
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA novel fluorescence phenomenon caused by amine induced ion-exchange between Cd2+ and Fe3+ ions†
Lei Jiaab,
Zhe Xueab,
Rong-Rong Zhuab,
Tong Yanab,
Yu-Na Wangab,
Suo-Shu Zhangab,
Xiao Gengab,
Lin Du*ab and
Qi-Hua Zhao *ab
*ab
aKey Laboratory of Medicinal Chemistry for Natural Resource Education Ministry, Yunnan University, Kunming 650091, People's Republic of China. E-mail: lindu@ynu.edu.cn; qhzhao@ynu.edu.cn
bSchool of Chemical Science and Engineering, School of Pharmacy, Yunnan University, Kunming 650091, People's Republic of China
First published on 2nd December 2019
Abstract
A 3D metal–organic framework {[Cd(5-Brp)(dpa)]·0.5DMF·H2O}n (1) was successfully synthesized and characterized, which markedly recognized iron ions under the induction of an amino group. With the concentration of Fe3+ increasing, the emission of 1 first declined, then enhanced with a red shift and was finally quenched, which was different from the reference compound [Cd(5-Brp)(bpp)(H2O)]n (2). This result drew our attention to amine induced ion-exchange. This peculiar phenomenon inspired us to construct an effective ion detector.
1. Introduction
Metal–organic frameworks (MOFs), also known as porous coordination polymers, differ from inorganic porous materials and general organic complexes. MOFs combine the rigidity of inorganic materials with the flexible characteristics of organic materials, offering great development potential and attractive development prospects in modern materials research. In order to change or improve the properties of MOFs, much effort has been made to change the structure by varying the reaction conditions,1,2 replacing the metal centers3 or ligands,4 and introducing functional groups such as carboxyl or amino groups.5,6 In recent years, with the deepening of research on metal–organic frameworks, people have discovered their wide applications such as gas adsorption and separation,7 catalysis,8 electromagnetic9 and ion detection,10,11 etc.For ion detection, analyzing the change of fluorescence is a immediate and effective method. There are mainly five kinds of detection mechanisms: the collapse of the framework,12 the ion-exchange between the targeted ions and central metal ions of MOFs,13 the resonance energy transfer,14 the absorption competition,15 and the weak interaction between metal ions and the heteroatom within the organic ligands.16 Of course different mechanisms can also exist in the same detection process.15 Since lots of MOFs have the fluorescence excitation or emission wavelength which overlaps with the ultraviolet absorption wavelength range of iron ions, these complexes are often used to detect iron ions with the mechanism of absorption competition and resonance energy transfer.15,17 However, the detection of iron ions with the mechanism of ion-exchange is rarely reported up to now, especially caused by inducing group. In addition, the quenching process of iron ions by most metal–organic frameworks rarely contains any peak shifts or enhances, as is the case with the quenching process of reference complex 2 herein (Fig. S10, ESI†). Complex 1 and 2 both conform with the rule of absorption competition (Fig. S13, ESI†), but their fluorescent phenomena during iron ion detection were very different. We also analyzed the quenching process of complex 1 and found an obvious distinction between these two complexes, although their ancillary ligands were only slightly different.
In order to study this striking phenomenon in depth, 1 and 2 were characterized by XRD, PXRD, TGA, elemental analyses (Fig. S2 and S3, ESI†), etc. And the possible amine induced ion-exchange mechanism was discussed in this paper.
2. Results and discussion
2.1 Structure of 1 and 2
Single-crystal X-ray diffraction analysis illustrated that 1 crystallizes in the monoclinic crystal system with the C2/c space group. The asymmetric unit consists of one six-coordinated Cd2+ ion, one 5-bromoisophthalic acid (5-Brp2−) ligand, and one di-4-pyridinamine (dpa) ligand. As revealed in Fig. 1a, Cd1 is coordinated with two N donors from two pyridine rings of different dpa ligands, four O atoms from two carboxylate groups of different 5-Brp2− ligands and one water O atom to form a coordination configuration of pentagonal bipyramidal. There are two carboxylate groups bridging the adjacent Cd2+ ions to form an unit of [Cd(COO)]2 cluster, which is linked by 5-Brp2− ligands to generate a ribbon 1D chain in the ab-plane. And in the bc-plane, Cd2+ ions are further linked by dpa ligands to form other 1D chain (Fig. 1b). A 3D structure consisting of two kinds of above 1D chains is generated. These two chains are intertwined like warp and weft, forming a three-dimensional structure whose topology with a point symbol of {65·8}.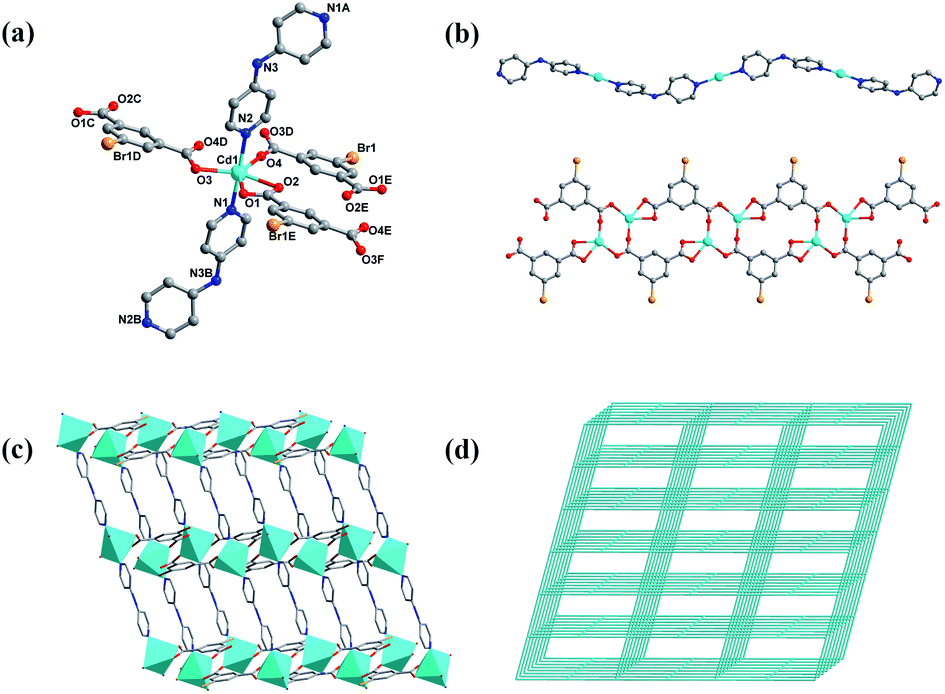 | ||
| Fig. 1 (a) Coordination environment of Cd(II) in 1 (symmetry code: (A) −0.5 + x, 0.5 + y, −0.5 + z; (B) 0.5 + x, 0.5 − y, 0.5 + z; (C) 0.5 + x, −0.5 + y, z; (D) 0.5 − x, 0.5 − y, −z; (E) −x, 1 − y, −z; (F) −0.5 + x, 0.5 + y, z) (b) 1D chain generated by dpa and Cd2+ ions (top) and 1D chain generated by 5-Brp and Cd2+ ions (bottom) (c) polyhedral illustration of the linkage through two kinds of 1D chains in 1 (d) view of 4-connected CdSO4 topology with the point symbol being {65·8} in 1. H atoms and solvent molecules are omitted for clarity. | ||
The complex 2 belonging to C2/c space group also crystallizes in the monoclinic crystal system. In addition, 2 was synthesized through a similar way with 1 except the bpp ligand substituted dpa ligand. Therefore complex 2 was selected as a comparison and its structure was shown in Fig. S1, ESI.†
2.2 Fluorescence
We collected the fluorescence of complex 1 in ethanol at room temperature, as well as ligand 5-Brp and dpa. From the Fig. S4, ESI† we found that the largest emissions of 5-Brp and complex 1 were both equal to 441 nm, which meant the emission of complex 1 was mainly from the ligand 5-Brp. 5 mg complex 1 was weighed and dissolved in 10 mL of ethanol. We obtained the supernatant of complex 1 by means of ultrasound, centrifugation, etc. The solution of Fe(NO3)3 (0.01 M) was added to 2 mL above supernatant and an interesting phenomenon occurred. The fluorescence intensity of complex 1 was declined when the concentration of Fe3+ ions was from 0 to 25 μM. While the concentration of Fe3+ ions increasing to 25 μM, an interesting new peak at about 525 nm arised and then the emission peak of complex 1 began to red shift to 480 nm and gradually enhanced until the concentration reached to 125 μM. Subsequently, it could be observed that the peak with red shift was weaken when the concentration of Fe3+ ions increased from 125 μM to 1 mM (Fig. 2).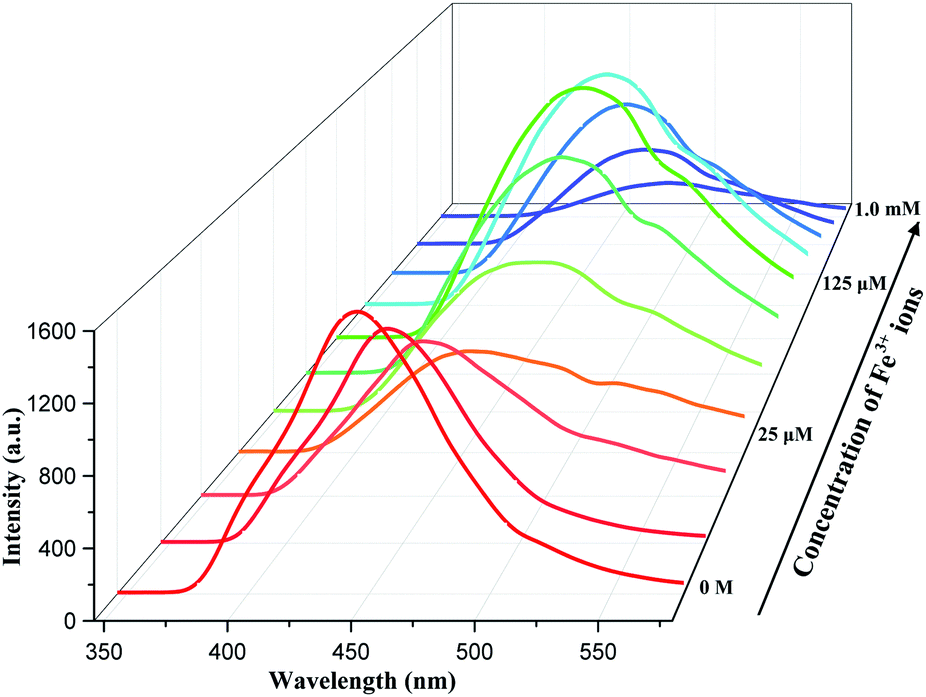 | ||
| Fig. 2 Fluorescence of complex 1 with different [Fe3+] in ethanol. | ||
What did cause the special luminescence phenomenon of complex 1? According to the mechanisms mentioned in the introduction, we have verified them separately. To further study the above interesting phenomenon, 5 mg complex 1 was immersed into 2 mL solution of Fe3+ (0.1 M) for 10 minutes. It was easy to find that the color of complex 1 turned from white to brown quickly and obviously (Fig. S5, ESI†). The residue was separated by centrifugation and washed by deionized water. The PXRD of obtained residue was collected and compared with complex 1. From the Fig. S6, ESI† we found the structure of complex 1 was not broken after immersed in Fe3+ solution for 10 minutes. First, we could exclude the redox reaction between complex 1 and iron ions because the Fe 2p XPS spectra remained unchanged after complex 1 immersed in Fe3+ ions (Fig. S7, ESI†). SEM with EDS was carried out and the result showed that there was no Fe(III) at the place where Cd(II) located and vice versa (Fig. 3). ICP-MS also showed that Cd2+ ions were released to solution after adding Fe3+ ions (Table S1†). Then we conjectured there might be a competition between Fe3+ and Cd2+ ions on the surface of complex 1, instead of the absorption of Fe3+ ions. We have also designed a test to prove the capture of Fe3+ ions. The powder of complex 1, 5-Brp and dpa were added into the mixed solution of Fe(NO3)3 and NaSCN, respectively. For dpa, the color of mixed solution turned to pale yellow from red rapidly. For complex 1, the color red also faded rapidly. But the color of mixed solution hardly changed for 5-H2Brp (Fig. S8, ESI†). The above result indicated that the ligand dpa and dpa in complex 1 both had a rather obvious interaction with Fe3+ ions. Then the maximum emission of dpa with different [Fe3+] in ethanol was recorded and we found a large shift from 425 to 525 nm (Fig. S9b, ESI†). Interestingly, this large shifted peak (at 525 nm) matched the new peak easily observed at about 525 nm in Fig. 2, which meant there was an interaction between Fe3+ ions and dpa in complex 1. Besides, the arising of this new peak implied the beginning of ion-exchange. Compared with the fluorescence perform of complex 2, it could be inferred that the secondary amine group rather than pyridine in ligand dpa played a decisive role. The FT-IR showed that there was no change at the peak of secondary amine, implying the connection between iron ion and the nitrogen atom from pyridine ring (Fig. S11, ESI†). And this kind of connection style was reported by Andrew Yeh and his co-authors in 1995.18 The PXRD of complex 1 immersed in different concentration of Fe3+ solution indicated the whole exchange process (Fig. S12, ESI†). Partial ion-exchange process can still retain the framework structure while the complete exchange can lead to destruction of the framework.13,19 Herein it could be indicated that the whole quenching process was divided into three stages. At first step low concentration of Fe3+ ions led to an absorption competition. With the increasing concentration of Fe3+ ions, exchange happened between Cd2+ and Fe3+ ions on the surface of complex 1. Finally, the above two metal ions completed the exchange. Then there occurred an absorption competition between the exchange product and surplus Fe3+ ions, leading the shift emission to fade.
 | ||
| Fig. 3 Element maps of 1 immersed in Fe3+ ions, (a) SEM image; (b) Cd (green); (c) Fe (blue); (d) overlap of above three images. | ||
3. Experimental
3.1 Materials and methods
All reagents and solvents (AR grade) purchased directly for synthesis were used without any further purification. The PXRD data of all samples were collected over a scanning ranges from 5 to 50° on a Rigaku TTRIII-18 kW diffractometer at ambient temperature. Thermogravimetric analyses (TGA) of all samples were carried out on a NETSCHZ STA-449C thermoanalyzer over a heating range from room temperature to 800 °C at a rate of 10 °C min−1 in N2 atmosphere. The Fourier-transform infrared spectra were recorded with a FT-IR Thermo Nicolet Avatar 360 FTIR spectrometer. The UV/Vis absorption spectra were recorded using a Hitachi U-4100 UV/Vis spectrophotometer. The fluorescence spectra were measured with an Edinburgh Instrument F920 spectrometer at room temperature. The XPS tests were performed using a Thermo Scientific K-Alpha XPS spectrometer. The elemental analyses (C, H, and N) were performed with a PerkinElmer analyzer. The ICP-MS analyses were acquired with Agilent 7700X.The XRD data were collected with a Bruker SMART AXS APEX II single crystal diffractometer at 298 K with Mo-Kα radiation (λ = 0.71073 Å). Absorption correction was based on symmetry equivalent reflections by using the SADABS program.20 The crystal structures of 1 and 2 were solved by direct methods and refined on F2 by full-matrix least-squares methods with the SHELXL-2014 program and Olex2 program.21,22 All non-hydrogen atoms were refined anisotropically. The crystalline structure data of all samples are displayed in Table S2,† and details of bond lengths and angles are recorded in Tables S3–S6.†
3.2 Preparation of {[Cd(5-Brp)(dpa)]·0.5DMF·H2O}n (1) and [Cd(5-Brp)(bpp)(H2O)]n (2)
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in a 5 mL glass tube. The glass tube was sealed and heated at 120 °C for 48 hours in oven and then cooled slowly to room temperature at a rate of 5 °C per hour. The resulting colourless crystals were collected and washed with DMF and water. The yield of 1 was 52% based on 5-H2Brp. C19.5H16.5BrCdN3.5O5.5 (580.17): calcd C 40.33%, H 2.84%, N 8.45%; found C 40.30%, H 2.86%, N 8.43%. FTIR (KBr pellets): ν∼ = 3438.97 (s), 1602.55 (vs.), 1557.82 (s), 1350.38 (s), 1210.33 (s), 1060.35 (m), 1027.61 (m), 821.09 (m), 772.54 (m), 724.53 (m), 606.54 (w), 533.01 (w).:7) in a 5 mL glass tube. The glass tube was sealed and heated at 120 °C for 48 hours in oven and then cooled slowly to room temperature at a rate of 5 °C per hour. The resulting colourless crystals were collected and washed with DMF. The yield of 2 was 37% based on 5-H2Brp. C21H19BrCdN2O5 (571.71): calcd C 44.08%, H 3.32%, N 4.90%; found C 43.99%, H 3.40%, N 4.92%. FTIR (KBr pellets): ν∼ = 3442.96 (s), 1612.80 (vs.), 1560.71 (m), 1349.79 (s), 1226.26 (m), 1099.00 (m), 1022.81 (m), 845.00 (m), 795.04 (m), 732.57 (m), 572.72 (w).
:1) in a 5 mL glass tube. The glass tube was sealed and heated at 120 °C for 48 hours in oven and then cooled slowly to room temperature at a rate of 5 °C per hour. The resulting colourless crystals were collected and washed with DMF and water. The yield of 1 was 52% based on 5-H2Brp. C19.5H16.5BrCdN3.5O5.5 (580.17): calcd C 40.33%, H 2.84%, N 8.45%; found C 40.30%, H 2.86%, N 8.43%. FTIR (KBr pellets): ν∼ = 3438.97 (s), 1602.55 (vs.), 1557.82 (s), 1350.38 (s), 1210.33 (s), 1060.35 (m), 1027.61 (m), 821.09 (m), 772.54 (m), 724.53 (m), 606.54 (w), 533.01 (w).:7) in a 5 mL glass tube. The glass tube was sealed and heated at 120 °C for 48 hours in oven and then cooled slowly to room temperature at a rate of 5 °C per hour. The resulting colourless crystals were collected and washed with DMF. The yield of 2 was 37% based on 5-H2Brp. C21H19BrCdN2O5 (571.71): calcd C 44.08%, H 3.32%, N 4.90%; found C 43.99%, H 3.40%, N 4.92%. FTIR (KBr pellets): ν∼ = 3442.96 (s), 1612.80 (vs.), 1560.71 (m), 1349.79 (s), 1226.26 (m), 1099.00 (m), 1022.81 (m), 845.00 (m), 795.04 (m), 732.57 (m), 572.72 (w).4. Conclusions
In summary, we synthesized two complexes and found an unusual enhanced red shift during iron ion detection of 1. After a series of studies, we drew a conclusion: It was the amine induced ion-exchange between Cd2+ and Fe3+ ions, which resulted the enhanced red shift while Fe3+ ions were added into the alcohol solution of complex 1. This phenomenon was rarely reported before and the possible mechanism suggested that the amine group in pyridine ligand might enhance the response of pyridine ligand to Fe3+ ions, which may induce ion-exchange and lead to emission shift in detection of ions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We express appreciate the support of this work by the National Natural Science Foundation of China (project no. 21461029 and 21561033), and the Yunnan University's Research Innovation Fund for Graduate Students (no. 2018Z069).Notes and references
- K. Shen, J. Chen, X. Chen, J. Long, R. Q. Lu, L. Zhang, Y. Li and B. Chen, Science, 2018, 359, 206 CrossRef CAS PubMed
.
- Y. Zhao, L. Wang, N. N. Fan, M. L. Han, G. P. Yang and L. F. Ma, Cryst. Growth Des., 2018, 18, 7114 CrossRef CAS
- C. Marquez, F. G. Cirujano, S. Smolders, C. Van Goethem, I. Vankelecom, D. De Vos and T. De Baerdemaeker, Dalton Trans., 2019, 48, 3946–3954 RSC
- Z.-F. Wu, B. Tan, J.-Y. Wang, C.-F. Du, Z.-H. Deng and X.-Y. Huang, Chem. Commun., 2014, 51, 157 RSC
- J. F. Feng, S. Y. Gao, T. F. Liu, J. L. Shi and R. Cao, ACS Appl. Mater. Interfaces, 2018, 10, 6014–6023 CrossRef CAS PubMed
- T.-Y. Luo, C. Liu, S. V. Eliseeva, P. F. Muldoon, S. Petoud and N. L. Rosi, J. Am. Chem. Soc., 2017, 139, 9333–9340 CrossRef CAS PubMed
- K. Adil, Y. Belmabkhout, R. S. Pillai, A. Cadiau, P. M. Bhatt, A. H. Assen, G. Maurin and M. Eddaoudi, Chem. Soc. Rev., 2017, 46, 3402–3430 RSC
- J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C. Y. Su, Chem. Soc. Rev., 2014, 43, 6011–6061 RSC
- Z. Xiang, Y. Song, J. Xiong, Z. Pan, X. Wang, L. Liu, R. Liu, H. Yang and W. Lu, Carbon, 2019, 142, 20–31 CrossRef CAS
- A. Pankajakshan, D. Kuznetsov and S. Mandal, Inorg. Chem., 2019, 58, 1377–1381 CrossRef CAS PubMed
- P. Chandra Rao and S. Mandal, Inorg. Chem., 2018, 57, 11855–11858 CrossRef CAS PubMed
- Y.-Y. Cao, X.-F. Guo and H. Wang, Sens. Actuators, B, 2017, 243, 8–13 CrossRef CAS
- D. Maity and T. Govindaraju, Chem. Commun., 2012, 48, 1039–1041 RSC
- S. M. Sheta, S. M. El-Sheikh, M. M. Abd-Elzaher and A. R. Wassel, Appl. Organomet. Chem., 2019, 33, e4777 CrossRef
- L.-S. Li, X. Wang, Y.-Y. Jia, S.-X. Xu, M.-H. Yu and Y.-H. Zhang, Eur. J. Inorg. Chem., 2018, 9, 1068–1072 CrossRef
- Q. Tang, S. Liu, Y. Liu, J. Miao, S. Li, L. Zhang, Z. Shi and Z. Zheng, Inorg. Chem., 2013, 52, 2799–2801 CrossRef CAS PubMed
- R. R. Zhu, T. Wang, T. Yan, L. Jia, Z. Xue, J. Zhou, L. Du and Q. H. Zhao, Dalton Trans., 2019, 48, 12159–12167 RSC
- C.-L. Lin, K. Hung, A. Yeh, H.-T. Tsen and C.-C. Su, Inorg. Chem., 1999, 38, 411–414 CrossRef CAS
- S. Dang, E. Ma, Z.-M. Sun and H. Zhang, J. Mater. Chem., 2012, 22, 16920–16926 RSC
- R. H. Blessing, Acta Crystallogr., Sect. A: Found. Crystallogr., 1995, 51, 33–38 CrossRef PubMed
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: PXRD, SEM, XPS, ICP-MS, FTIR, fluorescence, crystal structure data and photograph. CCDC 1916377 and 1916379 (for 1 and 2 respectively) contain the supplementary CIF data for this paper. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra07559c |
| This journal is © The Royal Society of Chemistry 2019 |