conjugated with Dex through disulfide bonds as the hydrophobic segments. When the mitochondria-targeted stimulus-responsive micelle entered blood circulation, the Dex, as the hydrophilic end of the micelles, endowed the micelles with long blood circulation. As shown in Scheme 1, after the TPP-Dex-ss-SPCL@DOX/EVO micelles accumulate in the tumor tissues through EPR effect, they would be internalized by the tumor cells. Then, TPP on the micelles help them target the mitochondria. Moreover, under the stimulation of GSH in the tumor cells, the micelles gradually disintegrate and release the chemotherapeutic drugs DOX and EVO around the mitochondria. EVO would destroy the mitochondrial membrane potential, increase the permeability of mitochondrial membrane, and further reduce the metastasis and invasiveness of tumors. When permeability of the mitochondrial membrane increases, more DOX could enter the mitochondria and effectively kill cancer cells.
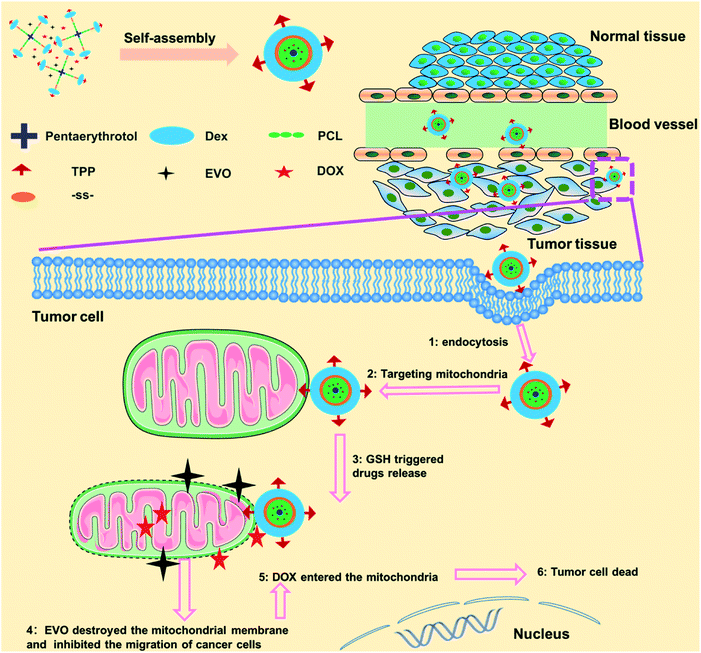 |
| | Scheme 1 Schematic of the fabrication of mitochondria-targeted stimulus-responsive micelle co-encapsulating DOX and EVO and the process of drugs-loaded micelles entering and acting on tumor cells. | |
2. Experimental section
2.1 Materials
Triphenylphosphine, 4-dimethylaminopyridine (DMAP), 3,3′-dithiodipropionic acid, ethylenediamine, 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC), pentaerythrotol, ethyl 4-bromobutyrate and glutathione (GSH) were purchased from Adamas-beta Reagent, Ltd. (Shanghai, China). Dextran (Dex, Mn = 3000 Da) was purchased from Yuanye Bio-Technology Co., Ltd. (Shanghai, China). MitoTracker-Green, 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-imidacarbocyanine iodide (JC-1) and LysoTracker-Green were purchased from Beyotime Biotechnology (Shanghai, China). All other reagents were obtained from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China) and used without further processing.
2.2 Cells and animals
Mouse breast cancer cell lines (4T1) and endothelial cells (EC) were obtained from Cell Bank of Chinese Academy of Sciences. Cells were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS) at 37 °C in a 5% CO2 atmosphere and 100% humidity. Specified-pathogen-free (SPF) female mice, Balb/c (20 ± 2 g) at 5–6 weeks of age, were purchased from the Experimental Animal Center of Sichuan University and fed under conditions of 25 °C and 55% humidity. Animal experiments were approved by the Institutional Animal Care and Use Committee of Sichuan Academy of Medical Sciences and all protocols for this animal study conformed to the Guide for the Care and Use of Laboratory Animals.
2.3 Synthesis and characterization of copolymers
2.3.1 Synthesis of SPCL. SPCL was synthesized according to a previous report with some changes.39 First, pentaerythrotol (0.227 g, 1.7 mmol), ε-CL (10 g, 87 mmol), and SnCl2 (0.10 g) were added to a 50 mL round-bottom flask equipped with a stopcock. After holding in vacuum for 5 h at room temperature (RM), the flask was placed at 140 °C for 6 h. After the reaction flask cooled, 5 mL dichloromethane was added to dissolve the polymer. Then, excess ethanol was added to precipitate the polymer, followed by drying in vacuum for 24 h. At last, the SPCL was collected as white powder (yield: 82%).
2.3.2 Synthesis of disulfide bond-modified star-shaped polycaprolactone (SPCL-ss-COOH). First, SPCL (5.00 g), DMAP (0.18 g, 1.5 mmol) and 3,3′-dithiodipropionic acid (0.32 g, 1.5 mmol) were added to 25 mL dichloromethane, then the reaction was stirred in an ice bath. After that, DCC (0.41 g, 2 mmol) in 20 mL of dichloromethane was added dropwise. When DCC was added, the reaction naturally returned to RM and was stirred until the 3,3′-dithiodipropionic acid completely reacted under monitoring with TLC. Subsequently, 20 mL ethyl acetate was resolved, the filtrate was concentrated under vacuum, and the residue was dissolved with a small amount of dichloromethane and precipitated with ice ethanol to obtain SPCL-ss-COOH (3.95 g, 74.2% yield).
2.3.3 Synthesis of (4-ethoxy-4-oxobutyl)triphenylphosphonium bromide (TPP-COOH). TPP-COOH was synthesized according to a previous report with some changes.40 Triphenylphosphine (2.62 g, 10 mmol) and ethyl-4-bromobutanoate (2.34 g, 12 mmol) were dissolved in CH3CN (20 mL). The reaction mixture was heated to reflux for 8 h under a nitrogen atmosphere. After that, it was cooled to RM and filtered, then 100 mL ethyl acetate was added to the reaction to give the corresponding phosphonium bromide salt. The product was directly used for the next reaction without further purification.
2.3.4 Synthesis of TPP-Grafted Dex (TPP-Dex). Dex (3.00 g, MW 3000), TPP-COOH (0.86 g, 2 mmol) and NHS (0.29 g, 2.5 mmol) were dissolved in 10 mL DMSO and stirred at room temperature. Then, 0.77 g EDC·HCl (4 mmol) was added gradually and the mixture was reacted for another 48 h. Next, 5 mL deionized water was added to quench the reaction and the solid impurities were filtered out. After that, 50 mL deionized water was added to the filtrate to precipitate the product. Finally, the product was filtered out and vacuum dried to obtain TPP-Dex (2.25 g, 68.4% yield).
2.3.5 Synthesis of TPP-Dex-ss-SPCL. TPP-Dex (3.30 g), SPCL-ss-COOH (6.36 g) and NHS (0.14 g, 1.2 mmol) were dissolved in 25 mL DMSO and stirred at room temperature. Then, 0.46 g EDC·HCl (2.4 mmol) was added gradually and the reaction was reacted for another 72 h. Then, 5 mL deionized water was added to quench the reaction and the solid impurities were filtered out. After that, 50 mL deionized water was added to the filtrate to precipitate the product. Finally, the product was filtered out and vacuum dried to obtain TPP-Dex-ss-SPCL (7.85 g, 81.8% yield).
2.4 Characterization of the polymers
1H nuclear magnetic resonance (1HNMR) spectra were measured using a Bruker AM 300 apparatus. (CD3)2SO was used as the solvent and tetramethylsilane (TMS) was used as the internal reference. The molecular weight of each polymer was analysed by gel permeation chromatography (GPC) system (Water 2695 pump and Styragel HT4DMF column).
2.5 Fabrication and characterization of micelles
2.5.1 Fabrication of micelles. The blank and drug-loaded micelles were fabricated by the dialysis method. Briefly, the blank micelles were fabricated as follows: first, 10 mg TPP-Dex-ss-SPCL was added into 10 mL DMSO to get TPP-Dex-ss-SPCL solution (1 mg mL−1). Then, the TPP-Dex-ss-SPCL solution was added into a dialysis bag (MWCO 1000) and the DMSO was further removed by dialysis in water. At the same time, the black micelles were formed by the self-assembly of copolymers. The preparation process of drug-loaded micelles is slightly different from that of blank micelles. When fabricating the drug-loaded micelles, drug and TPP-Dex-ss-SPCL were all added into DMSO to create the solution. The final drug-loaded micelles were lyophilized and stored at −20 °C.
2.5.2 Characterization of micelles. The micelles were prepared to a concentration of 1 mg mL−1. The freeze-dried samples (10 mg each) were re-suspended in 10 mL (0.1 M, pH 7.4) phosphate buffered solution (PBS). Then, the sizes of micelles were measured by dynamic light scattering (DLS) (ZetaSizer, Malvern Nano-ZS90, Malvern, UK) at 25 °C. The morphologies of micelles were observed by transmission electron microscopy (JEOL 2010F instrument, JEOL Ltd, Japan). All TEM samples were prepared by adding a drop of the micellar solution (1 mg mL−1) on a copper grid coated with amorphous carbon and staining with a drop of phosphotungstic acid solution (2 wt%). Before TEM observation, the copper grid was dried in vacuum. Drug loading content (LC) and encapsulation efficiency (EE) were measured with a HPLC (1260 Infinity II LC System, Agilent, USA). The critical micelle concentration (CMC) was measured by fluorescence spectrophotometer (F-7000, Hitachi, Japan) at 25 °C with pyrene as a fluorescent probe. The stability of micelles was investigated both in PBS and cell culture medium (containing 10% FBS, v/v) by measuring the size change of micelles with DLS. Briefly, the dry micelle powders (3 mg) were dispersed in 3 mL of PBS or cell culture medium at 37 °C. Then, the changes in average sizes of the micelles were monitored as a function of time by DLS measurement.
2.6 GSH-triggered disassembly behaviors of the micelles
The change of micellar size distribution in response to PBS (pH 7.4) containing GSH (10 mM) was measured by DLS. As comparison, the changes of micellar size distribution in response to acetate buffered solution (ABS) (pH 5) containing GSH (10 mM), PBS (pH 7.4) containing GSH (10 μM), and PBS (pH 5.0) containing GSH (10 μM) were all detected.
2.7 In vitro drug release
In vitro DOX and EVO release from TPP-Dex-ss-SPCL@DOX/EVO micelles were investigated. The freeze-dried drug-loaded micelles were dissolved in water (1 mg mL−1) and 1.0 mL of micellar solution was transferred into a dialysis bag (MWCO 2000). Then, the dialysis bag was immersed into a tube containing 30 mL of different incubation media, including PBS (pH 7.4) with 10 μM or 10 mM GSH and ABS (pH 5.0) with 10 μM or 10 mM GSH. All samples were kept in a shaking bed (100 rpm and 37 °C). The amounts of released DOX and EVO in the incubation medium were quantified by HPLC (1260 Infinity II LC System, Agilent, USA).
2.8 Cytocompatibility evaluation
Alamar blue assay was used to evaluate the cytocompatibility of the blank micelles (TPP-Dex-ss-SPCL and Dex-ss-SPCL) by co-culture of blank materials and 4T1 cells or EC cells. Cells were seeded in a 48-well plate (1 × 104 cells per well) and cultured for 24 h. Then, the blank micelle solutions (ranging from 20 mg mL−1 to 100 mg mL−1) were added and co-cultured with cells for another 48 h. After that, the medium was removed and the cells were washed three times with PBS. Then, 300 mL Alamar blue solution (10% Alamar blue, 80% DMEM and 10% FBS, v/v) was added and incubated for another 3 h. Finally, the Alamar blue solution was transferred to a 96-well plate and read by a microplate spectrophotometer (ELX800 Biotek, USA). For live/dead staining, 4T1 and EC cells (1 × 104 cells per well) were seeded in 24-well plates. Different concentrations of TPP-Dex-ss-SPCL and Dex-ss-SPCL (ranging from 20 mg mL−1 to 100 mg mL−1) were added to co-culture with cells for another 48 h. Then, the cells were stained by 2 mM calcein acetoxymethylester (Calcein-AM) for 10 min and propidium iodide (PI) for 5 min. All the cells were visualized by fluorescence microscopy.
2.9 Cellular uptake, intracellular distribution and co-location analysis
To evaluate whether the TPP could promote micelles targeting mitochondria, 4T1 cells (10 × 104 cells per well) were seeded in 6-well plates and treated with free DOX, Dex-ss-SPCL@DOX/EVO, TPP-Dex-SPCL@DOX/EVO and TPP-Dex-ss-SPCL@DOX/EVO for 6 h (DOX-equivalent dose: 1 μg mL−1). Then, cells were rinsed with PBS three times and Mito-Tracker Green (Beyotime Biotech, China) was added to the plates and incubated with cells for 0.25 h. All the cells were visualized by fluorescence microscopy (CKX41, Olympus, Japan) and the co-localization analyses were completed with Image-Pro Plus 6.0.
2.10 In vitro cytotoxicity assay
To evaluate the in vitro anticancer effect of different agents, including Dex-SPCL@DOX/EVO, Dex-ss-SPCL@DOX/EVO, TPP-Dex-SPCL@DOX/EVO, TPP-Dex-ss-SPCL@DOX/EVO and free DOX/EVO (DOX dosages varied from 0.01 to 5 μg mL−1 and EVO dosages were calculated according to the ratio of the two drugs in micelles), we performed a tumor cell inhibition test with 4T1 cells using the Alamar blue assay. In brief, 4T1 cells were seeded in a 48-well plate (1 × 104 cells per well) and incubated for 24 h with 5% CO2 at 37 °C. After incubation with the respective agents for another 24 h, the cell viabilities were evaluated by Alamar blue assay.
2.11 Detection of increased permeability of mitochondrial membrane
Fluorescent probe JC-1 was employed to detect mitochondrial membrane potential changes. 4T1 cells were seeded at 1 × 105 cells per well in a 6-well plate. After culturing for 24 h, control solution (PBS), TPP-Dex-ss-SPCL@EVO, Dex-SPCL@EVO, Dex-ss-SPCL@EVO, TPP-Dex-SPCL@EVO and free EVO were added to co-culture with cells for another 12 h, then all the plates were washed three times with PBS. After that, fluorescence microscopy was used to detect the fluorescence of JC-1 in cells.
2.12 Wound scratch assay
The effect of material on 4T1 cells was studied by scratch test. The cells (6 × 105 cells per well) were seeded in a 6-well plate and cultured at 37 °C in humidified atmosphere equipped with 5% CO2 for 24 h. Then the medium was removed, and a scratch in each well was scored by the same pipette tip against a steel ruler. Each well was washed with PBS three times. PBS was replaced with a fresh medium of free EVO, Dex-SPCL@EVO, Dex-ss-SPCL@EVO or TPP-Dex-ss-SPCL@EVO (EVO-equivalent dose: 0.5 μg mL−1) and incubated with cells for another 24 h. Afterward, the medium was removed and washed with PBS three times. Then calcein-AM was used for 10 min to stain the live cells. After that, cells were observed with a fluorescence microscope.
2.13 In vivo antitumor activity
4T1 tumor bearing Balb/c mice were treated with saline, TPP-Dex-ss-SPCL blank micelles, free DOX/EVO or drug-loaded micelles (TPP-Dex-ss-SPCL@DOX/EVO and Dex-ss-SPCL@DOX/EVO) at a dosage of 5 mg DOX equiv. kg−1. The formulations were intravenously injected on days 0, 3, 6 and 9. The tumor sizes were measured every three days and the volume was calculated according to the formula: V = 0.5 × a × b2, where a is the length of the tumor and b is the width of the tumor. The mice were weighed every 3 days and survival was recorded.
2.14 Histological assessment
Hematoxylin and eosin (H&E) and terminal deoxynucleotidyltransferase mediated UTP end labeling (TUNEL) staining were used to examine histological changes and apoptosis. Balb/c mice were sacrificed at day 21. The tumors were removed, fixed in 10% formalin, then embedded in paraffin blocks to prepare tumor sections at a thickness of 5 μm. After deparaffinization, the tumor sections were stained with H&E and TUNEL. The tumor sections were visualized by an optical microscope (Olympus, Japan).
2.15 Statistical analysis
SPSS software 18.0 was used for the statistical data analysis. Data were expressed as means ± SD. One-way ANOVA was performed to test for statistical significance of the data. The statistical significances were considered significant for p values ** < 0.01, *** < 0.001.
3. Results and discussion
3.1 Preparation and characterization of copolymer
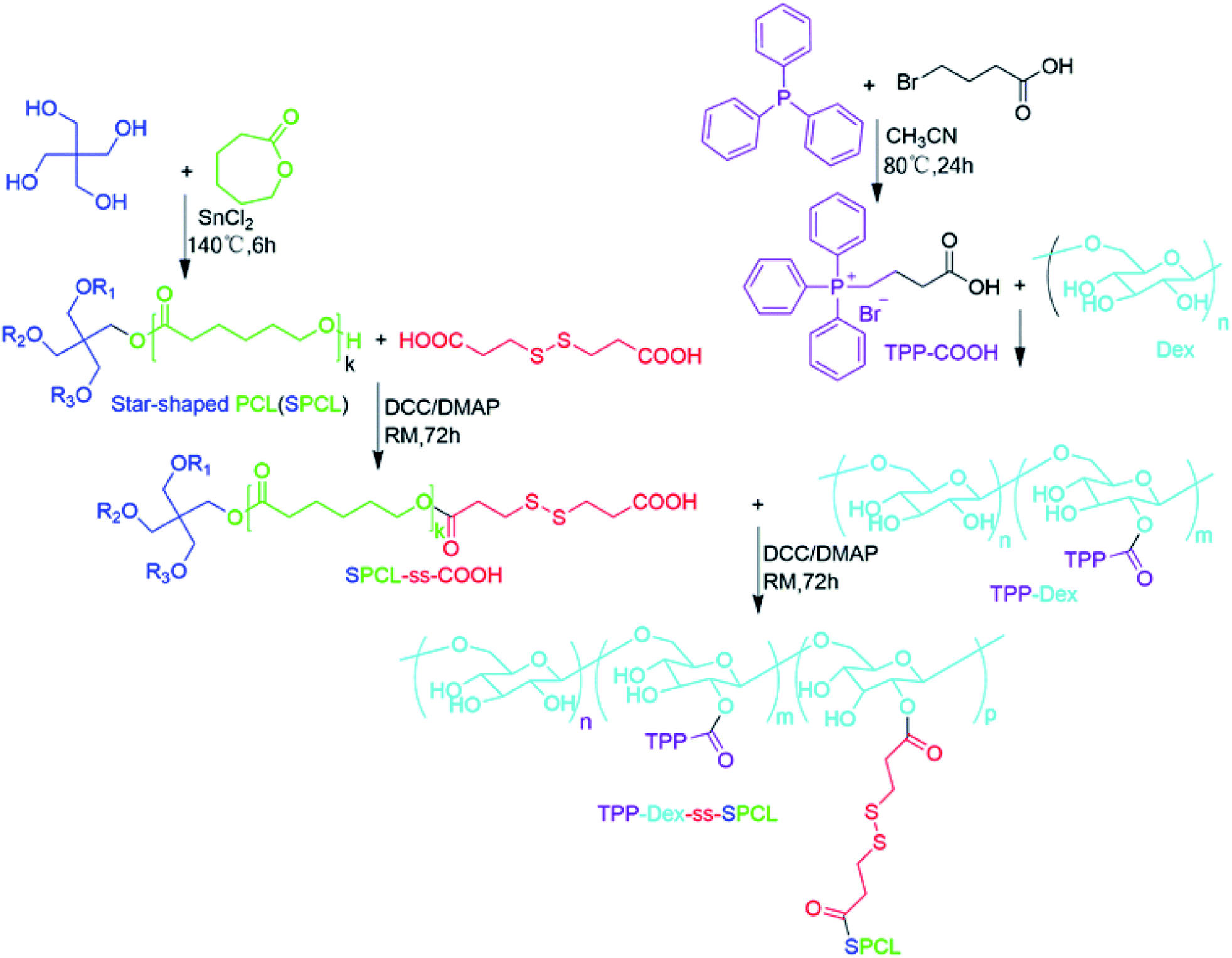
The process for the preparation of mitochondria-targeted stimulus-responsive micelles is shown in Fig. 1. First, triphenylphosphine (TPP) was reacted with ethyl-4-bromobutanoate to obtain (4-ethoxy-4-oxobutyl) triphenylphosphonium bromide (TPP-COOH). After that, TPP-COOH was grafted onto Dex to prepare TPP-Dex. Then, we obtained SPCL through the ring-opening polymerization (ROP) of ε-caprolactone that is initiated by pentaerythrotol. Further, the disulphide-bond-modified PCL (SPCL-ss-COOH) was prepared by reaction of SPCL and 3,3′-dithiodipropionic acid. Finally, the mitochondria-targeted stimulus-responsive amphiphilic copolymer (TPP-Dex-ss-SPCL) was synthesized by reacting SPCL-ss-COOH with TPP-Dex. The structures of the resulting compounds, including TPP-COOH, SPCL, TPP-Dex and TPP-Dex-ss-SPCL, were ascertained by 1H nuclear magnetic resonance (1H NMR) (Fig. S1–S4†). From Fig. S4,† with the molecular weight of Dex (Mn = 3 kDa) used as the standard, the 1H NMR analysis showed that the graft ratio of TPP on Dex was 13%. At the same time, the molecular weight of TPP-Dex-ss-SPCL was 4.2 kDa by 1H NMR, which was less than the GPC result of 6.8 kDa. From GPC analysis, the Mn of the SPCL-ss-COOH was 5.8 kDa.
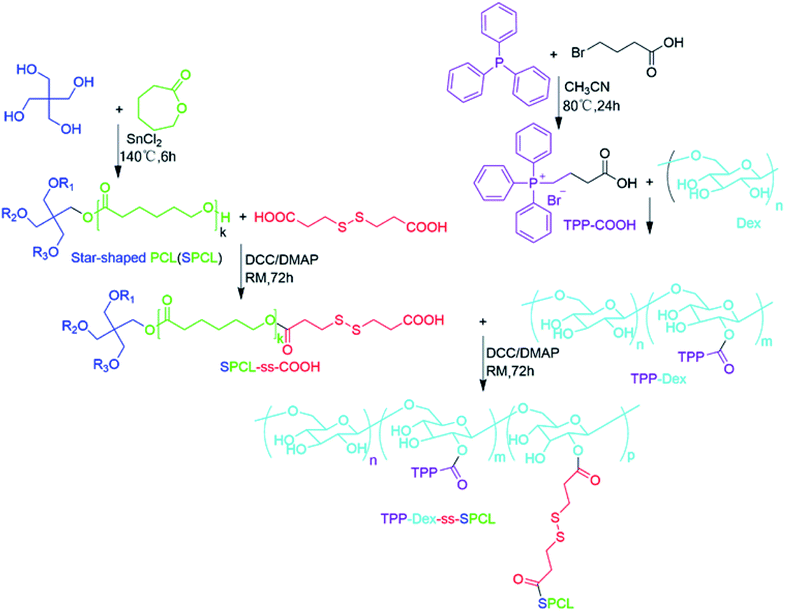 |
| | Fig. 1 Synthesis route of the mitochondria-targeted stimulus-responsive copolymer TPP-Dex-ss-SPCL. | |
3.2 Characterization of blank micelles
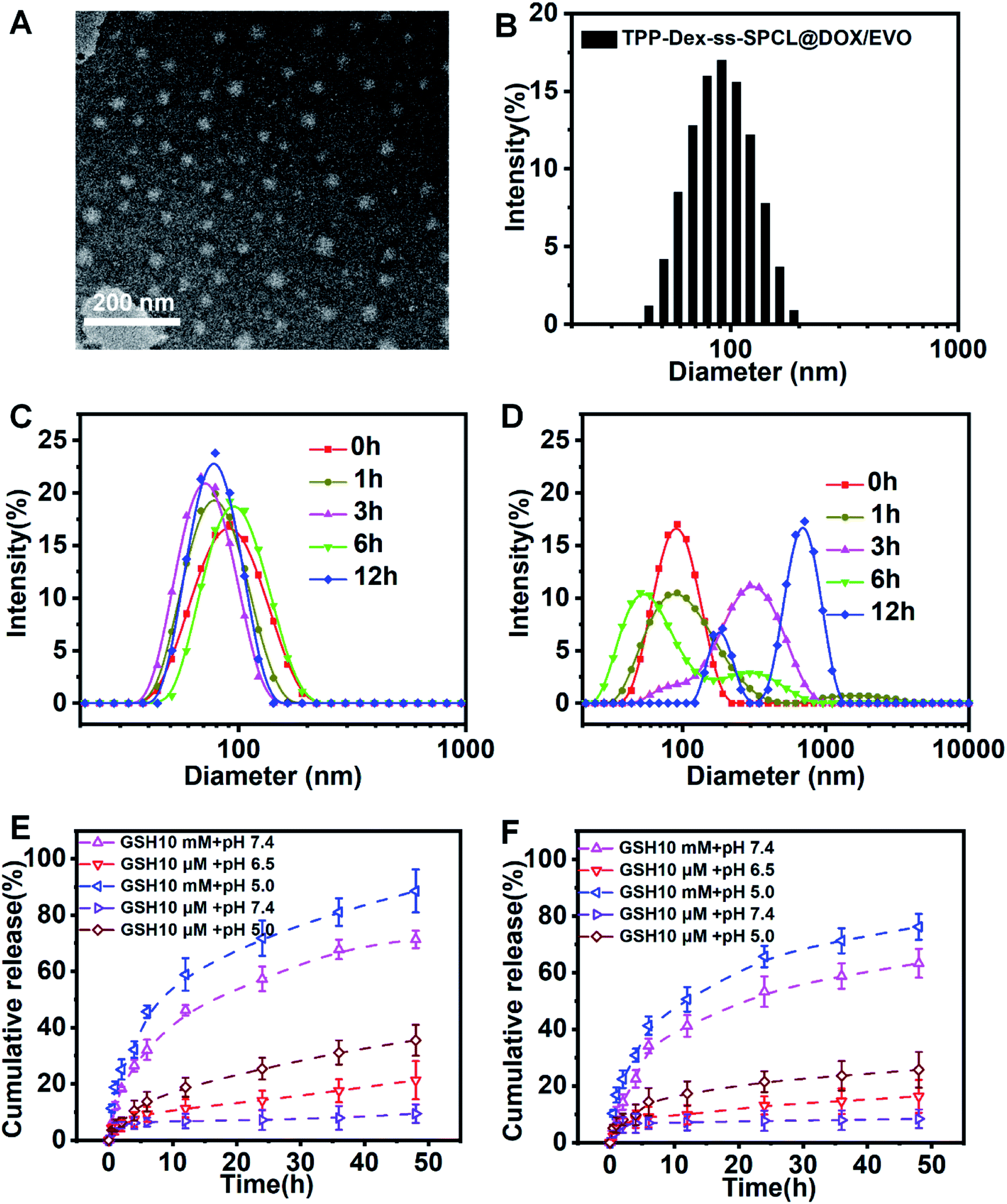
The blank micelles were formed from the mitochondria-targeted stimulus-responsive copolymers (TPP-Dex-ss-SPCL) by the dialysis method. Transmission electron microscopy (TEM) measurements indicated that the TPP-Dex-ss-SPCL micelles had a uniform spherical shape (Fig. 2A). The size distribution of the TPP-Dex-ss-SPCL micelles is displayed in Fig. 2B, which indicates the hydrodynamic diameter is approximately 85.7 nm with PDI of 0.176. The zeta potential of TPP-Dex-ss-SPCL micelles is +22.7 mV (Fig. S5†). The size changes of the blank micelles in different microenvironments were also detected by DLS. When the blank micelles were in the condition of pH 7.4 and 10 μM GSH (mimicking blood circulation environment), the size of micelles remained almost the same, indicating that the structures of micelles would be stable in blood circulation (Fig. 2C). However, when the concentration of GSH was adjusted to 10 mM and the pH was changed to 5.0, the size distribution of the micelles increased significantly over the incubation time (Fig. 2D), which may be due to the rapid cleavage of disulfide bonds in the micelles. In order to more realistically mimic the tumoral microenvironment, the size changes of blank micelles in a simulated late lysosome environment (pH 5.0 and 10 μM GSH) and simulated cytoplasmic environment (pH 7.4 and 10 mM GSH) were also detected (Fig. S6†). When micelles were exposed to the late lysosome environment, the size of the blank micelles increased slightly, which may be due to the slow breakage of ester bonds in an acidic environment. However, in the cytoplasmic environment, the micelle size varied greatly due to the role of GSH. The CMC values of blank micelles at natural condition (0.74 mg L−1) and acidic condition (0.98 mg L−1) indicated that they would possess a stable structure in the blood circulation and in late lysosomes (Fig. S7†). At the same time, there was almost no size change in micelles after incubation in PBS or in cell culture medium (containing 10% FBS, v/v) at 37 °C for 5 days (Fig. S8†). This result showed that the micelles have good stability.
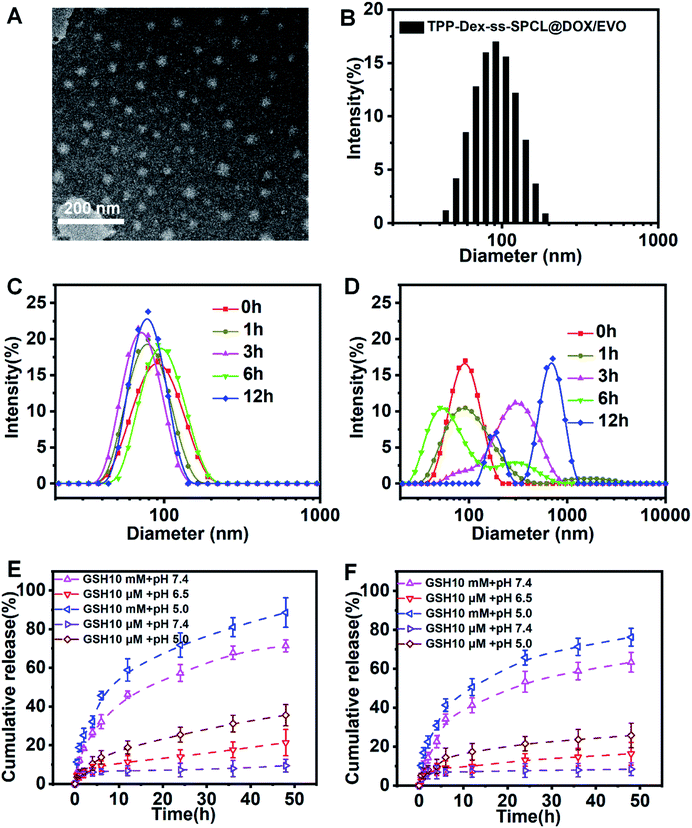 |
| | Fig. 2 (A) Transmission electron microscope image of TPP-Dex-ss-SPCL. (B) Size distribution of TPP-Dex-ss-SPCL determined by dynamic light scattering. (C) Change in size distribution profiles of the micelles in the presence of 10 μM GSH in PBS at pH 7.4. (D) Change in size distribution profiles of TPP-Dex-ss-SPCL triggered by 10 mM GSH at pH 5.0. (E) In vitro DOX release from the TPP-Dex-ss-SPCL@DOX/EVO micelles triggered with GSH (10 μM or 10 mM) in PBS (pH 7.4) or ABS (pH 5.0 and 6.5) at 37 °C. (F) In vitro EVO release from the TPP-Dex-ss-SPCL@DOX/EVO micelles triggered with GSH (10 μM or 10 mM) in PBS (pH 7.4) or ABS (pH 5.0 and 6.5) at 37 °C. | |
3.3 Characterization of drug-loading micelles
After DOX and EVO were encapsulated in these micelles, the size and potential of the micelles changed slightly compared with blank micelles. The DOX-loading content (LC) and encapsulation efficiency (EE) of TPP-Dex-ss-SPCL@DOX/EVO micelles were 6.8 ± 0.2% and 76 ± 3%, respectively, while the EVO-loading content (LC) and encapsulation efficiency (EE) of TPP-Dex-ss-SPCL@DOX/EVO micelles are 5.6 ± 0.1% and 62 ± 2%, respectively (Table S1†). For comparison, micelles, Dex-SPCL@DOX/EVO, Dex-ss-SPCL@DOX/EVO and TPP-Dex-SPCL@DOX/EVO were also prepared and their properties are also summarized in Table S1.†
3.4 In vitro GSH-triggered drug release
In order to determine whether drug-loaded micelles can be effectively released under the stimulation of simulated tumor cell microenvironment, in vitro pH- and GSH-triggered drug release from the TPP-Dex-ss-SPCL@DOX/EVO micelles were investigated. From Fig. 2E and F, we can see that the cumulative releases of DOX and EVO in a simulated blood circulation environment (pH 7.4, 10 mM GSH) were 18% and 6% in 36 h, respectively. The results showed that the drugs-loaded micelles would be stable in blood circulation. In contrast, the cumulative releases of DOX and EVO were all over 40% when triggered with 10 mM of GSH at pH 7.4. Moreover, when the TPP-Dex-ss-SPCL@DOX/EVO micelles were placed in the condition of pH 5.0 and 10 mM GSH, the cumulative releases of DOX and EVO were the highest. These results were obtained mainly due to the disulfide bond breakage induced by GSH stimulation. In addition, an acidic environment may help to increase DOX solubility.
3.5 The cytotoxicity of these blank micelles
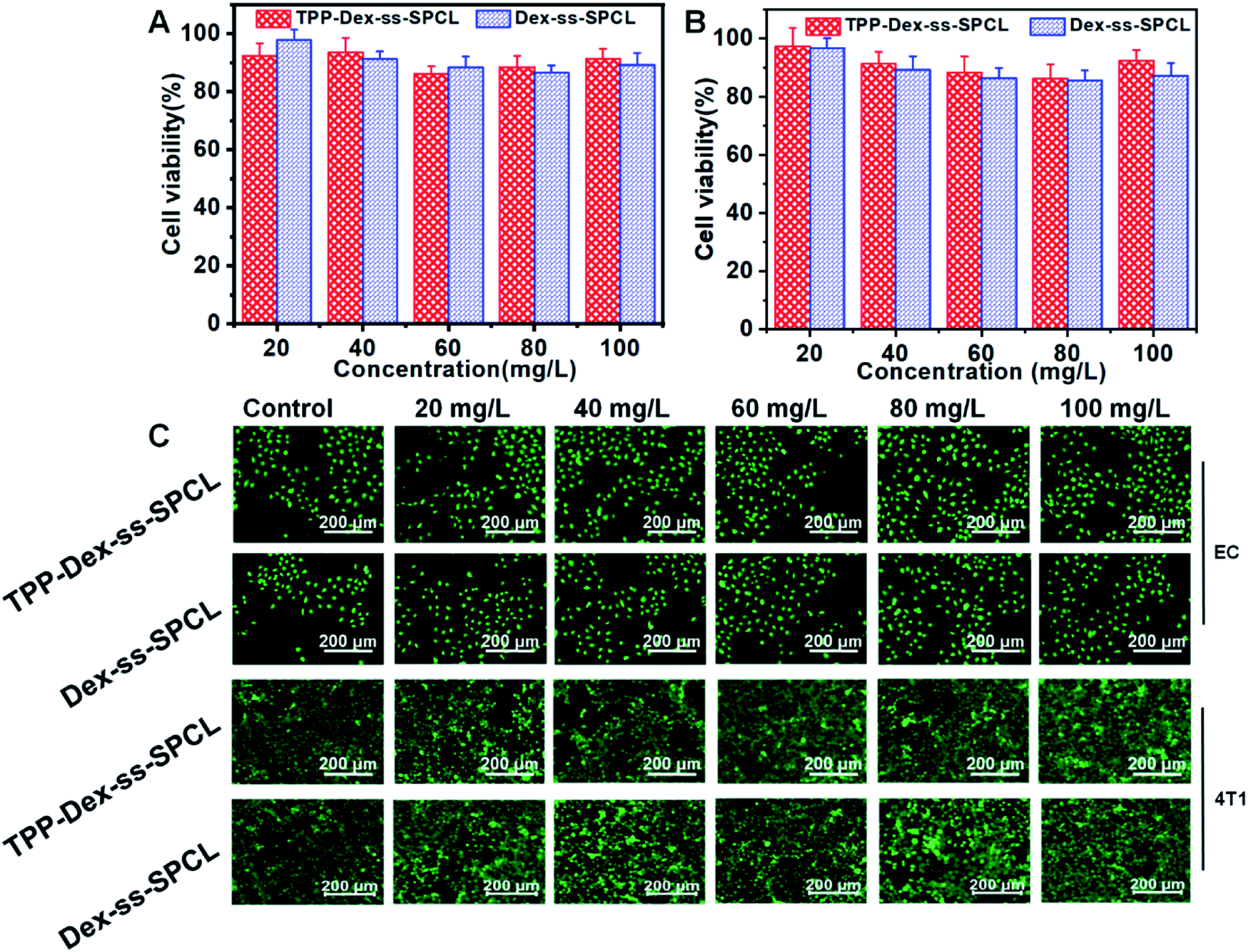
First, the cytotoxicities of the blank micelles (TPP-Dex-ss-SPCL and Dex-ss-SPCL) were evaluated. Different concentrations of blank micelles were incubated with 4T1 and EC cells. After that, the viability of cells was investigated by Alamar blue (AB) assay and live/dead staining. The AB results in Fig. 3A and B show that when the concentration of blank material reached up to 100 mg mL−1, the survival rates of both normal and cancer cells were still above 85%. Those results proved that the blank material had low toxicity and good biocompatibility. The live/dead staining also agreed with the AB assay results: when the concentration of the micelles increased to 100 μg mL−1, the cells grew healthily and few dead cells could be seen (Fig. 3C).
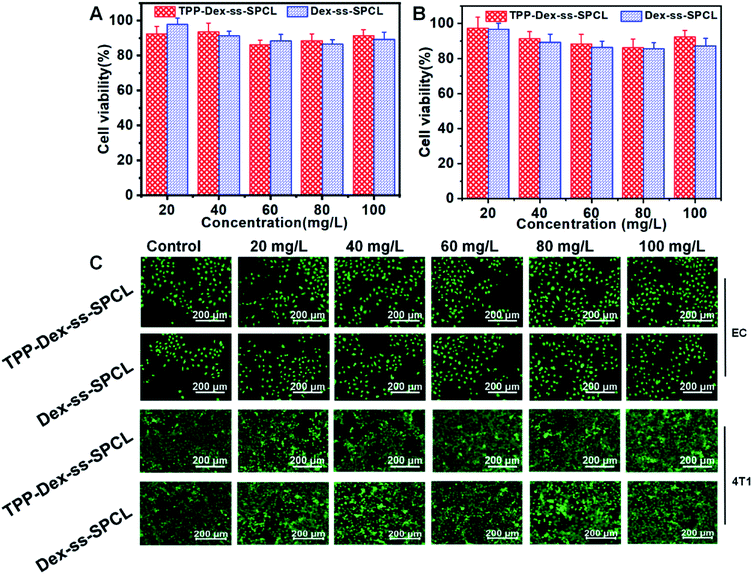 |
| | Fig. 3 Cell viabilities of (A) EC cells and (B) 4T1 cells after incubation with blank micelles (TPP-Dex-ss-SPCL and Dex-ss-SPCL) at different concentrations over 48 h. (C) Fluorescence images of EC cells and 4T1 cells after treatments with blank micelles at different concentrations for 48 h. The live cells were stained green and red indicates dead cells. | |
3.6 Mitochondrial targeting and damage analysis
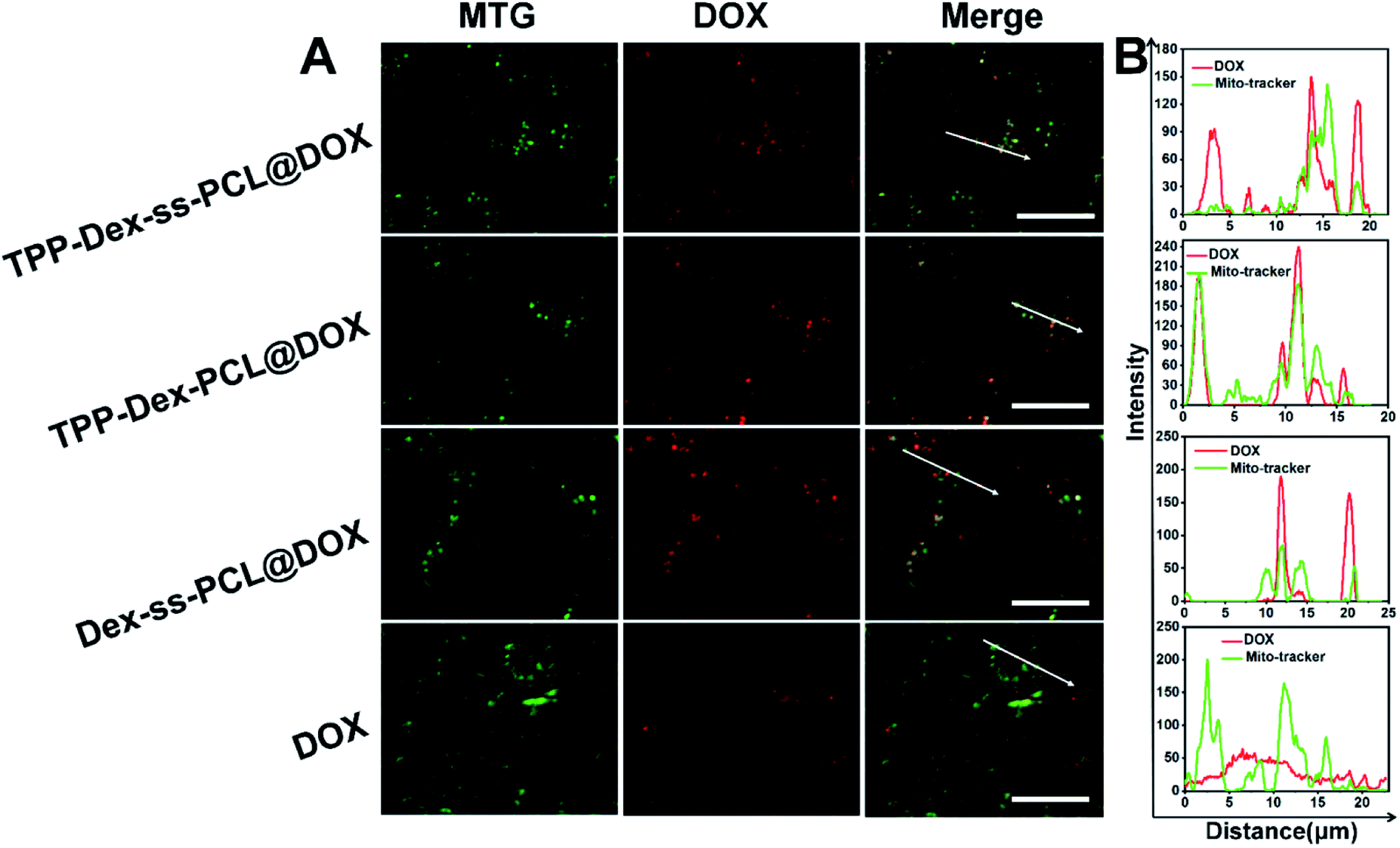
To investigate the targeting efficiency of the drug-loaded micelles to mitochondria, co-localization analysis was used to compare free DOX and the corresponding DOX-loaded micelles. DOX showed red fluorescence and the mitochondria of 4T1 cells were stained in green by Mito-Tracker Green (MTG). When yellow fluorescence appears through merging green and red fluorescence, it implies DOX and mitochondria overlap (Fig. 4A).
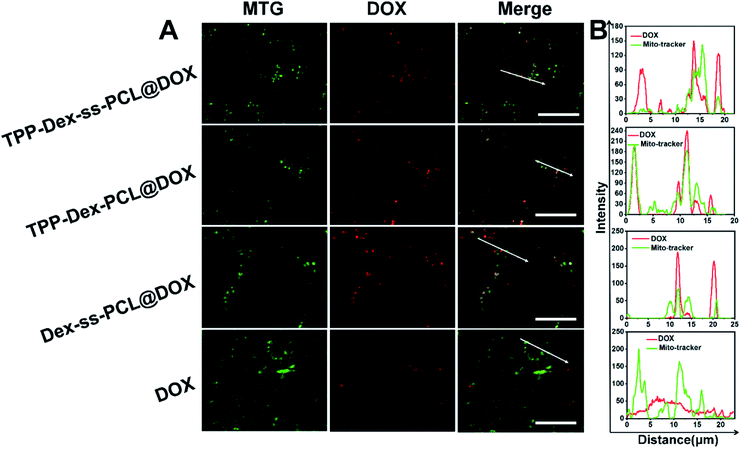 |
| | Fig. 4 Mitochondrial targeting analysis. (A) Mitochondrial targeting of TPP-Dex-ss-SPCL@DOX, Dex-ss-SPCL@DOX and free DOX (scale bar: 20 μm, red fluorescence represents DOX, green fluorescence represents MitoTracker Green (MTG), white arrows in the merge are the co-localization of the selected linear region); (B) co-localization analysis of DOX and MTG in 4T1 cells. | |
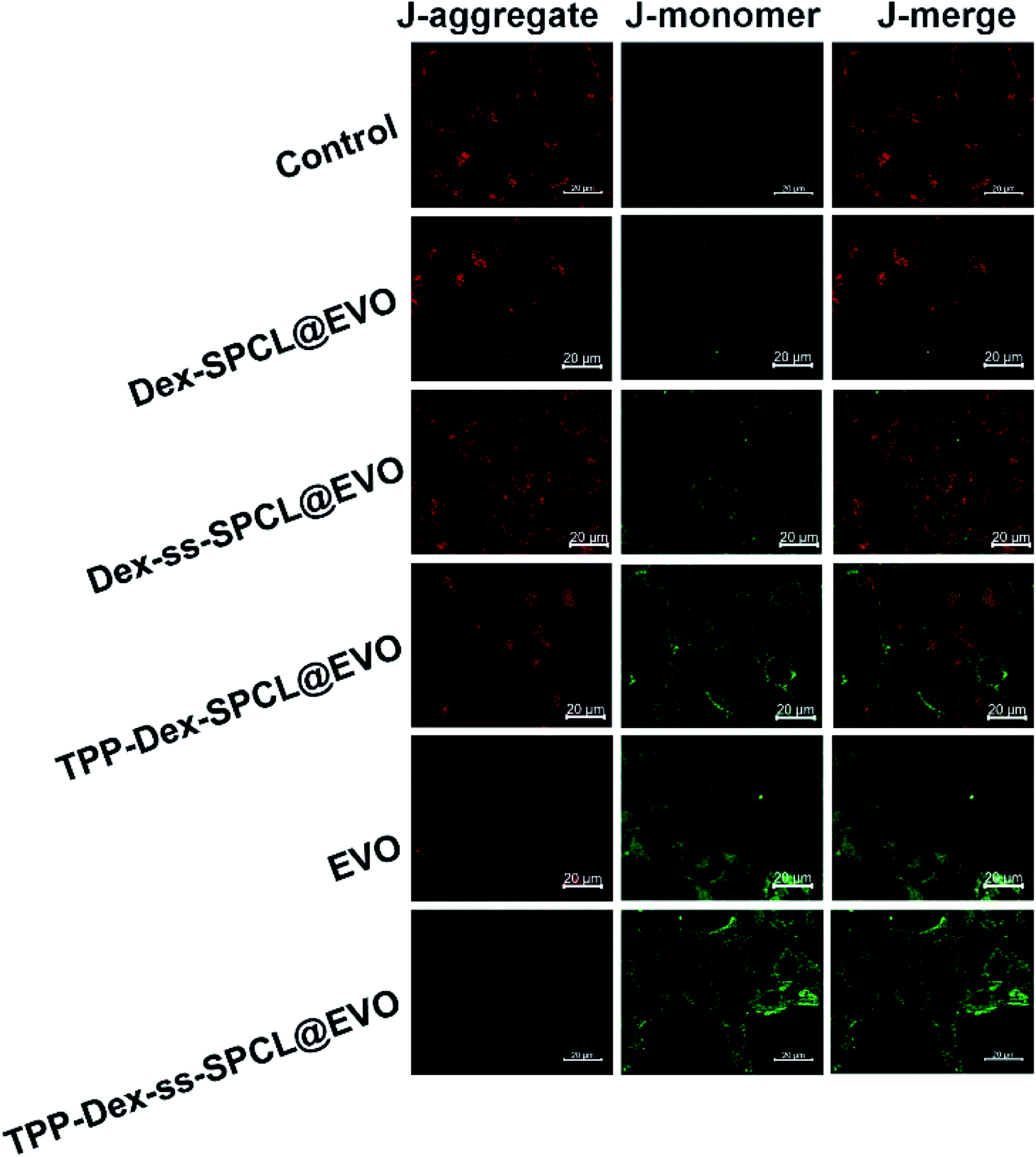
Further, linear regions of interest intensity profiles were used to evaluate the co-localization of the selected linear region (Fig. 4B). In the free DOX group, the DOX and MTG were barely overlapped. At the same time, the DOX was distributed all over the cells. For the Dex-ss-SPCL@DOX micelles group, the red fluorescence was aggregated, but red fluorescence and green fluorescence overlapped very little, which proved that the co-localization of DOX and mitochondria is very weak. In contrast, the DOX and MTG of the TPP-Dex-SPCL@DOX micelles group overlapped very well: DOX was not distributed in cells except in mitochondria. It was noteworthy that the DOX and MTG in TPP-Dex-ss-SPCL@DOX group also overlapped well. However, unlike TPP-Dex-SPCL@DOX micelles group, here DOX was also distributed in a small amount in sites other than mitochondria, which may be due to the DOX released from the disulfide bond breakage. All these results suggested that after modifying micelles with TPP, the micelles specifically accumulated in the mitochondrial region in live cells and the introduction of disulfide bonds could promote drug release. After the micelles targeted mitochondria, the loaded DOX and EVO should be released. The disulfide bonds of the TPP-Dex-ss-SPCL@DOX micelles were cleaved due to the high concentration GSH in cytoplasm and the micelles were disassembled quickly, resulting in the DOX being released rapidly; this is consistent with the in vitro release result in Fig. 2E. In order to evaluate the DOX distribution in organelles, Pearson's correlation coefficient (Rr) was utilized to quantitatively evaluate the correlation between red and green fluorescence and was also used for the fluorescence co-localization analysis. The values for Rr range from 1 to −1. A value of “1” represents perfect correlation, while “−1” represents perfect exclusion and “zero” represents random localization. When the value of Rr is close to 1, it means that the co-location of two types of fluorescence is better. Compared to the free DOX group (Rr = 0.67), the TPP-Dex-SPCL@DOX group possessed the higher Rr (0.97), which means the TPP-Dex-SPCL@DOX group could target the mitochondria well. Additionally, the Rr of TPP-Dex-ss-SPCL@DOX was 0.89, which indicated the DOX was partially released. Further, after releasing around mitochondria, DOX needed to enter the mitochondria and act on mitochondrial DNA to induce cell apoptosis. The large amount of DOX entering mitochondria required increased permeability of mitochondrial membrane. To verify whether the EVO can increase the mitochondrial membrane depolarization and disrupt mitochondrial structure, mitochondrial membrane potential dye JC-1 was employed to stain the mitochondrial membrane. When the mitochondrial membrane was intact, the JC-1 was aggregated into red fluorescence, while in depolarized mitochondria, JC-1 forms green fluorescence. From Fig. 5, both free EVO and TPP-Dex-ss-SPCL@DOX/EVO showed weaker JC-1 red fluorescence than other groups. Compared with the TPP-Dex-ss-SPCL@DOX/EVO micelles group, although TPP-Dex-SPCL@DOX/EVO can target mitochondria, they could not release EVO quickly; hence, TPP-Dex-SPCL@DOX/EVO group had stronger red fluorescence. These results demonstrated that after modifying micelles with TPP, the micelles could target mitochondria well. Then, micelles containing disulfide bonds could further achieve the effective release of drugs after targeting mitochondria. EVO released around mitochondria could quickly destroy the function of mitochondrial membrane and increase its permeability, contributing to the entry of DOX into mitochondria and helping exert a stronger anti-tumor effect for DOX.
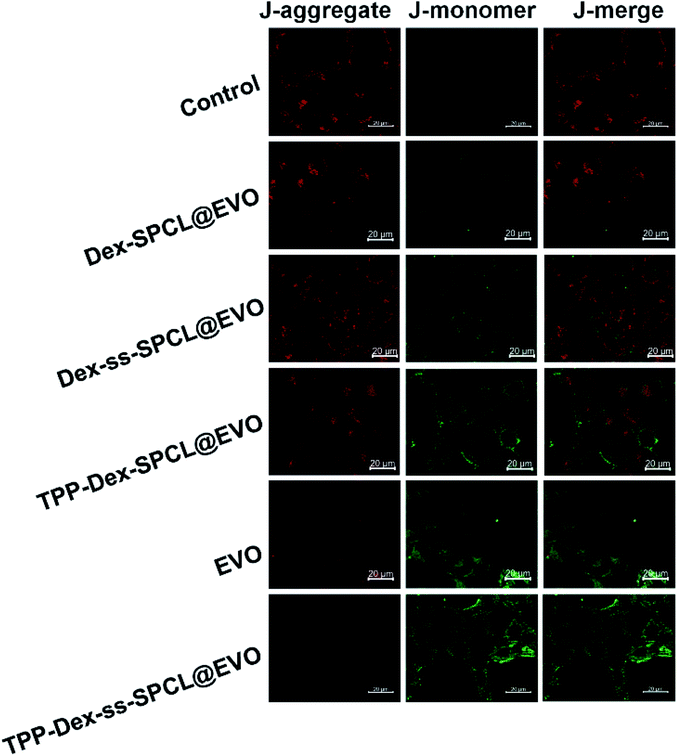 |
| | Fig. 5 Mitochondrial membrane potentials (Δψm) measured by JC-1 staining of 4T1 cells after incubation with control solution (PBS), TPP-Dex-ss-SPCL@EVO, Dex-SPCL@EVO, Dex-ss-SPCL@EVO, TPP-Dex-SPCL@EVO and free EVO (scale bar = 20 μm). | |
3.7 In vitro cytotoxicity analysis and anticancer effect
In order to verify the anti-tumor effect of DOX after the targeted destruction of the mitochondrial membrane, the cytotoxicity of free DOX/EVO, Dex-SPCL@DOX/EVO, Dex-ss-SPCL@DOX/EVO, TPP-Dex-ss-SPCL@DOX/EVO and TPP-Dex-SPCL@DOX/EVO were further investigated. As shown in Fig. S9,† the cytotoxicity of the TPP-Dex-ss-SPCL@DOX/EVO group was the highest among all the groups, with a half maximal inhibitory concentration (IC50) of 1.45 μg mL−1, which was 1.14-fold more potent than the free drug group (DOX/EVO) (1.66 μg mL−1). At the same time, the IC50 of TPP-Dex-ss-SPCL@DOX/EVO group was 1.52-fold more potent than the Dex-ss-SPCL@DOX/EVO group, which indicated that the micelles targeting mitochondria can effectively kill tumor cells. The Dex-SPCL@DOX/EVO group possessed the lowest cytotoxicity with an IC50 of 4.77 μg mL−1, which means that if micelles cannot target mitochondria and release drugs effectively in cancer cells, they will not kill tumors well. In addition, the wound-healing assay result showed that, compared to the control, free EVO has weak ability to inhibit tumor migration. The TPP-Dex-ss-SPCL@EVO treatment significantly delayed the wound closure, as wounds retained their width for 24 h after the scratch (Fig. S10†).
3.8 In vivo antitumor effect
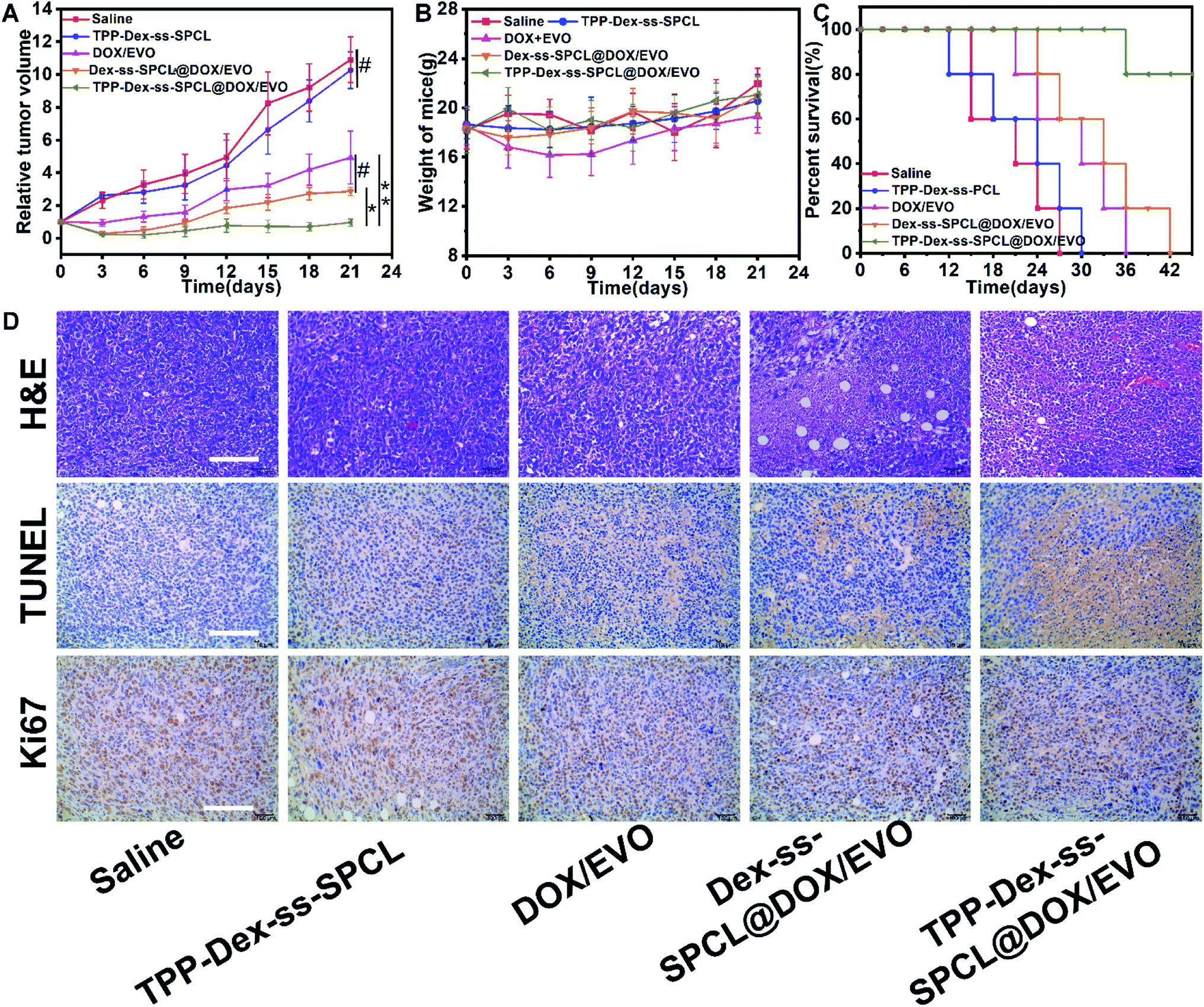
To establish whether mitochondrial targeted dual-drug-loaded responsive micelles can also have good anti-tumor effect in vivo, we performed therapeutic studies using a 4T1 tumor model on Balb/c mice by tail-vein injection of the drug-loaded micelles. The antitumor efficacies of various micelles and free drug are illustrated in Fig. 6. As shown in Fig. 6A, after 21 days treatment, the final tumor volumes were ∼513 mm3 (saline group), 545 mm3 (blank TPP-Dex-ss-SPCL group), 246 mm3 (free DOX/EVO group), 143 mm3 (Dex-ss-SPCL@DOX/EVO group) and 48 mm3 (TPP-Dex-ss-SPCL@DOX/EVO group). The mean tumor volume of the TPP-Dex-ss-SPCL@DOX/EVO group was the smallest, suggesting that this group possessed the best therapeutic effect. This result may be because the material TPP-Dex-ss-SPCL possessed the ability to target mitochondria and, after the co-delivery of DOX and EVO into tumor cells and targeting to mitochondria, the released EVO would destroy mitochondrial membrane, leading to more released chemotherapeutic DOX entering into mitochondria, destroying mitochondrial DNA, and finally effectively killing cancer cells. Moreover, the survival rate of mice treated with the TPP-Dex-ss-SPCL@DOX/EVO group was the highest among all groups (Fig. 5C).
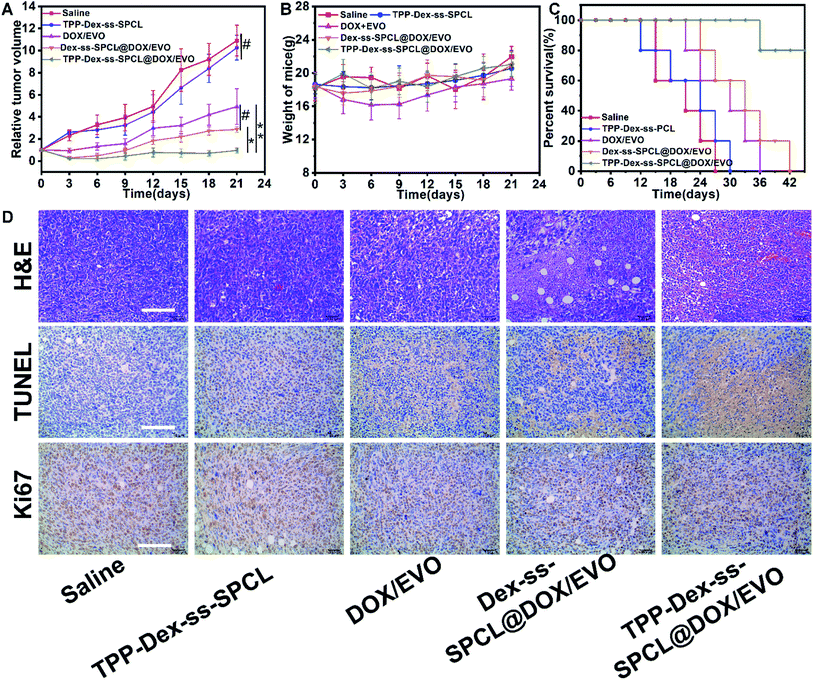 |
| | Fig. 6 In vivo antitumor efficacy towards 4T1 tumor-bearing Balb/c mice. (A) Relative tumor volume (n = 5), (B) body weight changes (n = 5), and (C) survival curves of tumor-bearing mice after treatment with different formulations. (D) H&E, TUNEL and Ki67 staining of tumor sections after different treatments (scale bar: 100 μm) (#) p > 0.05, (*) p < 0.05, (**) p < 0.01. | |
3.9 Histological analysis
The antitumor efficacy was further confirmed by histological evaluation of tumor tissues stained with hematoxylin and eosin (H&E) and terminal deoxynucleotidyltransferase-mediated UTP end labeling (TUNEL) and immunostained with a rabbit polyclonal antibody for Ki-67. Fig. 6D shows extensive nuclear shrinkage and fragmentation and massive tumor necrosis in TPP-Dex-ss-SPCL@DOX/EVO group, further indicating its best antitumor efficacy. Additionally, except for the free DOX group, no significant weight loss was observed in the other groups (Fig. 6B), suggesting that these drug-loaded micelles have almost no systemic toxicity and the side effects of free DOX can be overcome through the encapsulation of the micelles.
4. Conclusions
In summary, we presented dual drug-loaded mitochondria-targeted and reduction-sensitive micelles. These drugs-loaded micelles had good stability during blood circulation. After being internalized by the tumor cells, the micelle could target mitochondria through its TPP groups. Then, the high content of intracellular GSH would break the disulfide bonds in the micelles, leading to rapid release of EVO and DOX. The released EVO further induced the depolarization of mitochondrial membrane with decrease of the ΔΨ and increase of the mitochondrial membrane permeability. Consequently, more released DOX would directly diffuse into mitochondria, resulting in mitochondrial DNA damage. This work provides a novel strategy for the design and engineering of smart micelles for highly effective and accurate cancer therapy.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was partially supported by Natural Science Foundation project of science and technology research program of Chongqing Education Commission of China. (No. KJ1602506).
References
- R. L. Siegel, K. D. Miller and A. J. Jemal, Ca-Cancer J. Clin., 2019, 69, 7–34 CrossRef PubMed.
- S. Halvaei, S. Daryani, Z. Eslami-S, T. Samadi, N. Jafarbeik-Iravani, T. O. Bakhshayesh, K. Majidzadeh-A and R. Esmaeili, Mol. Ther.–Nucleic Acids, 2018, 10, 131–141 CrossRef CAS PubMed.
- C. Twelves, M. Jove, A. Gombos and A. Awada, Crit. Rev. Oncol.-Hematol., 2016, 100, 74–87 CrossRef PubMed.
- Q. Liu, M. Das, Y. Liu and L. Huang, Adv. Drug Delivery Rev., 2018, 127, 208–221 CrossRef CAS PubMed.
- W. Shao, X. Liu, G. Sun, X.-Y. Hu, J.-J. Zhu and L. Wang, Chem. Commun., 2018, 54, 9462–9465 RSC.
- S. Xing, X. Zhang, L. Luo, W. Cao, L. Li, Y. He, J. An and D. Gao, Nanotechnology, 2018, 29, 405101 CrossRef PubMed.
- R. Singh and J. W. Lillard Jr, Exp. Mol. Pathol., 2009, 86, 215–223 CrossRef CAS PubMed.
- J. S. Suk, Q. Xu, N. Kim, J. Hanes and L. M. Ensign, Adv. Drug Delivery Rev., 2016, 99, 28–51 CrossRef CAS PubMed.
- R. Tietze, J. Zaloga, H. Unterweger, S. Lyer, R. P. Friedrich, C. Janko, M. Pöttler, S. Dürr and C. Alexiou, Biochem. Biophys. Res. Commun., 2015, 468, 463–470 CrossRef CAS PubMed.
- V. Torchilin, Adv. Drug Delivery Rev., 2011, 63, 131–135 CrossRef CAS PubMed.
- H. Maeda, J. Controlled Release, 2012, 164, 138–144 CrossRef CAS PubMed.
- H. Maeda, H. Nakamura and J. Fang, Adv. Drug Delivery Rev., 2013, 65, 71–79 CrossRef CAS PubMed.
- Y. S. Youn and Y. H. Bae, Adv. Drug Delivery Rev., 2018, 130, 3–11 CrossRef CAS PubMed.
- S. Liu, W. Wei, B. Xie, H. Yue, D. Ni, Y. Bao, G. Ma and Z. G. Su, Adv. Healthcare Mater., 2016, 5, 2872–2881 CrossRef CAS PubMed.
- J. Chen, L. Liu, S. M. Motevalli, X. Wu, X. H. Yang, X. Li, L. Han, A. Magrini, W. Guo and J. Chang, Adv. Funct. Mater., 2018, 28, 1707291 CrossRef.
- Y. Dai, C. Xu, X. Sun and X. Chen, Chem. Soc. Rev., 2017, 46, 3830–3852 RSC.
- M. E. Fichera and D. S. Roos, Nature, 1997, 390, 407 CrossRef CAS PubMed.
- N. M. Sakhrani and H. Padh, Drug Des., Dev. Ther., 2013, 7, 585 CAS.
- R. Elliott, X. Jiang and J. Head, Breast Cancer Res. Treat., 2012, 136, 347–354 CrossRef CAS PubMed.
- S. Gingis-Velitski, D. Loven, L. Benayoun, M. Munster, R. Bril, T. Voloshin, D. Alishekevitz, F. Bertolini and Y. Shaked, Cancer Res., 2011, 71, 6986–6996 CrossRef CAS PubMed.
- H. Xiong, S. Du, J. Ni, J. Zhou and J. Yao, Biomaterials, 2016, 94, 70–83 CrossRef CAS PubMed.
- J.-n. Liu, W. Bu, L.-m. Pan, S. Zhang, F. Chen, L. Zhou, W. Peng and J. Shi, Biomaterials, 2012, 33, 7282–7290 CrossRef CAS PubMed.
- J. G. Huang, T. Leshuk and F. X. Gu, Nano Today, 2011, 6, 478–492 CrossRef CAS.
- L. Yang, Z. Wang, J. Wang, W. Jiang, X. Jiang, Z. Bai, Y. He, J. Jiang, D. Wang and L. Yang, Nanoscale, 2016, 8, 6801–6809 RSC.
- Y. Yuan, B. Guo, L. Hao, N. Liu, Y. Lin, W. Guo, X. Li and B. Gu, Colloids Surf., B, 2017, 159, 349–359 CrossRef CAS PubMed.
- Q. Qu, X. Ma and Y. Zhao, Nanoscale, 2015, 7, 16677–16686 RSC.
- S. Chen, Q. Lei, W.-X. Qiu, L.-H. Liu, D.-W. Zheng, J.-X. Fan, L. Rong, Y.-X. Sun and X.-Z. Zhang, Biomaterials, 2017, 117, 92–104 CrossRef CAS PubMed.
- S. R. Jean, D. V. Tulumello, C. Riganti, S. U. Liyanage, A. D. Schimmer and S. O. Kelley, ACS Chem. Biol., 2015, 10, 2007–2015 CrossRef CAS PubMed.
- L. Kazak, A. Reyes and I. J. Holt, Nat. Rev. Mol. Cell Biol., 2012, 13, 659 CrossRef CAS PubMed.
- J. Wang, X. Yang and J. Zhang, Cell. Signal., 2016, 28, 1099–1104 CrossRef CAS PubMed.
- A. B. Claiborne, R. A. English and J. P. Kahn, Science, 2016, 351, 668–670 CrossRef CAS PubMed.
- S. R. Jean, M. Ahmed, E. K. Lei, S. P. Wisnovsky and S. O. Kelley, Acc. Chem. Res., 2016, 49, 1893–1902 CrossRef CAS PubMed.
- F. S. Sjöstrand and V. Hanzon, Exp. Cell Res., 1954, 7, 393–414 CrossRef.
- Z.-F. Zhong, W. Tan, S.-P. Wang, W.-A. Qiang and Y.-T. Wang, Sci. Rep., 2015, 5, 16415 CrossRef CAS PubMed.
- Y. Lv, G. Ding, J. Zhai, Y. Guo, G. Nie and L. Xu, Colloids Surf., B, 2013, 110, 411–418 CrossRef CAS PubMed.
- X. Hu, D. Li, C. Chu, X. Li, X. Wang, Y. Jia, H. Hua and F. Xu, Int. J. Mol. Sci., 2018, 19, 3403 CrossRef PubMed.
- V. Mohan, R. Agarwal and R. P. Singh, Biochem. Biophys. Res. Commun., 2016, 477, 1065–1071 CrossRef CAS PubMed.
- S. H. Kim, J. G. Kang, C. S. Kim, S.-H. Ihm, M. G. Choi and S. J. Lee, Anticancer Res., 2018, 38, 6339–6352 CrossRef PubMed.
- M. R. Nabid, S. J. T. Rezaei, R. Sedghi, H. Niknejad, A. A. Entezami, H. A. Oskooie and M. M. Heravi, Polymer, 2011, 52, 2799–2809 CrossRef CAS.
- M. Liu, P. Yang, M. K. Karunananda, Y. Wang, P. Liu and K. M. Engle, J. Am. Chem. Soc., 2018, 140, 5805–5813 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra07096f |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Yanlin Zhou‡,
Li Shen,
Han Jia and
Xiaorong Tan
*,
Yanlin Zhou‡,
Li Shen,
Han Jia and
Xiaorong Tan