
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBacteriophage nanofiber fabrication using near field electrospinning†
Ryota Sugimotoab,
Ju Hun Lee‡
ab,
Ju-Hyuck Lee§
ab,
Hyo-Eon Jin¶
ab,
So Young Yoo *de and
Seung-Wuk Lee*abc
*de and
Seung-Wuk Lee*abc
aDepartment of Bioengineering, University of California at Berkeley, Berkeley, CA 94720, USA. E-mail: leesw@berkeley.edu
bBiological Systems and Engineering, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA
cTsinghua Berkeley Shenzhen Institute, Berkeley, USA
dBIO-IT Foundry Technology Institute, Pusan National University, Busan 609-735, Republic of Korea. E-mail: yoosy@pusan.ac.kr
eResearch Institute for Convergence of Biomedical Science and Technology, Yangsan 626-770, Republic of Korea
First published on 28th November 2019
Abstract
M13 bacteriophage (phage) nano- and microfibers were fabricated using electrospinning. Using liquid crystalline suspension of the phage, we successfully fabricated nano- and microscale pure phage fibers. Through a near field electrospinning process, we fabricated the desired phage fiber pattern with tunable direction and spacing. In addition, we demonstrated that the resulting phage fibers could be utilized as an electrostatic-stimulus responsive actuator. The near field electrospinning would be a very useful tool to design phage-based chemical sensors, tissue regenerative materials, energy generators, metallic and semiconductor nanowires in the future.
Introduction
Organized functional nanomaterials are very useful in many applications in material synthesis, sensing, energy harvesting, and biomedical engineering.1–6 Conventionally, self-assembly,1,2,6–9 photolithography micro- and nanofabrication, atomic layer deposition,10–14 and electrospinning have been utilized to fabricate desirable nano- and microstructures.15–20 Through the coupling with functional biomaterials with active proteins and peptides, these technologies have been applied to a variety of biomaterial fields including tissue engineering,21–24 drug delivery,25 biosensors5,26 and energy harvesting.2,27–30 Recently, M13 bacteriophage (phage) has been used to construct various functional nanomaterials and devices due to its self-assembly properties and chemical flexibility to display functional peptides.31 In order to create one-dimensional nanofibers of M13 phage, electrospinning (ES) has been demonstrated.4,32–35 However, in previous studies, due to the low viscosity of the phage suspension, viscosity enhancing polymers, such as polyvinyl-pyrrolidone (PVP), have been utilized, which significantly block the biochemical functions of the phages through the non-specific binding to the M13 phage. Alternatively, 1,1,1,3,3,3-hexafluoro-2-propanol, a toxic ES solvent has been adapted to optimize evaporation of solvent to induce fiber formation during the ES process. The resulting fiber exhibited non-woven fiber mesh structures useful for filters, fabrics, and tissue engineering material design.36 More recently, the near field electrospinning (NFES) technique has been developed by coupling the ES process with writing capability with stage translators.37–40 NFES is an advanced ES process to enable to deposit electrospun nanofibers in desired patterns with a translating stage. Through combination of the phage engineering and NFES, we can greatly expand our design capability of phage nanomaterials for multiple new applications. Here, we fabricated pure M13 phage nano- and microfibers using the NFES approaches. We first utilized a highly concentrated liquid crystalline solution of the phage and successfully span it into non-woven nano- and microfiber mesh structures. We then utilized NFES to make them organize in desired patterns using a translating stage controller. Furthermore, we demonstrated that electrospun phage fibers could be utilized as mechanical actuators using oscillating electric field. The resulting phage electrospun fiber might be useful to construct various desired nano- and micropatterns for electric and biomedical sensors, actuators, and tissue engineering materials in the future.Experimental
Genetic engineering of phage
We used four glutamates engineered phage for electrospinning that were constructed previously.1 We amplified the single clone of the phage in a large scale using Escherichia coli cultures and purified, and confirmed the DNA sequences as previously reported.1Phage electrospinning
To conduct electrospinning (ES), phage solutions (40–80 mg ml−1) were prepared in 12.5 and 25 mM Tris-buffered saline (TBS), and deionized water (DIW). Prepared phage solution was filled in a micro nozzle (108 and 210 μm inner and outer diameter respectively; TSK laboratory). We used ITO coated glass plates for optical characterization samples and gold coated silicon wafer for AFM characterization samples as a collector. One electrode was connected to the nozzle to apply a high voltage (4–6 kV) using a high voltage supplier (Glassman High Voltage, NJ) and the other electrode was connected to a gold coated silicon substrate as a ground. The distance between the nozzle and the collector was controlled at ∼4 cm.Phage near field electrospinning
We used a home-built translator by modifying 3D printer (Da Vinci, XYZ-printing, San Diego, CA) where both of the needle and the collector were installed. We controlled three dimensional (x-, y-, and z-axis) movement of spinning nozzle by G-code editing software (Repetier-Host and Repetier-Firmware, Hot-World GmbH & Co. KG, Germany). Distance between the needle and the collector was controlled by the 3D printer, while being monitored and confirmed by an attached camera (USB Digital microscope, XCSOURCE, Tseung Kwan O, Hong Kong). NFES was conducted using 500 μm nozzle-collector distance, and 600–700 V potential to produce electrospun fibers. The phage electrospun fibers were produced with wide range of concentrations (40–80 mg ml−1, solvent: 50 mM and Di water). The distance between the nozzle and the collector was controlled at ∼5 mm.Microscopy characterization
Optical microscopy images were taken using an IX71 Inverted Fluorescence Microscope (Olympus, Tokyo, Japan) equipped with a digital CCD camera (QImaging, Surrey, Canada) and with polarizing filters. SEM images were collected using a scanning electron microscope (Quanta™ 3D FEG, FEI™, Hillsboro, OR). Atomic force microscopy images were collected using an MFP3D AFM (Asylum Research, Santa Barbara, CA) and analyzed using Igor Pro 6.0 (WaveMetrics, Inc. Lake Oswego, OR) and the Asylum software package (Asylum Research, Santa Barbara, CA). All images were taken in air using tapping mode.Mechanical characterization of electrospun phage fibers
The Asylum MFP-3D (Asylum Research, Santa Barbara, CA), Igor Pro 6.0, and Asylum software package was used to measure the mechanical properties of electrospun phage fibers through AFM nanoindentation method. In this method, the probe was a cone shaped silicon tip (Tap300Al-G, Ted Pella, Inc., Redding, CA) with <10 nm radius, and 62 N m−1 spring constant. The force-indentation loading and unloading curves of 50 different points in several phage electrospun fibers were taken with AFM nanoindentation method. According to the Oliver–Pharr model,41 a line with slope S = dF dl−1 (S: unloading stiffness, F: force, l: indentation length) was fit in the upper portion of the unloading curves in the data to calculate Young's modulus.42 We used dehydrated electrospun phage fibers on the gold-coated silicon substrate under environmental conditions (25 °C, 40% relative humidity).Phage-based actuator fabrication and characterization
To fabricate self-standing fibers, near field electrospinning was conducted over two separated pieces of copper electrode both of which were grounded, and the gap between two copper electrodes was an approximately 1 mm gap. After conducting near field electrospinning, the copper electrodes were pealed from the substrate, and the fibers were observed using USB Digital microscope, (XCSOURCE, Tseung Kwan O, Hong Kong). Potential response experiment has been conveyed in the 3D printer and images were obtained by the USB camera aforementioned. The distance between the fiber and the electrode was confirmed by the image from web camera. Several shapes of electric potential waves were applied by a function generator (BK PRECISION 4003A, BK Precision, Yorba Linda, CA), and the applied waves were verified by an oscilloscope (Tektronix, TDS 1001B, Beaverton, OR).Results and discussion
We utilized liquid crystalline solution of M13 phage for fabricating phage electrospun fibers. M13 phage is a long-rod shape bacterial virus (Fig. 1A). It has 880 nm in length and 6.6 nm in diameter. It is covered by ∼2700 copies of coat protein (pVIII), which encapsulate ∼7000 base pair phage genome. Due to its anisotropic shape, we can easily form lyotropic liquid crystalline structures with nematic, cholesteric, and smectic alignment (Fig. 1B).43–46 We amplified phage in a large scale using bacterial amplification, purified through polyethylene glycol precipitation, and suspended in buffered solutions. We prepared viscous liquid crystalline phage solution (40–80 mg ml−1) suspended in various solvents. We filled the phage solution in a reservoir connected to a micro nozzle. One electrode was connected to the nozzle to apply a high voltage (4–6 kV) using a high voltage supplier and the other electrode was connected to a gold-coated silicon substrate as a ground. The distance between the nozzle and the collector was controlled at ∼4 cm (Fig. 1C). Because of the liquid crystalline properties of the phage suspension, the phage solution exhibit bright cholesteric liquid crystalline morphology under the cross polarized light (Fig. 1B and C).46 When we applied the electric field, the Taylor cone was immediately developed and began to spin the fibers. During the ES process, the cholesteric phage solution was pinched to form Taylor cone and broke to spin phage fiber. Near the tearing surface of the Taylor cone (red arrow in Fig. 1C), the solution exhibited brighter light scattering indicating that the phages are more directionally aligned on the Taylor cone surface. The electrospun fibers from the Taylor cone also exhibited very bright light under the cross-polar, indicating that they have a higher crystallinity with directional alignment parallel to the long axis of the fiber (Fig. 1C).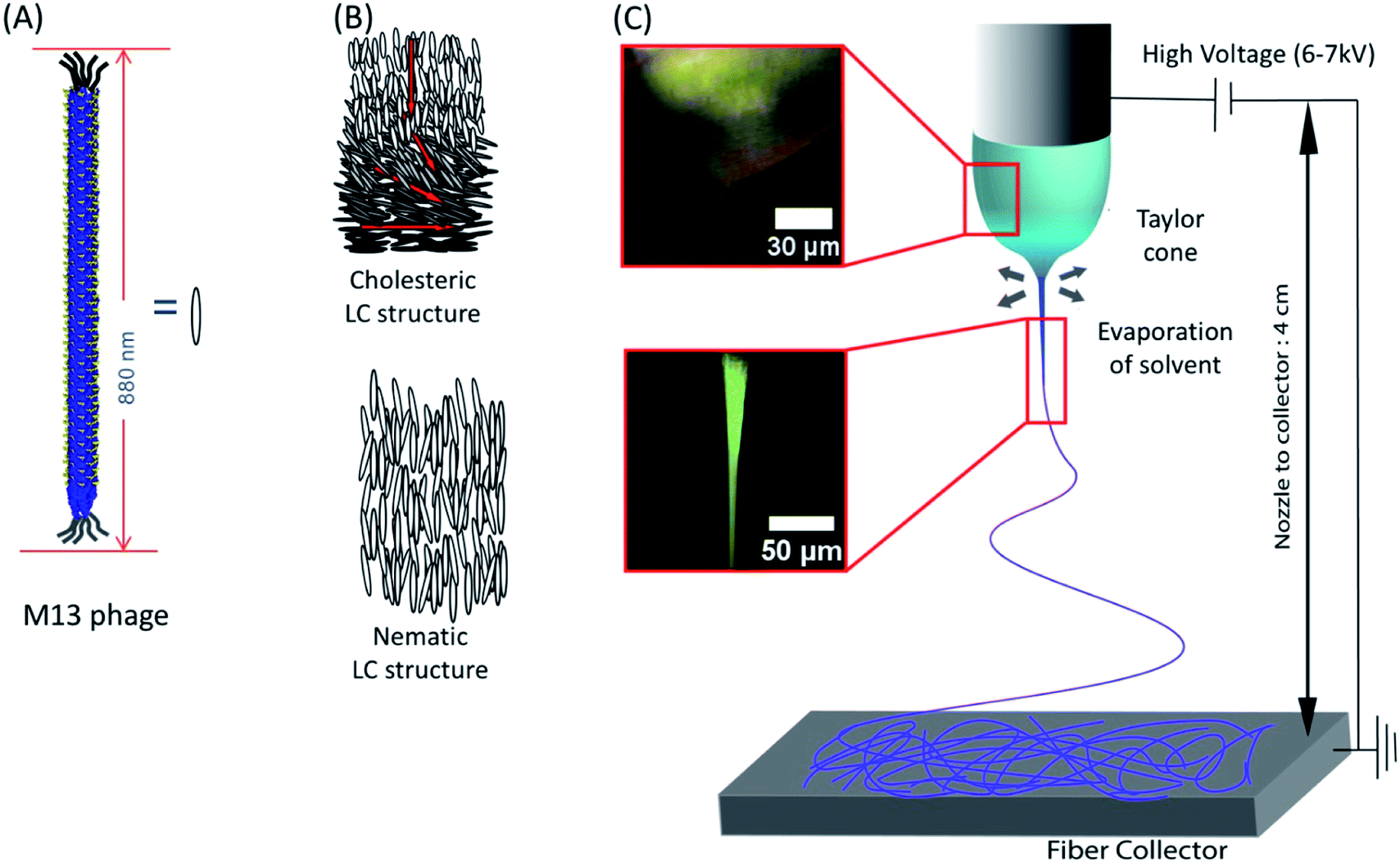 | ||
| Fig. 1 Schematic of the phage electrospinning. M13 phage has (A) rod like structure, and its aggregation typically forms (B) nematic or cholesteric structure. (C) POM image of phage droplet electrospinning showing a Taylor cone transitioning from cholesteric phage solution, which was typically observed in 30–80 mg ml−1 phage concentration, into a highly crystalized state (68.4 mg ml−1 12.5 mM, TBS). | ||
Optical microscopy images (Fig. 2A) of the electrospun phage showed that the resulting phage fiber formed continuous non-woven fiber network structures. POM image analysis (Fig. 2B) shows that the electrospun phage fiber exhibited specifically oriented bright light scattering, which indicates that fibers have highly crystalline ordered structure. SEM image (Fig. 2C) analysis shows that the resulting phage fiber thickness and appearance varied in the range from tens nanometers to a few micrometers. The AFM image of the electrospun fibers (Fig. 2D) shows that the fibers composed of multiple smaller phage fibrils (40–60 nm in diameter), which are oriented through the long axis of the fibers. When we increased the applied voltages during the electrospinning, the diameter of the phage fibers gradually decreased (Fig. 2E). The resulting fiber morphologies and shapes showed relatively high distribution possibly due to instability of Taylor cone and phage solution inhomogeneity.
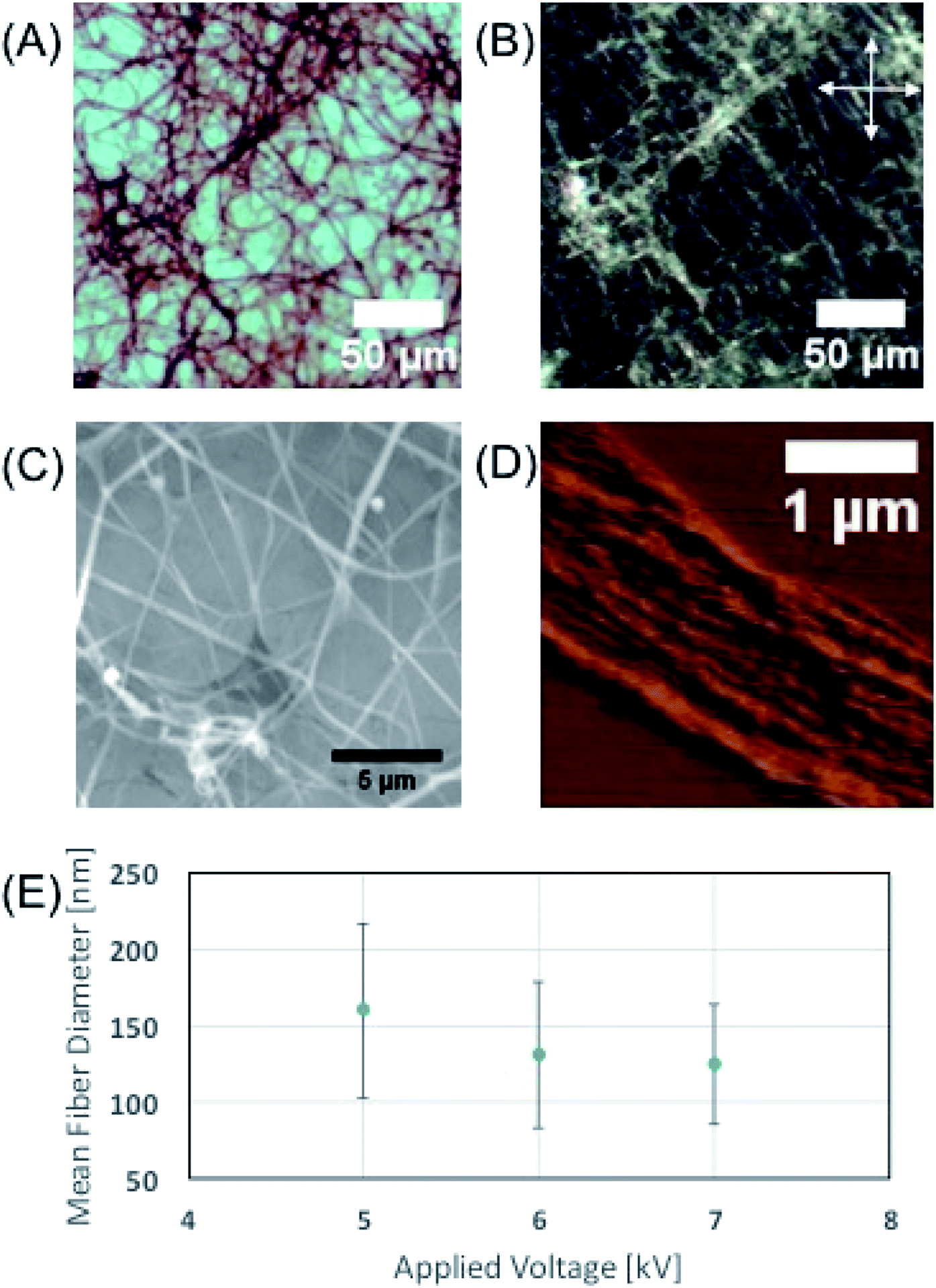 | ||
| Fig. 2 Characterization of phage fibers fabricated by electrospinning. (A) Optical microscope image of (4E, 56.5 mg ml−1, DI water) phage fibers. (B) Polarized Optical Microscope image of image (A). Allows are indicating the direction of the cross polar. (C) Scanning Micro Scope image of phage fibers (4E, 75.6 mg ml−1, 25 mM TBS). (D) Atomic force microscope image of an unfolded phage fiber (4E, 75.6 mg ml−1, 25 mM TBS). (E) Experimental result of applied voltage vs. phage fiber diameter (4E, 75.6 mg ml−1, 25 mM TBS). | ||
We created desired phage fiber patterns using NFES process. We constructed home-built NFES apparatus by coupling the ES equipment with conventional 3D printer (Da Vinci, XYZ-printing, San Diego, CA). The NFES utilizes a short needle–collector distance, a low applied voltage and rapid and precise movement of the spinning nozzle using translating stage controller so that it can produce highly controlled continuous fiber with desired pattern, orientation and position.37–40 We used 500 μm nozzle–collector distances, and 600–700 V potential to produce fibers continuously during the NFES (Fig. 3A). For NFES, we used similar liquid crystalline solution concentration (40–80 mg ml−1) to that of the ES previously described. By manipulating the translating stage, we produced highly aligned phage fibers (Fig. 3B). The resulting phage fiber exhibited strong light scattering. Dark-field imaging of the phage fiber exhibited iridescent light scattering (Fig. 3C). POM images showed the electrospun phage fiber exhibited highest brightness at 45° to the cross polar, while disappearing at vertical or parallel orientation to the cross polar direction (Fig. 3D). We compared these optical properties with a wet spun fiber, which was fabricated with 30 mg ml−1 and 260 μm diameter capillary by being crosslinked with 2.5% glutaraldehyde solution in accordance with the previously reported method.4 The resulting near field electrospun fibers have highly crystalline ordered structure while the wet spun fibers exhibited bright light scattering regardless of the orientation under the cross polar. This is mainly due to that the wet spun fibers have multiple domains that have a nematic liquid crystalline phage structure but it has loosely oriented through the long axis of the wet spun fibers (ESI Fig. S1†). However, the NFES fibers have much higher orientation than those of the wet spun fibers. We could write various desired pattern such as a right-angled line, triangle, parallel line, and mesh patterns by optimizing movement and writing speed of the translating stage (Fig. 3E–G).
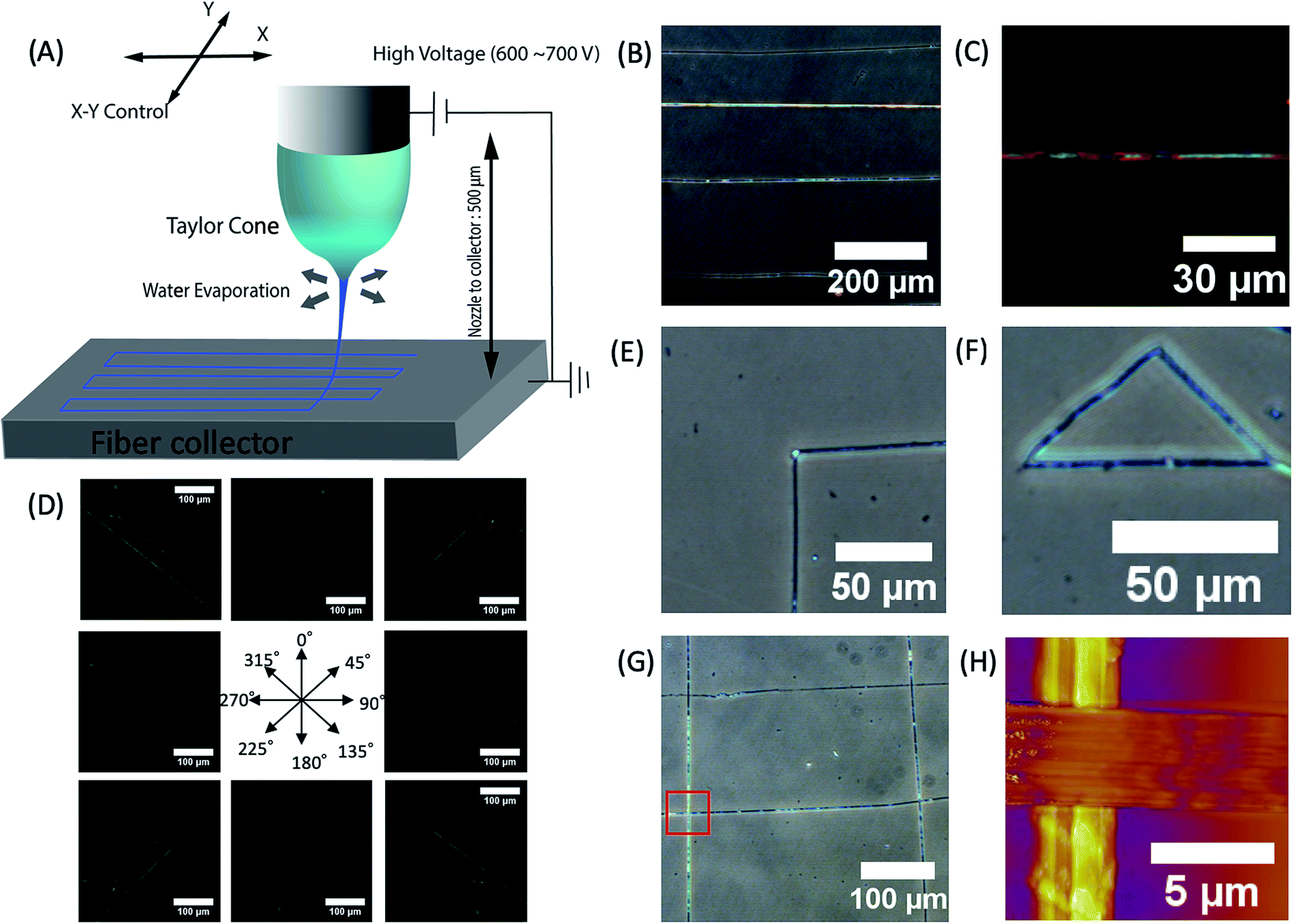 | ||
| Fig. 3 Characterization of phage fibers fabricated by near field electrospinning (A) schematic diagram of phage near field electrospinning. (B) Optical image of straight line patterns fabricated by phage near field electrospinning (56.5 mg ml−1, DIW). (C) Dark field optical microscope image of phage fiber (56.5 mg ml−1, DIW). (D) POM images of aligned phage fibers showing high birefringent (56.5 mg ml−1, DIW). (E–G) Optical microscope mages of ordered patterns written by near field electrospinning such as (E) 90° corner, (F) triangle and (G) grid (4E, 56.5 mg ml−1, DIW). (H) AFM image of crossing fiber showing parallel alignment of phages (56.5 mg ml−1, DIW). | ||
In order to investigate the effect of the writing velocity of translator on the fiber morphology, the writing velocity of the fiber has been characterized between 500–1250 cm min−1. We observed that the microfiber orientation could be more easily controlled by increasing the writing velocity (Fig. 4A and B) in the given voltage ranges. Moreover, in order to see the controllability of fiber position, variance of actual fiber–fiber distance from the desired distance was characterized in terms of writing velocity (Fig. 4C). We observed that position variation of electrospun fiber decreases with increase of writing velocity (Fig. 4D).
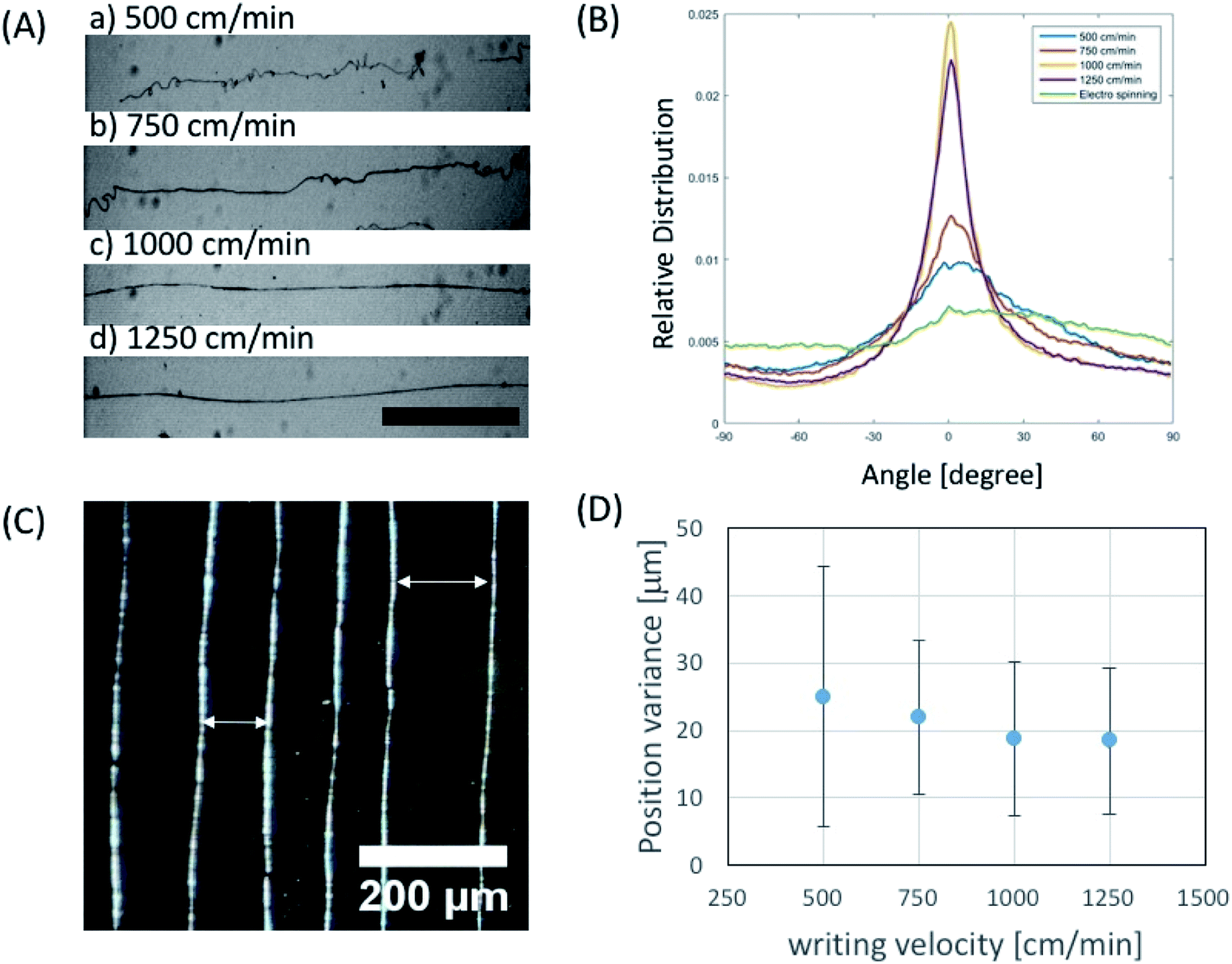 | ||
| Fig. 4 Characterization of near field electrospun fiber depending on writing velocity. (A) Optical microscope images of phage fibers written by several writing velocities (40 mg ml−1 in 50 mM TBS, scale bar: 500 μm). (B) Characterization of phage fiber orientation vs. writing velocity. (C) Phase contrast image of squares written by near field electrospinning at the writing velocity of 1000 cm min−1 (40 mg ml−1 in 50 mM TBS). (D) Experimental result of positioning variance from aimed place vs. writing velocity. | ||
We demonstrated the electrospun microfibers as an actuator using alternating electric field. Due to the intrinsic charged surface of the phage, the M13 phage can exhibit good material properties as an electric resonator. In order to test this hypothesis, we first characterized the mechanical properties of the electrospun phage using AFM nanoindentation. Using Oliver–Pharr model,41 which enables to evaluate mechanical parameters of elastic–plastic materials, we measured the elastic modulus of electrospun phage fibers. To calculate Young's modulus, a line with unloading stiffness slope was fit in the upper regime of the unloading curves in AFM nanoindentation results. Base on unloading stiffness value, the calculated Young's modulus of electrospun phage fiber was 1.7 ± 0.4 GPa (n = 50) (ESI Fig. S2†). We found that a self-standing phage microfiber showed highly sensitive mechanical bending response toward applied electric potentials (Fig. 5A). The mechanical bending of the phage against electrostatic-stimulus is depending on the fiber stiffness, electric potential, and the distance between the fiber and the electrode. When we applied electric potentials ranging from 1 to 20 V, we observed a mechanical bending actuation of the phage fiber (Fig. 5B and C). To characterize the sensitiveness of the actuation, we measured the response of the fiber which was fabricated by NFES and located ∼100–200 μm away from the electrode. Electric potentials were applied by a function generator and the applied electric field were verified by an oscilloscope. Upon application of 20 V square waves to the fiber, the phage fiber exhibited prompt response (Fig. 5D). When we applied an alternate sine wave potential, the deflection of the phage fiber faithfully followed the applied sine wave shapes (Fig. 5E). To evaluate the potential response quantitatively, we measured the deflection corresponding to alternating sign wave voltages ranging from 1 to 20 V. The phage fiber linearly increased its deflection with increase of electric potential. The fiber could sensitively distinguish single voltage difference in the response of the phage fiber deflection (Fig. 5F). When we repeated the deflection 3000 times, we observed there is little hysteresis on the actuation motion (Fig. 5G). We believe that the highly sensitive response of the phage fiber to the electric field is originated due to negatively charged surface of the phages and its outstanding mechanical properties.
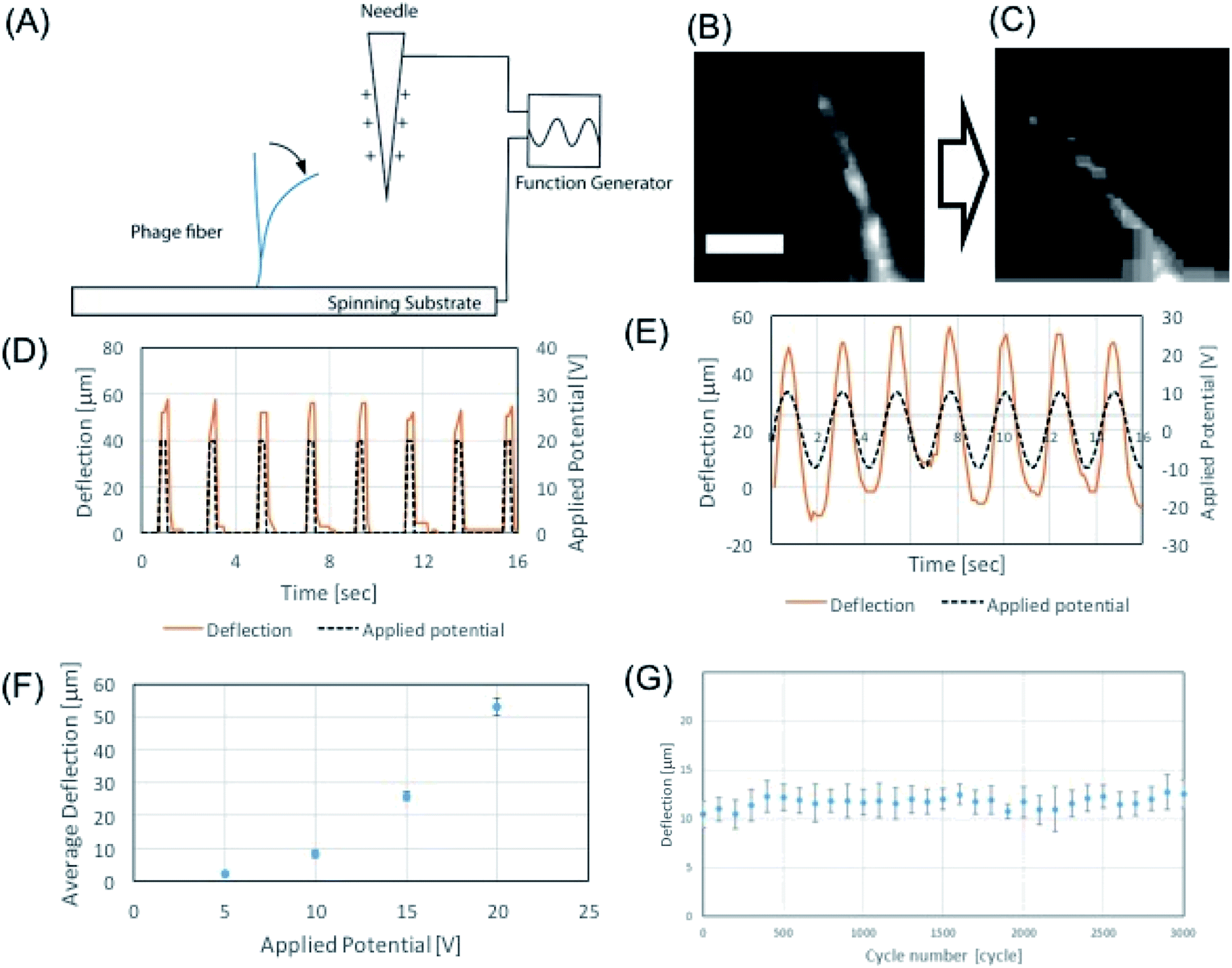 | ||
| Fig. 5 Phage-based electric resonator. (A) Schematics of potential response of phage resonator. (B and C) Standing phage fiber responding to 20 V applied potential (56.5 mg ml−1, DI water) (scale bar 40 μm). (D and E) Phage fiber responses to square potential (D) and sinewave potential (E). (F) Phage fiber deflection corresponding to 5–20 V applied potential in 200 μm distance. (G) Experimental result of cyclic virus fiber deflection test with 0–20 V sinewave for 3000 cycles. | ||
Conclusions
We constructed phage-based nanofibers using ES. We prepared liquid crystalline suspension which is viscous enough to spin the phage fibers and fabricated the phage nano- and microfibers solely made of the M13 phage without any additives. Through the NFES process, we also constructed desired phage patterns with directional orientation and spacing. Furthermore, we constructed the electrostatic-stimulus responsive actuator using the resulting phage fibers. Our approaches to fabricate phage nano- and microfibers without any function inhibitors such as polymers and cross-linkers will be very useful to create many phage-based materials and applications. Furthermore, through genetic modification of functional peptides on the M13 phage and writing desired patterns, the phage NFES would be very useful to design chemical sensor,1,5,26,51 tissue regenerative medicine,21,22,24,47–52 energy generation, harvesting,2,27–30 metal reduction and metal nanowires,53 and water purification54 in the future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Tsinghua–Berkeley Shenzhen Institute, the National Research Foundation of Korea grant funded by the Korean Government (NRF-2014S1A2A2027641) and supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HI16C1067). Further support by Yuhan Inc. is appreciated.References
- W.-J. Chung, J.-W. Oh, K. Kwak, B. Y. Lee, J. Meyer, E. Wang, A. Hexemer and S.-W. Lee, Nature, 2011, 478(7369), 364–368 CrossRef CAS PubMed
.
- B. Y. Lee, J. Zhang, C. Zueger, W.-J. Chung, S. Y. Yoo, E. Wang, J. Meyer, R. Ramesh and S.-W. Lee, Nat. Nanotechnol., 2012, 7(6), 351–356 CrossRef CAS PubMed
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318(5849), 426–430 CrossRef CAS PubMed
- S. W. Lee and A. M. Belcher, Nano Lett., 2004, 4(3), 387–390 CrossRef CAS
- J.-W. Oh, W.-J. Chung, K. Heo, H.-E. Jin, B. Y. Lee, E. Wang, C. Zueger, W. Wong, J. Meyer, C. Kim, S.-Y. Lee, W.-G. Kim, M. Zemla, M. Auer, A. Hexemer and S.-W. Lee, Nat. Commun., 2014, 5, 3043 CrossRef PubMed
- S. H. Yang, W. J. Chung, S. McFarland and S. W. Lee, Chem. Rec., 2013, 13(1), 43–59 CrossRef CAS PubMed
- A. Lomander, W. Hwang and S. Zhang, Nano Lett., 2005, 5(7), 1255–1260 CrossRef CAS PubMed
- D. J. Lipomi, R. V. Martinez, M. A. Kats, S. H. Kang, P. Kim, J. Aizenberg, F. Capasso and G. M. Whitesides, Nano Lett., 2011, 11(2), 632–636 CrossRef CAS PubMed
- D. C. Watson, R. V. Martinez, Y. Fontana, E. Russo-Averchi, M. Heiss, A. Fontcuberta I Morral, G. M. Whitesides and M. Lončar, Nano Lett., 2014, 14(2), 524–531 CrossRef CAS PubMed
- Y. M. Lee, B. Jung, Y. H. Kim, A. R. Park, S. Han, W. S. Choe and P. J. Yoo, Adv. Mater., 2014, 26(23), 3899–3904 CrossRef CAS PubMed
- J. S. Lee, Y. J. Lee, E. L. Tae, Y. S. Park and K. B. Yoon, Science, 2003, 301(5634), 818–821 CrossRef CAS PubMed
- C. Guan, X. Wang, Q. Zhang, Z. Fan, H. Zhang and H. J. Fan, Nano Lett., 2014, 14(8), 4852–4858 CrossRef CAS PubMed
- S. M. George, Chem. Rev., 2010, 110(1), 111–131 CrossRef CAS PubMed
- R. M. Costescu, D. G. Cahill, F. H. Fabreguette, Z. A. Sechrist and S. M. George, Science, 2004, 303(5660), 989–990 CrossRef CAS PubMed
- A. Greiner and J. H. Wendorff, Angew. Chem., Int. Ed., 2007, 46(30), 5670–5703 CrossRef CAS PubMed
- D. Li, Y. Wang, Y. Xia, D. Li, Y. Wang, Y. Wang, Y. Xia and Y. Xia, Adv. Mater., 2004, 16(4), 361–366 CrossRef CAS
- D. Li, Y. Wang and Y. Xia, Nano Lett., 2003, 3(8), 1167–1171 CrossRef CAS
- D. Li and Y. Xia, Adv. Mater., 2004, 16(14), 1151–1170 CrossRef CAS
- W. Liu, S. Thomopoulos and Y. Xia, Adv. Healthcare Mater., 2012, 1(1), 10–25 CrossRef CAS PubMed
- J. Xie, M. R. MacEwan, A. G. Schwartz and Y. Xia, Nanoscale, 2010, 2(1), 35–44 RSC
- H. Wang and S. C. Heilshorn, Adv. Mater., 2015, 1–20 Search PubMed
- G. A. Silva, C. Czeisler, K. L. Niece, E. Beniash, D. A. Harrington, J. A. Kessler and S. I. Stupp, Science, 2004, 303, 1352–1355 CrossRef CAS PubMed
- J. D. Hartgerink, Science, 2001, 294(5547), 1684–1688 CrossRef CAS PubMed
- M. A. Bokhari, G. Akay, S. Zhang and M. A. Birch, Biomaterials, 2005, 26(25), 5198–5208 CrossRef CAS PubMed
- C. Westwater, L. M. Kasman, D. A. Schofield, P. A. Werner, J. W. Dolan, M. G. Schmidt and J. S. Norris, Antimicrob. Agents Chemother., 2003, 47(4), 1301–1307 CrossRef CAS PubMed
- J. Jaworski, K. Yokoyama, C. Zueger, W. J. Chung, S. W. Lee and A. Majumdar, Langmuir, 2011, 27(6), 3180–3187 CrossRef CAS PubMed
- D. Oh, J. Qi, B. Han, G. Zhang, T. J. Carney, J. Ohmura, Y. Zhang, Y. Shao-Horn and A. M. Belcher, Nano Lett., 2014, 14(8), 4837–4845 CrossRef CAS PubMed
- S. H. Nam, H. S. Shim, Y. S. Kim, M. A. Dar, J. G. Kim and W. B. Kim, ACS Appl. Mater. Interfaces, 2010, 2(7), 2046–2052 CrossRef CAS
- K. T. Nam, D. Kim, P. J. Yoo, C. Chiang, N. Meethong, P. T. Hammond, Y. Chiang and A. M. Belcher, Science, 2008, 312, 885–889 CrossRef PubMed
- J. P. Breen, F. C. Meunier, M. Haneda, W. Sun, Y. Kindaichi, H. Hamada, Y. Yu, J. Saussey, M. Daturi, C. Hedouin, T. Seguelong, A. Simon, J. M. Weil, G. W. Harris, H. Frei, B. Weckhuysen, S. Diego, E. Fridell, M. Wolf, D. A. King, Z. P. Liu, H. Arnolds, I. M. Lane, D. C. Papageorgopoulos, A. Rushton, M. Gill, P. Fox, A. Daunois, M. Yamaguchi, I. Goto, M. Kumagai, K. C. Patil, M. S. Hegde, S. J. Jenkins, P. Bazin, O. Marie, D. Astruc, T. Yoshinari, Y. Kintaichi, Y. J. Lee, H. Yi, W.-J. Kim, K. Kang, D. S. Yun, M. S. Strano, G. Ceder and A. M. Belcher, Science, 2009, 324, 1051–1055 Search PubMed
- T. Bratkovič, Cell. Mol. Life Sci., 2010, 67(5), 749–767 CrossRef PubMed
- L. Wu, J. Zang, L. A. Lee, Z. Niu, G. C. Horvatha, V. Braxtona, A. C. Wibowo, M. A. Bruckman, S. Ghoshroy, H.-C. zur Loye, X. Li and Q. Wang, J. Mater. Chem., 2011, 21(24), 8550–8557 RSC
- W. Salalha, J. Kuhn, Y. Dror and E. Zussman, Nanotechnology, 2006, 17(18), 4675–4681 CrossRef CAS PubMed
- R. Korehei and J. Kadla, J. Appl. Microbiol., 2013, 114(5), 1425–1434 CrossRef CAS PubMed
- R. Korehei and J. F. Kadla, Carbohydr. Polym., 2014, 100, 150–157 CrossRef CAS PubMed
- C. Y. Chiang, C. M. Mello, J. Gu, E. C. C. M. Silva, K. J. Van Vliet and A. M. Belcher, Adv. Mater., 2007, 19(6), 826–832 CrossRef CAS
- G. Zheng, W. Li, X. Wang, D. Wu, D. Sun and L. Lin, J. Phys. D: Appl. Phys., 2010, 43, 415501 CrossRef
- D. Sun, C. Chang, S. Li and L. Lin, Nano Lett., 2006, 6(4), 839–842 CrossRef CAS PubMed
- J. Chang, Y. Liu, K. Heo, B. Y. Lee, S. W. Lee and L. Lin, Small, 2014, 10(10), 1920–1925 CrossRef CAS PubMed
- C. Chang, K. Limkrailassiri and L. Lin, Appl. Phys. Lett., 2008, 93(12), 2–5 CrossRef
- W. C. Oliver and G. M. Pharr, J. Mater. Res., 1992, 7(6), 1564–1583 CrossRef CAS
- B. R. Neugirg, S. R. Koebley, C. Schniepp and A. Fery, Nanoscale, 2016, 8, 8414–8426 RSC
- G. P. Smith and V. a. Petrenko, Chem. Rev., 1997, 97(96), 391–410 CrossRef CAS PubMed
- Z. Dogic and S. Fraden, Langmuir, 2000, 16(20), 7820–7824 CrossRef CAS
- M. Adams, Z. Dogic, S. L. Keller and S. Fraden, Nature, 1998, 393(1997), 349–352 CrossRef CAS
- S. W. Lee, C. Mao, C. E. Flynn and A. M. Belcher, Science, 2002, 296(5569), 892–895 CrossRef CAS PubMed
- K. Rajangam, H. A. Behanna, M. J. Hui, X. Han, J. F. Hulvat, J. W. Lomasney and S. I. Stupp, Nano Lett., 2006, 6(9), 2086–2090 CrossRef CAS PubMed
- S. Y. Yoo, J.-W. Oh and S.-W. Lee, Langmuir, 2012, 28(4), 2166–2172 CrossRef CAS PubMed
- S. Y. Yoo, W.-J. Chung, T. H. Kim, M. Le and S.-W. Lee, Soft Matter, 2011, 7(2), 363–368 RSC
- S. Y. Yoo, A. Merzlyak and S.-W. Lee, Soft Matter, 2011, 7(5), 1660–1666 RSC
- Q. P. Pham, U. Sharma and A. G. Mikos, Tissue Eng., 2006, 12(5), 1197–1211 CrossRef CAS PubMed
- S. Y. Yoo, M. Kobayashi, P. P. Lee and S. W. Lee, Biomacromolecules, 2011, 12(4), 987–996 CrossRef CAS PubMed
- J. P. Park, M. Do, H. E. Jin, S. W. Lee and H. Lee, ACS Appl. Mater. Interfaces, 2014, 6(21), 18653–18660 CrossRef CAS PubMed
- I. Xu, B. Xin, Z. Chen, Y. Liu, Y. Zheng and F. Zhang, RSC Adv., 2019, 9(29), 16754–16766 RSC
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra07510k |
| ‡ Present address: Department of Bionanoengineering, Hanyang University, Ansan 15588, South Korea. |
| § Present address: Department of Energy Science and Engineering, Daegu Gyeongbuk Institute of Science and Technology (DGIST), Daegu 42988, Republic of Korea. |
| ¶ Present address: College of Pharmacy, Ajou University, Suwon 16499, Republic of Korea. |
| This journal is © The Royal Society of Chemistry 2019 |