
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnalysis of antisense oligonucleotides with the use of ionic liquids as mobile phase modifiers†
Anna Kaczmarkiewicz ,
Judyta Zielak,
Łukasz Nuckowski and
Sylwia Studzińska*
,
Judyta Zielak,
Łukasz Nuckowski and
Sylwia Studzińska*
Chair of Environmental Chemistry and Bioanalytics, Faculty of Chemistry, Nicolaus Copernicus University, 7 Gagarin Str., PL-87-100 Toruń, Poland. E-mail: kowalska@chem.umk.pl; Fax: +48-56-6114837; Tel: +48-56-6114308
First published on 28th November 2019
Abstract
The main goal of this study was the investigation of the impact of several ionic liquids, commonly used as free silanol suppressors, on the retention and separation of phosphorothioate oligonucleotides. Three various stationary phases (octadecyl, octadecyl with embedded polar groups and pentafluorophenyl) as well as ionic liquids with the concentration range of 0.1–7 mM were used for this purpose. The results obtained during this study showed that the increase in concentration of ionic liquids results in increasing retention of the oligonucleotides. Such an effect was observed regardless of the stationary phase used. Moreover, elongation of the alkyl chain in the structure of ionic liquids caused an increase of antisense oligonucleotide retention factors. The results obtained during retention studies confirmed that addition of ionic liquids to the mobile phase influences antisense oligonucleotide retention in a way similar to the case of commonly used ion pair reagents such as amines. A method of oligonucleotide separation was also developed. The best selectivity was obtained for the octadecyl stationary phase since separation of mixtures of antisense oligonucleotides and their metabolites differing in sequence length was successful. It has to be pointed out that ionic liquids were used for the first time as mobile phase additives for oligonucleotide analysis.
1. Introduction
Ionic liquids (ILs) are organic salts, typically composed of large, asymmetrical organic cations such as 1-alkyl-3-methylimidazolium and inorganic anions, especially chloride, tetrafluoroborate or hexafluorophosphate.1,2 These compounds have several interesting physicochemical properties related to their structures, e.g. minor vapor pressure, which is a beneficial factor influencing the safety of using ILs as solvents.3 ILs are used in a variety of applications, e.g. as stationary phases in capillary electrophoresis and gas chromatography, and as additives to the mobile phase in liquid chromatography.4–6Application of ILs as mobile phase modifiers in liquid chromatography results from their donor–acceptor properties, whereby they are able to block free silanols present on the surface of silica stationary phases, similarly to amines which are commonly used silanol activity suppressors.3 Xiaohua et al.4 used four different ILs containing 3-methylimidazolium tetrafluoroborate anions in amines analysis. They proved that as concentration of ILs and the length of the alkyl chain in the imidazolium cation structure increase, the retention factors (k) of amines decrease, which is related to cation repulsion. Furthermore, the researchers postulated that addition of ILs to the mobile phase may effectively shield free silanol groups and improve analyte peak shapes. Similar conclusions were drawn by He et al.2 during the analysis of ephedrines. Moreover, the authors suggested that ILs are better additives for peak shape enhancement than amines commonly used as silanol blockers (e.g. triethylamine TEA).
Besides the ability of amines to suppress the activity of free silanol groups, they are also commonly used as ion pair reagents (IPRs) in ion pair chromatography (IPC) as they are adsorbed on the stationary phase surface, and form ion pairs with the negatively charged analytes. Thus, polar and ionized compounds, such as oligonucleotides, are retained due to electrostatic and hydrophobic interactions, as well as hydrogen bonding.7
Nowadays, IPC is the most popular technique for quantification and separation of antisense oligonucleotides (ASOs), which are nucleic acid analogs, used in the treatment of several diseases.8 These compounds are built of several dozen nucleotides and are characterized by a modified structure, which prevents digestion by intracellular nucleases. Several difficulties can be encountered during their analysis, e.g. selection of stationary phase and appropriate IPR, which could provide effective separation of ASOs within a reasonable time as well as high MS sensitivity.9–12 Commonly used mobile phase composed of triethylammonium acetate (TEAA) and acetonitrile causes significant ionization suppression in mass spectrometry (MS).13 Application of more hydrophobic alkylamine acetates caused enhancement of MS sensitivity, however, the main issue was cation adduction. For this reason, 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) was introduced to the mobile phase composed of alkylamine and methanol, providing high MS sensitivity, a small number of adducts and complete ASO separation.14 Nowadays, such mobile phases are the most popular for oligonucleotides analysis. On the other hand, Huber et al.15–17 obtained a significant reduction of cation adducts when triethylammonium bicarbonate and butyldimethylammonium bicarbonate were used as mobile phase for the analysis of various oligonucleotides. However, the separation efficiency for some of them (e.g. polythymidylic acids) was not satisfactory when a low concentration of IPR was applied. Consequently, there is a need for the development of other mobile phase additives (e.g. ILs) or other modes of liquid chromatography application. Ion exchange chromatography (IEC) and hydrophilic interaction liquid chromatography (HILIC) are also used in the analysis of oligonucleotides. IEC offers selective separation of N-deleted oligonucleotides, and accurate optimization of the chromatographic conditions allows for analysis of ASO in biological matrices with detection limits ranging from 40–250 ng ml−1.12 HILIC mode offers relatively high MS sensitivity, however, application of ammonium acetate or ammonium formate significantly influences separation selectivity.18,19
Until now, there have been no reports on application of ILs as mobile phase additives in ASO analysis. Therefore, an attempt has been made to examine the influence of IL type and concentration on the ASO retention for three different chromatographic columns. Moreover, the study included a comparison of the influence of ILs and conventional IPR on ASO retention. The methods for separation of modified ASOs and their metabolites were also developed for each tested column.
2. Experimental
2.1 Materials
ASOs standards with the different modification, length and sequence were used in the present investigation (Table 1). Standards were prepared by dissolution in deionized water to the concentration of 25 μM. Six phosphorothioate oligonucleotides, modified in each phosphate group, were selected for retention and separation studies (OL1–OL6) and they had been purchased from Sigma-Aldrich (Gillingham, Dorset, UK). The remaining ASOs were modified in each sugar moiety and were obtained from Eurogentec (Seraing, Liege, Belgium).| Name | Sequence 5′ → 3′ | Modification | Molecular mass | Mixture |
|---|---|---|---|---|
| OL1 | GCCCAAGCTGGCATCCGTCA | Phosphorothioate | 6368 | MIX 1 |
| OL2 | GCCCAAGCTGGCATCCG | 5413 | ||
| OL3 | GCCCAAGCTGGCAT | 4458 | ||
| OL4 | GGGGAAGCTGGCATCCGTCA | 6488 | MIX 2 | |
| OL5 | GCCCAAGCTGGCATGGGTCA | 6448 | ||
| OL6 | GGGGAAGCTGGCATGGGTCA | 6568 | ||
| ME20 | GCCCAAGCTGGCATCCGTCA | 2′-O-Methyl | 6621 | MIX 3 |
| ME19 | GCCCAAGCTGGCATCCGTC | 6278 | ||
| ME18 | GCCCAAGCTGGCATCCGT | 5959 | ||
| MOE20 | GCCCAAGCTGGCATCCGTCA | 2′O-Methoxyethyl | 7657 | MIX 4 |
| MOE19 | GCCCAAGCTGGCATCCGTC | 7269 | ||
| MOE18 | GCCCAAGCTGGCATCCGT | 6892 |
Mobile phases were made with the use of deionized water (Milli-Q system, Millipore, El Paso, TX, USA), methanol (MeOH) (Merck KGaA, Darmstadt, Germany) and ILs such as 1-ethyl-3-methylimidazolium chloride [EMIM][Cl], 1-butyl-3-methylimidazolium chloride [BMIM][Cl], and 1-hexyl-3-methylimidazolium chloride [HMIM][Cl] (Sigma-Aldrich, Gillingham, Dorset, UK). The structures of ILs cations were presented on Fig. S1 in ESI.† Moreover, N,N-dimethylbutylamine and triethylamine (Sigma-Aldrich, Gillingham, Dorset, UK) were used in order to compare the influence of ILs and alkylamines on ASO retention. 5 mM amine chlorides were prepared by dilution of amine in deionized water and adjusting the pH to 6.8 by using of 5% hydrochloric acid (Merck KGaA, Darmstadt, Germany). Sodium chloride solution of the same concentration was also used as a mobile phase (POCH S.A., Gliwice, Poland). The mixture of phenol/chloroform/isoamyl alcohol (25/24/1, v/v/v) (VWR International, Poland) was used during liquid–liquid extraction (LLE) of ASO from human serum enriched with ASO.
2.2 Oligonucleotide selection
Six phosphorothioate oligonucleotides were selected for the retention studies. One oxygen atom in each phosphate group in these ASOs is replaced with the sulfur atom. Three of them (OL1–OL3) differed in the length and other ones (OL4–OL6) had the same length, but different position of nucleotides (Table 1). The remaining ASOs (ME18–20 and MOE18–20) are modified in each sugar moiety with the use of 2-O-methyl and 2-O-methoxyethyl groups. Such modifications are introduced in order to increase the stability of these compounds with regard to the activity of intracellular enzymes activity. OL2 and OL3 are synthetic metabolites of OL1, shorter by three and six nucleotides respectively from 3′ end, while ME19, ME18 as well as MOE18 and MOE19 are metabolites of ME20 and MOE20 respectively (Table 1). Mixtures of synthetic ASO metabolites and positional isomers (OL4–OL6) differing in the sequence were selected in order to study the method selectivity. In the case of large molecules such as ASOs, separation of mixture of compounds with small differences in sequences and masses demonstrates the selectivity of the developed method.2.3 Apparatus and chromatographic conditions
All chromatographic analyses were performed with the use of the UltiMate® 3000 Binary Rapid Separation LC (RSLC) system with a DAD-3000RS Diode Array Detector (Dionex, Sunnyvale, CA, USA). The data were collected with the Thermo Scientific Dionex Chromeleon 7.0 software.Three various chromatographic columns with the same dimensions (100 × 2.1 mm, 1.7 μm) were tested in the investigations: Syncronis aQ (octadecyl with embedded polar group, Thermo, Waltham, USA), Kinetex F5 (pentafluorophenyl, Phenomenex, Torrance, USA) and Kinetex C18 (octadecyl, Phenomenex, Torrance, USA).
The concentrations of the studied ILs were in the range of 0.1–7 mM for octadecyl column, while for remaining stationary phases 3–7 mM of ILs were tested. Mobile phase flow rate was 0.2 ml min−1 (due to the limited apparatus pump pressure) for aQ column and 0.3 ml min−1 for the remaining ones. The following gradient elution program was used: 25–60% v/v MeOH in 15 minutes. The temperature of the autosampler and column was 30 °C. The UV detection wavelength was selected as λ = 260 nm, while the injection volume was 1–3 μl depending of the IL type.
For separation of the tested mixtures, the concentration of [HMIM][Cl] was 7 mM and 10 mM. In order to obtain the best possible separation, some parameters such as the flow rate, column temperature and gradient elution programs were adjusted.
In order to compare ASO retention for ILs and alkylamines, the following gradient elution program was applied: 20–40% v/v MeOH in 15 minutes. The injection volume equaled 2 μl, while the autosampler and column temperature was 30 °C.
2.4 Determination of OGN in human serum with the developed method
Human serum was obtained from Ludwik Rydygier Voivodship Polyclinical Hospital of Torun, Poland. Serum was collected from healthy volunteers and all required consent was obtained for any experimentation with human subject. Diluted human serum at the ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 was used during ASO quantification. A sample was enriched with OL4 at final concentration of 10 μM and mixed with phenol:chloroform:isoamyl alcohol (25:24:1 v/v/v) mixture at the ratio of 1:1. Next, the suspension was centrifuged at 14000 rpm for 35 min. The resulting supernatant was washed with chloroform in order to remove phenol residue. This extraction was repeated five times with chloroform at the ratio of 1:3. The calibration curve was prepared by spiking the serum after extraction with the following concentrations of OL4: 8 μM, 10 μM, 13 μM, 17 μM and 20 μM.
:10 was used during ASO quantification. A sample was enriched with OL4 at final concentration of 10 μM and mixed with phenol:chloroform:isoamyl alcohol (25:24:1 v/v/v) mixture at the ratio of 1:1. Next, the suspension was centrifuged at 14000 rpm for 35 min. The resulting supernatant was washed with chloroform in order to remove phenol residue. This extraction was repeated five times with chloroform at the ratio of 1:3. The calibration curve was prepared by spiking the serum after extraction with the following concentrations of OL4: 8 μM, 10 μM, 13 μM, 17 μM and 20 μM.
3. Results and discussion
3.1 The impact of IL concentration on ASO retention
It is supposed that the excess amount of ionized IL molecules may adsorb on the stationary phase ligands, and therefore hydrophobic interactions between alkyl chain in IL cations and stationary phase ligands occur.4,20–22 For this reason different concentrations of three various ILs were selected for the retention studies of phosphorothioate ASOs. For all the tested ILs the anion was Cl−; thus only the impact of IL cation was tested. It needs to be emphasized it was the first time ILs were used as mobile phase modifiers in ASO analysis. Application of gradient elution was necessary in order to directly compare the k′ values of ASOs for each IL and column due to different strength of interactions between analytes, ILs and the tested stationary phase ligands.Firstly, [BMIM][Cl] in the wide range of concentrations (0.1–7 mM) was investigated with respect to the ASOs retention on octadecyl column. Such IL concentrations were selected based on the literature.2,20,21,23 ASO k′ values obtained during this step of study are presented in Table 2.
| k′ ± SD | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| [EMIM][Cl] | [BMIM][Cl] | [HMIM][Cl] | |||||||||
| OGN | 3 mM | 5 mM | 7 mM | 0.1 mM | 0.5 mM | 3 mM | 5 mM | 7 mM | 3 mM | 5 mM | 7 mM |
| aQ | |||||||||||
| OL1 | 5.988 ± 0.166 | 6.590 ± 0.331 | 10.036 ± 0.045 | n.a. | n.a. | 11.797 ± 0.011 | 12.347 ± 0.036 | 12.757 ± 0.091 | 19.717 ± 0.343 | 20.453 ± 0.077 | 21.431 ± 0.066 |
| OL2 | 6.445 ± 0.036 | 6.492 ± 0.127 | 6.658 ± 0.000 | n.a. | n.a. | 10.449 ± 0.011 | 11.193 ± 0.597 | 11.641 ± 0.254 | 18.301 ± 0.089 | 20.010 ± 0.048 | 20.239 ± 0.050 |
| OL3 | 5.425 ± 0.123 | 5.695 ± 0.027 | 7.021 ± 0.011 | n.a. | n.a. | 9.330 ± 0.066 | 10.388 ± 0.054 | 10.900 ± 0.620 | 17.025 ± 0.143 | 18.905 ± 0.054 | 19.170 ± 0.077 |
| OL4 | 6.081 ± 0.034 | 6.794 ± 0.023 | 8.226 ± 0.034 | n.a. | n.a. | 10.646 ± 0.089 | 11.538 ± 0.066 | 11.663 ± 0.111 | 18.208 ± 0.022 | 20.101 ± 0.000 | 20.203 ± 0.077 |
| OL5 | 6.090 ± 0.000 | 6.833 ± 0.011 | 8.124 ± 0.022 | n.a. | n.a. | 10.630 ± 0.497 | 11.804 ± 0.320 | 11.944 ± 0.199 | 18.237 ± 0.000 | 20.251 ± 0.011 | 20.494 ± 0.111 |
| OL6 | 5.260 ± 0.0658 | 6.215 ± 0.045 | 7.599 ± 0.011 | n.a. | n.a. | 10.183 ± 0.054 | 11.139 ± 0.034 | 12.180 ± 0.066 | 18.106 ± 0.100 | 20.196 ± 0.000 | 20.317 ± 0.000 |
|
|||||||||||
| C18 | |||||||||||
| OL 1 | 8.308 ± 0.008 | 9.016 ± 0.047 | 10.492 ± 0.49 | tm | 6.971 ± 0.082 | 15.892 ± 0.130 | 17.645 ± 0.130 | 18.683 ± 0.136 | 27.417 ± 0.014 | 31.808 ± 0.195 | 39.105 ± 0.833 |
| OL 2 | 6.494 ± 0.148 | 7.369 ± 0.305 | 8.331 ± 0.000 | tm | 6.167 ± 0.117 | 13.978 ± 0.006 | 15.917 ± 0.055 | 17.181 ± 0.026 | 26.752 ± 0.114 | 31.338 ± 0128 | 38.483 ± 0.075 |
| OL 3 | 0.954 ± 0.089 | 2333 ± 0.062 | 6.944 ± 0.053 | tm | 1.212 ± 0.135 | 12.593 ± 0.014 | 14.605 ± 0000 | 15.907 ± 0.028 | 26.762 ± 0.400 | 30.583 ± 0.128 | 37.285 ± 0209 |
| OL 4 | 6.408 ± 0.177 | 7.873 ± 0.095 | 8.466 ± 0.122 | n.a. | n.a. | 15.079 ± 0.061 | 16.855 ± 0.053 | 18.186 ± 0.014 | 28.980 ± 0.010 | 32.967 ± 0.169 | 40.388 ± 0.075 |
| OL 5 | 6.235 ± 0.055 | 7.456 ± 0.400 | 8.226 ± 0.067 | n.a. | n.a. | 14.733 ± 0.006 | 16.677 ± 0.020 | 18.138 ± 0.014 | 29.214 ± 0.020 | 32.904 ± 0.758 | 40.915 ± 0.171 |
| OL 6 | 5.445 ± 0.006 | 6.786 ± 0.548 | 7.130 ± 0.061 | n.a. | n.a. | 14.389 ± 0.047 | 16.473 ± 0.041 | 17.918 ± 0.012 | 29.515 ± 0.244 | 34.356 ± 0.670 | 40.464 ± 0.792 |
|
|||||||||||
| F5 | |||||||||||
| OL1 | 16.224 ± 0.295 | 18.545 ± 0.075 | 20.391 ± 1.306 | n.a. | n.a. | 25.085 ± 0.396 | 26.759 ± 0.443 | 29.255 ± 0.370 | 36.562 ± 0.198 | 41.360 ± 0.468 | 41.623 ± 0.000 |
| OL2 | 13.135 ± 0.075 | 15.210 ± 0.148 | 16.695 ± 0.223 | n.a. | n.a. | 23.688 ± 0.000 | 25.679 ± 0.000 | 28.172 ± 0.025 | 35.761 ± 0.395 | 39.686 ± 0.171 | 40.000 ± 0.121 |
| OL3 | 10.921 ± 0.097 | 13.207 ± 0.025 | 14.862 ± 0.000 | n.a. | n.a. | 21.666 ± 0.050 | 23.933 ± 0.050 | 26.341 ± 0.000 | 34.697 ± 0.122 | 37.768 ± 0.025 | 39.426 ± 0.199 |
| OL4 | 14.846 ± 1.532 | 15.892 ± 0.075 | 17.235 ± 0.050 | n.a. | n.a. | 24.684 ± 0.025 | 26.743 ± 0.025 | 29.080 ± 0.025 | 37.401 ± 0.50 | 41.238 ± 0.050 | 42.336 ± 0.075 |
| OL5 | 13.257 ± 0.050 | 15.490 ± 0.050 | 16.780 ± 0.050 | n.a. | n.a. | 24.282 ± 0.050 | 26.357 ± 0.025 | 28.677 ± 0.000 | 37.542 ± 0.000 | 41.222 ± 0.025 | 42.983 ± 0.345 |
| OL6 | 11.583 ± 0.148 | 13.973 ± 0.079 | 15.524 ± 0.000 | n.a. | n.a. | 23.585 ± 0.050 | 23.986 ± 2.738 | 28.313 ± 0.025 | 37.768 ± 0.075 | 41.291 ± 0.025 | 43.263 ± 0.050 |
ASOs were not retained on the stationary phase surface for the lowest concentration of IL cations (0.1 mM) in the mobile phase (Table 2). This is probably related to the fact that, the amount of [BMIM] cations adsorbed on the stationary phase ligands is probably insufficient to obtain ASO retention (Table 2).
Increasing of the ILs concentration ILs to 0.5 mM resulted in elution of ASOs after column void time; however, the k′ values obtained for these compounds are low (Table 2). Higher ASO k′ values were noted when [BMIM][Cl] concentration in the mobile phase was further increased (in the range between 3–7 mM) (Table 2 and Fig. 1). Such behavior of ASOs is similar to the case of IPC used in ASO analysis, where IPRs are used as mobile phase modifiers. Generally, the ASO retention mechanism in IPC results from simultaneous ion pair formation between negatively charged ASOs and cations of alkylamines, as well as IPRs adsorption on hydrophobic stationary phase ligands.9,11,24 Thus, both hydrophobic and electrostatic interactions take place in the retention of the analytes. One of the most important factors in IPC which influences ASOs retention is alkylamine concentration, since its increase results in greater ASOs retention, as in the case of our findings (Table 2).
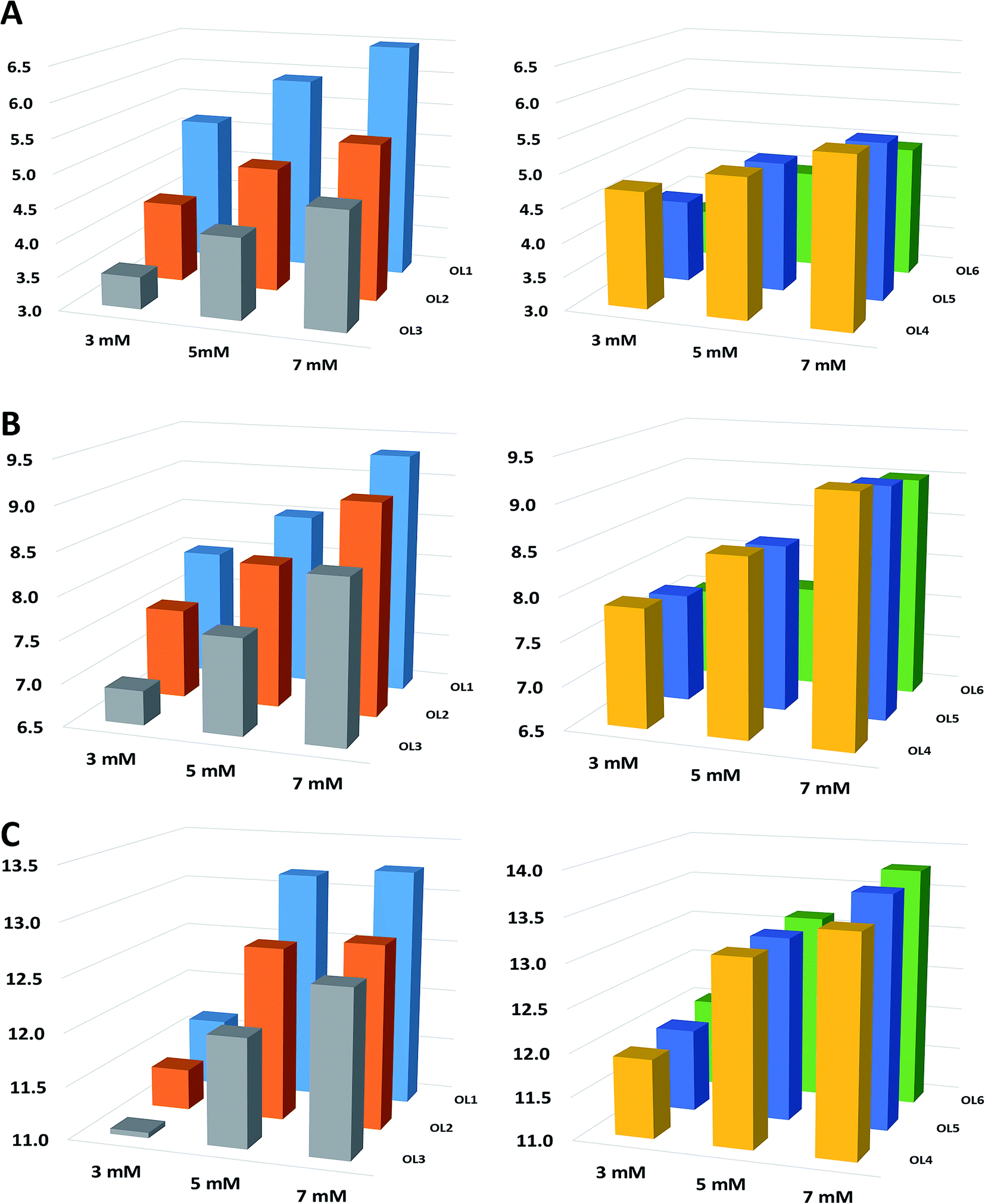 | ||
| Fig. 1 The dependence of the concentration of ionic liquid on ASOs retention coefficients (A) [EMIM][Cl], (B) [BMIM][Cl], (C) [HMIM][Cl]. Experimental conditions: Kinetex F5 column, column and autosampler temperature 30 °C, flow rate 0.3 ml min−1; gradient elution program: 25–60% v/v MeOH in 15 minutes. | ||
In order to confirm the tendencies noted for C18 stationary phase, investigation was extended to other stationary phases and two ILs ([EMIM][Cl], [HMIM][Cl]) for the concentration range 3–7 mM. Fig. 1 illustrates the results obtained for F5 column. The results for the remaining stationary phases are shown in Fig. S2 and S3 in the ESI.† As it can be seen in these figures, similar tendencies as in the case of C18 column were obtained for the other ILs and stationary phases. The observed effects are probably related to the fact that for higher IL concentration probably a greater amount of imidazolium cations adsorbs on the stationary phase ligands. Thus ion exchanger is dynamically formed on the packing material ligands, which consequently leads to more effective electrostatic interactions with ASOs (Fig. 1, S2, S3 and Table 2). Another possible cause of this effect may be related to the formation of ion pairs between IL cations and the studied compounds. The negative charge of ASOs is then neutralized and the adsorption on the hydrophobic stationary phase takes place during a chromatographic run, as a consequence of hydrophobic interactions.
Furthermore, for lower IL concentrations the observed peak shapes were poorer than for higher ones (data not shown).
3.2 The influence of IL type on ASO retention
Based on the data included in Table 2, it may be concluded that the highest ASO k′ values were obtained for the [HMIM][Cl], which contains the longest alkyl chain in the imidazolium cation, while the lowest ones were recorded for [EMIM][Cl] with the shortest alkyl chain in its structure. Such an effect was observed regardless of the tested column and ASO. Again, the observed tendencies are similar to those ones noted in IPC, where strength of interactions mentioned in Section 3.2 depends on the IPR structure – the longer the alkyl chain, the greater the ASO retention.7,9,11,25–27 This effect may be a consequence of stronger interactions between the hexyl chain in the [HMIM] cation and stationary phase ligands compared to ethyl chain in [EMIM][Cl] and butyl chain in [BMIM][Cl]. The greater the hydrophobicity of the imidazolium cation, the stronger its adsorption on the hydrophobic packing material and consequently greater ASOs retention.Berthod et al.22 postulated that ILs have a dual nature and both their anions and cations can adsorb on the stationary phase, depending on their position in lyotropic order. Chaotropic cations such as imidazolium ones are weakly hydrated, so they preferentially adsorb on the hydrophobic stationary phase ligands, since adsorption of cosmotropic anions (e.g. Cl−) is minor because they are strongly hydrated. Consequently, retention of basic solutes with positive charge is reduced when IL with a chaotropic cation and cosmotropic anion is added to the mobile phase.28 The k′ values of such analytes also decrease when alkyl chain in the imidazolium cation is longer.3 In the case of negatively charged ASOs, an opposite effect was observed (Table 2).
Furthermore, based on the data included in Table S1 in ESI† it may be concluded that, the longer the alkyl chain in the ILs structure is, the lower the symmetry of ASO peaks. Such an effect was observed regardless of tested stationary phase. It is probably related to stronger electrostatic interactions between ASOs and [HMIM] cation, compared to [BMIM] or [EMIM], which consequently leads to the peaks tailing. Moreover, with the increasing of given ILs concentration, there is slight decrease in the symmetry of tested compounds, which may be related with the adsorption of greater number of ILs cations and consequently, stronger electrostatic interactions (Table S1†). Although asymmetry factors obtained for [HMIM][Cl] are slightly greater, compared to the remaining ILs, peaks obtained for this IL were narrower, which may be crucial in terms of ASO mixtures separation.
Another important factor which may influence ASO retention is the sequence length. The longer the phosphodiester chain in the ASO structure, the stronger the retention, caused by the greater number of negatively charged phosphorothioate groups. Consequently, it is likely that a greater number of ion pairs (between ILs and ASOs) is formed, and they interact more effectively with the ILs adsorbed at the stationary phase structure (Table 2).
Another step of this investigation was performed in order to study the retention of imidazolium cations on the octadecyl ligands. The investigation was conducted for detection wavelength λ = 210 nm and the mobile phase consisted of methanol and water. Individual ionic liquids ([EMIM][Cl] and [BMIM][Cl]) were injected into the chromatographic system, together with the decreasing percentage of methanol in the mobile phase (from 95% to 5% v/v of MeOH changed in increments of 5%). For [EMIM][Cl] retention was observed only at low content of methanol in the mobile phase (5% and 10% v/v), which was insufficient to describe adsorption effects. For the [BMIM] cation, with the decrease of the MeOH content in the mobile phase, k′ values were increasing in the range of 0.87–7.12 for 5–25% v/v of MeOH (Table S2 in the ESI†). Stronger adsorption was obtained for the imidazolium cation with a longer alkyl chain in its structure ([BMIM]), compared to [EMIM] cations (Table S2†). Based on these results it was demonstrated that ILs adsorb on stationary phase ligands, similarly to alkylamine IPRs in IPC mode. Therefore we have proved that increasing IL concentration in the mobile phase makes them IPRs suitable for ASOs. Such an effect is in the agreement with the results obtained by Berthod et al.,22 who conducted a frontal analysis for this purpose.
3.3 Influence of stationary phases on ASO retention
Three different chromatographic columns with different functional groups (octadecyl, octadecyl with embedded polar groups and pentafluorophenyl) were selected for this study based on the literature, as well as on our previous experience in ASO analysis.9,24,29,30 As it can be seen in Fig. 2, S2, S3 and Table 2, the highest k′ values for ASOs were obtained for F5 column (Fig. 2). Such an effect was observed, regardless of the applied IL and its concentration. Besides hydrophobic and electrostatic interactions between ILs, ASOs and stationary phase ligands, π⋯π interactions probably taking place between aromatic ring in the stationary phase structure and nitrogen bases in the ASOs, as well as those in the imidazolium cation ring, may significantly influence the retention (Fig. 2 and Table 2). In the case of C18 and aQ packing materials, the k′ values were dependent on the IL cation structure. For the most polar [EMIM], lower k were obtained for C18 packing material compared to the aQ column (Fig. S2A and S3A in ESI†). This effect may be related to the interactions between polar groups embedded in the alkyl chain of aQ and [EMIM], compared to the other stationary phases (Fig. S2A†). For [BMIM] cation, differences in k′ values for C18 and aQ are small (Fig. S2B and S3B†), while for the most hydrophobic cation [HMIM], higher k′ values were obtained for C18 column compared to aQ (Fig. S2C and S3C†). Such phenomenon is probably related to greater hydrophobicity of C18 stationary phase, than that of aQ. Thus greater adsorption of hydrophobic [HMIM] is observed on this packing material than in the case of [EMIM] and [BMIM] cations (Fig. S3C†).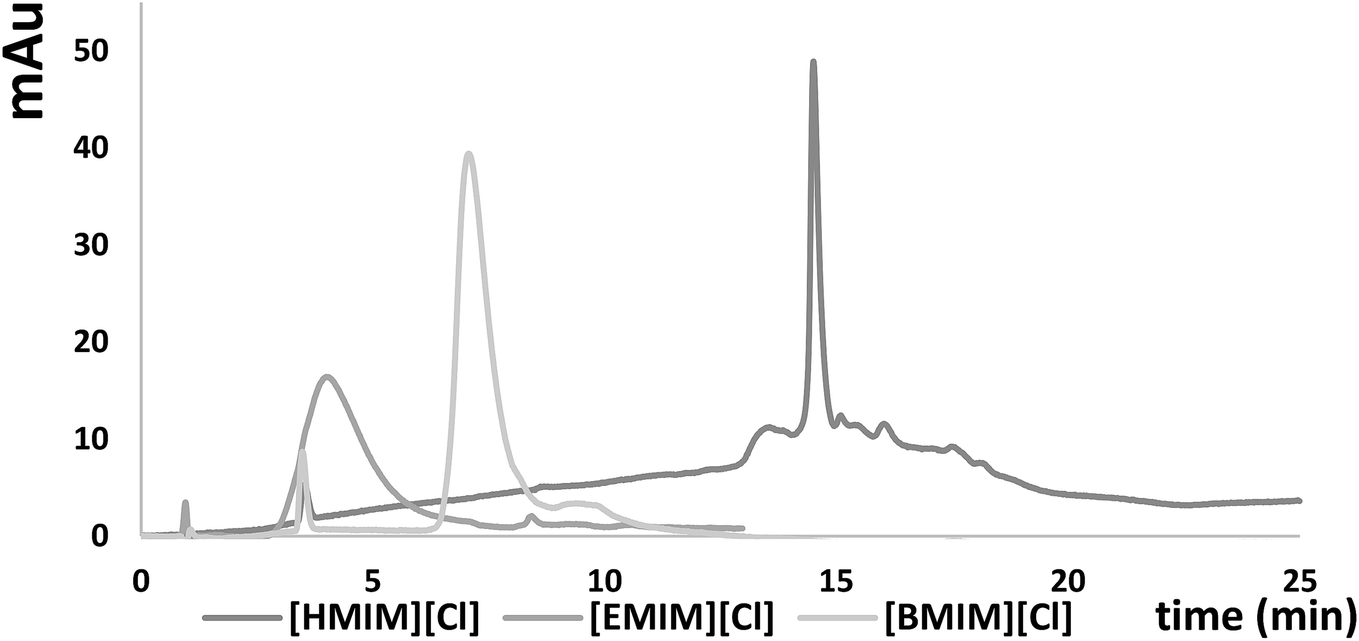 | ||
| Fig. 2 Chromatograms presenting the influence of the ILs type on the retention of OL2. Experimental conditions: Kinetex C18 column, autosampler and column temperature: 30 °C, mobile phase flow rate: 0.3 ml min−1, injection volume 2 μl, mobile phase composition: 7 mM of ILs and MeOH, gradient elution program 25–60% v/v MeOH in 15 minutes. | ||
3.4 Comparison of ASO retention for ILs and alkylamines
The comparison of the different mobile phase additives used as silanol suppressors and IPRs on the ASOs k′ values and peak shapes was performed. The following mobile phases in the mixture with MeOH were selected for this purpose: [BMIM][Cl], triethylamine chloride (pH = 6.8) (TEACl), [EMIM][Cl], N,N-dimethylbutylamine chloride (pH = 6.8) (DMBACl) and sodium chloride. TEACl and DMBACl were selected due to the alkyl chain length attached to the IL imidazolium cation (similar to the length of alkyl chains in EMIM and BMIM respectively) although we are aware that direct comparison of these compounds is not possible due to differences in their structures. The same anion (chloride) was used in case of all salts in order to study the impact of the cation only. The concentration of these salts was 5 mM. NaCl was used in order to compare it to the mobile phases with the presence of IPRs or ILs. Table 3 presents the results obtained during this part of study. Greater ASO k′ values were obtained for ILs than for the commonly used IPRs (Table 3). This effect is probably related to the fact that with the increasing length of the alkyl chain in the IPRs or imidazolium structure, the amount of cations adsorbed on the C18 stationary phase surface also increases. Thus, ASOs were more strongly retained when [EMIM][Cl] and [BMIM][Cl] with the larger cations were used as mobile phases additives, compared to TEACl and DMBACl. However, in the case of alkylamines more symmetrical peaks were obtained, as shown at Fig. 3. It can be also concluded that the longer the alkyl chain, both in the structure of DMBACl as well as in the [BMIM][Cl], the more symmetrical the peaks are compared to [EMIM][Cl] and TEACl, due to more effective shielding of the free silanol groups.| k′ ± SD | |||||
|---|---|---|---|---|---|
| [BMIM][Cl] | DMBACl | [EMIM][Cl] | TEACl | NaCl | |
| OL1 | 70.715 ± 0.078 | 58.237 ± 0.036 | 45.074 ± 0.812 | 35.241 ± 0.191 | 31.115 ± 0.155 |
| OL2 | 64.679 ± 0.191 | 52.554 ± 0.191 | 37.993 ± 0.024 | 30.405 ± 0.000 | 29.832 ± 0.036 |
| OL3 | 59.658 ± 0.424 | 49.031 ± 0.078 | 31.528 ± 0.967 | 25.844 ± 0.133 | 20.250 ± 0.310 |
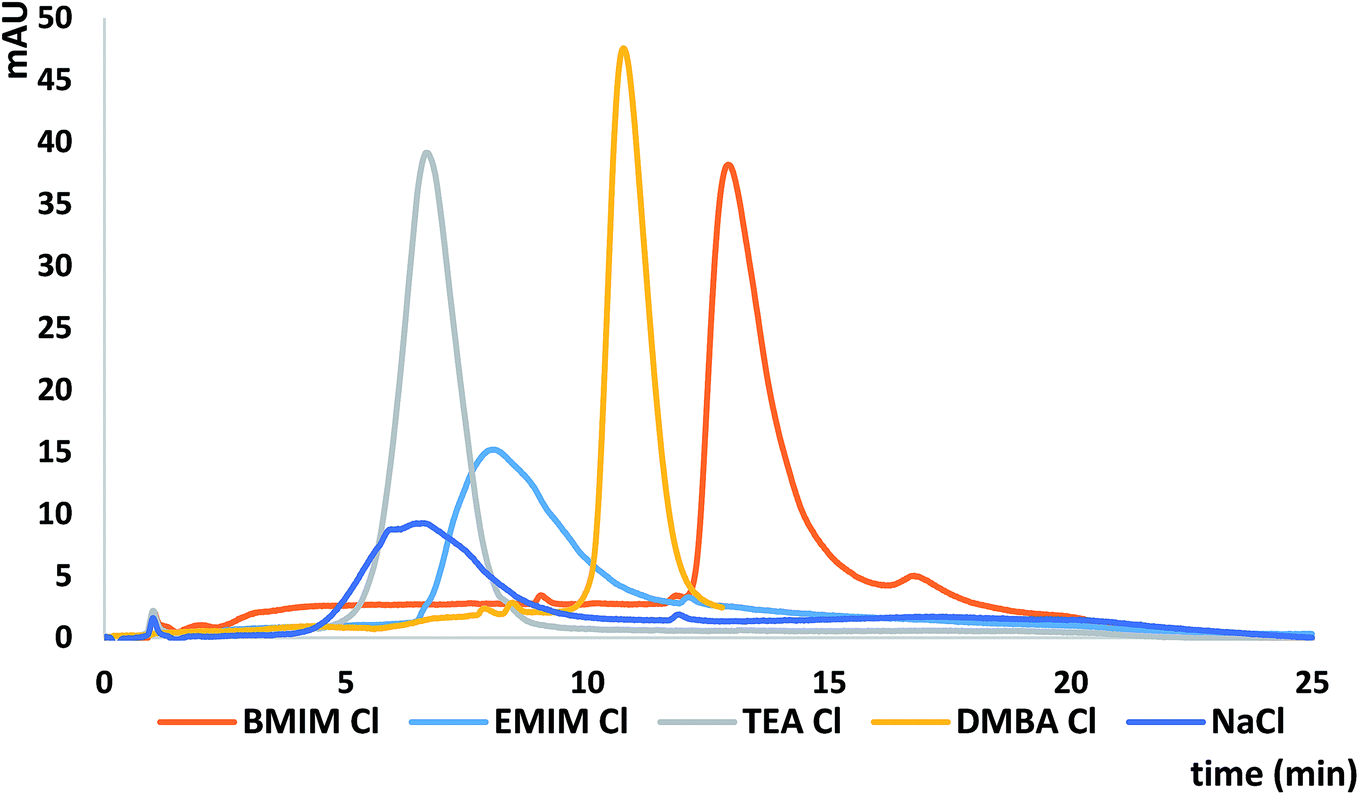 | ||
| Fig. 3 Chromatograms obtained for OL2 for [BMIM][Cl], [EMIM][Cl], TEACl, DMBACl and NaCl. Experimental conditions: Kinetex C18 column, gradient elution program: 20–40% v/v MeOH in 15 minutes; injection volume 2 μl, autosampler and column temperature 30 °C. | ||
Surprisingly, ASO k′ values obtained for NaCl and TEACl are similar (Table 3). Such an effect may be the result of interactions between sodium cations and negatively charged ASOs, causing reduction of their charge. Consequently, increase of their affinity to the stationary phase occurred, resulting in retention improvement. However, ASO peaks obtained with NaCl used as mobile phase additive were much broader than in the case of mobile phase modified with the TEACl. This may be a consequence of increased role of hydrophobic interactions between ASOs and stationary phase, when IPR is added to the mobile phase, contrary to NaCl.
3.5 Development of separation method for mixtures of ASOs
In order to evaluate the usefulness of ILs as additives to the mobile phase in chromatographic analysis of ASOs, an attempt was made to separate four mixtures with the use of each tested column. Mixture compositions are included in Table 1. [HMIM][Cl] was selected for the separation studies of tested mixtures, due to the greatest k′ values as well as lower ASOs peak width observed during retention studies. 7 mM and 10 mM of [HMIM][Cl] was used for the separation of the tested mixtures, because these conditions provided the greatest resolution of tested mixtures. Chromatograms obtained for MIX 1, 3 and 4 for each of tested column are presented in Fig. 4.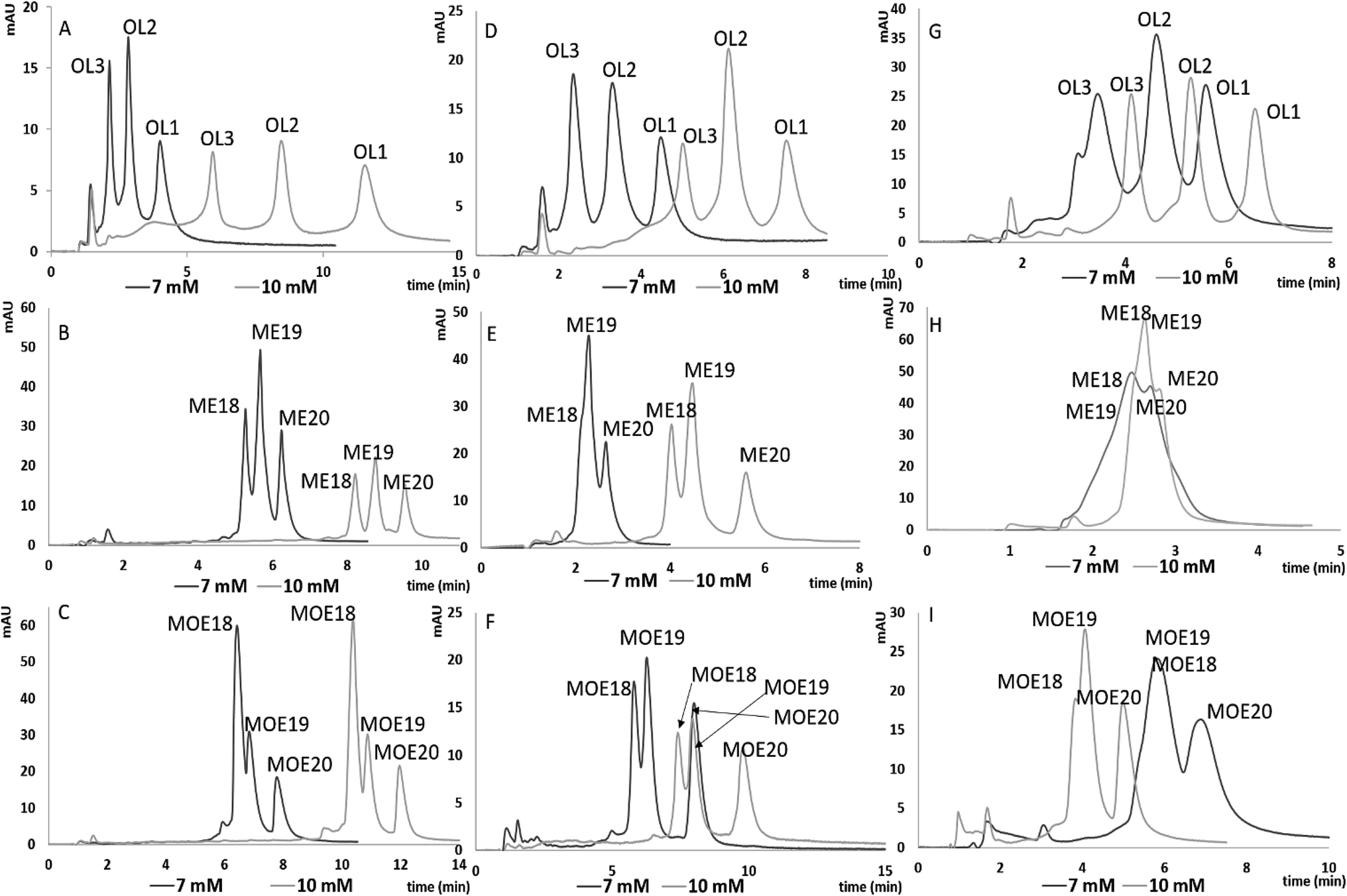 | ||
| Fig. 4 Chromatograms presented the separation of tested mixtures for C18 column (A–C), F5 (D–F) and aQ (G–I) and for 7 mM and 10 mM of [HMIM][Cl]. Experimental conditions: 50 °C column temperature, injection volume 2 μl, gradient elution: (A) for 7 mM 50–60% v/v MeOH in 15 minutes; for 10 mM 48–55% v/v MeOH in 15 minutes, flow rate 0.3 ml min−1 (B) 45–55% v/v MeOH in 15 minutes for both concentrations, flow rate 0.3 ml min; (C) 47–54% v/v MeOH in 15 minutes for both concentrations, flow rate 0.3 ml min; (D) 45–60% v/v MeOH in 15 minutes for both concentrations, flow rate 0.3 ml min; (E) 45–47% v/v MeOH in 15 minutes for both concentrations, flow rate 0.3 ml min; (F) 45–60% v/v MeOH in 15 minutes for both concentrations, flow rate 0.3 ml min; (G) for 7 mM 45–50% v/v MeOH in 15 minutes flow rate 0.2 ml min; for 10 mM 45–60% v/v MeOH in 15 minutes, flow rate 0.35 ml min; (H) for 7 mM 45–50% v/v MeOH in 15 minutes, flow rate 0.2 ml min; for 10 mM 47–52% v/v MeOH in 15 minutes, flow rate 0.35 ml min; (I) for 7 mM 45–50% v/v MeOH in 15 minutes, flow rate 0.2 ml min−1, for 10 mM 47–49% v/v MeOH in 15 minutes, flow rate 0.35 ml min−1. | ||
With increasing [HMIM][Cl] concentration, the resolution of MIX 1, 3, 4 also increased for all the tested stationary phases (Fig. 4). Moreover, an improvement in the shape and symmetry of ASO peaks was observed. Such tendencies were demonstrated by the asymmetry factors obtained for the analysis for both concentrations: in the case C18 for MIX1, asymmetry factors equaled 1.36, 1.43 and 1.26 for 7 mM of ILs, while for 10 mM these values were as follows: 0.81, 0.98 and 1.19 (Fig. 4A). For F5 and aQ stationary phases similar effects were observed regarding asymmetry factors. For the first packing material and 7 mM and 10 mM of ILs following asymmetry factors were noted: 1.59, 1.49, 1.49 and 1.01, 1.33, 1.23 (Fig. 4D), while for aQ one: 1.25, 1.17, 1.64 and 1.02, 0.90, 0.99 (Fig. 4G). This effect may be related to stronger adsorption of [HMIM][Cl] when higher concentration was applied. However, it may be also a result of the different gradient elution programs applied for 7 mM and 10 mM of MIX 1 (Fig. 4A). The same chromatographic parameters were used in the separation of MIX 3 and MIX 4 for C18 column and both IL concentrations. For higher [HMIM][Cl] concentration, an improvement of resolution for both ASO mixtures was noted.
Similar effects were observed for the F5 column, where the impact of concentration on resolution was observed in particular for MIX 3 (Fig. 4E). Coelution of metabolites was observed for 7 mM of [HMIM][Cl], while increasing concentration caused enhancement of separation efficiency (Fig. 4E). For the other mixtures (Fig. 4D and F) increasing IL concentration caused just elongation of analysis time, without affecting resolution.
In case of aQ column, the satisfactory resolution of ASOs was obtained only for MIX 1, contrary to MIX 3 and MIX 4 (Fig. 4G). MIX 1 is composed of ASOs which are built of 20, 17 and 13 nucleotides, whereas MIX 3 and MIX 4 contain 20-mer ASOs and their n − 1 and n − 2 synthetic metabolites (Table 1). Consequently, sequence differences are greater for MIX 1 components, which allows rapid separation. Increasing of the mobile phase flow rate for the 10 mM of [HMIM][Cl] allowed reduction of ASO k′ values (compared to lower IL concentration) without affecting their resolution. Regardless of the tested ILs concentration, separation of MIX 3 and MIX 4 was not satisfactory since coelution of shorter ASOs was observed (Fig. 4H and I). Such phenomenon may be related to insufficient selectivity of this stationary phase.
To summarize, the best resolution of tested mixtures was obtained for C18; however, for F5 the ASO mixture separation was also satisfactory, so both of them may be used in further studies.
It has to be pointed out, however, that the separation of MIX 2, consisting of ASOs differing by their sequence (Table 1) has been unsuccessful due to the coelution of the mixture components. Such an effect was observed regardless of the stationary phase used and [HMIM][Cl] concentration. This indicates that the developed method is not sufficiently selective with respect to positional isomers (MIX 2), because of small differences in their molecular masses and position of nucleotides (Table 1). Thus, they interact with the stationary phases with similar strength.
3.6 Application of ILs as mobile phase additives for oligonucleotide quantification in fortified serum and for determination of ASOs impurities
Since ASOs have therapeutic potential in the treatment of several diseases, there is a need for their quantification, especially in biological matrices (e.g. serum, tissues, urine), because it provides information about their pharmacokinetics and metabolic pathways.10 Another critical aspect concern their purification from sequence related impurities. Thus, in order to verify the applicability of ILs as mobile phase additives, 10 mM [HMIM][Cl] was used for separation and determination of OL1 and its potential impurities as well as quantification of OL4 in enriched serum samples. F5 column was selected for quantification in serum samples, while aQ one for separation of ASO from potential impurity. Fig. 5A and B presents chromatograms obtained for OL4 extracted from enriched serum sample and separation of OL1 from potential impurities.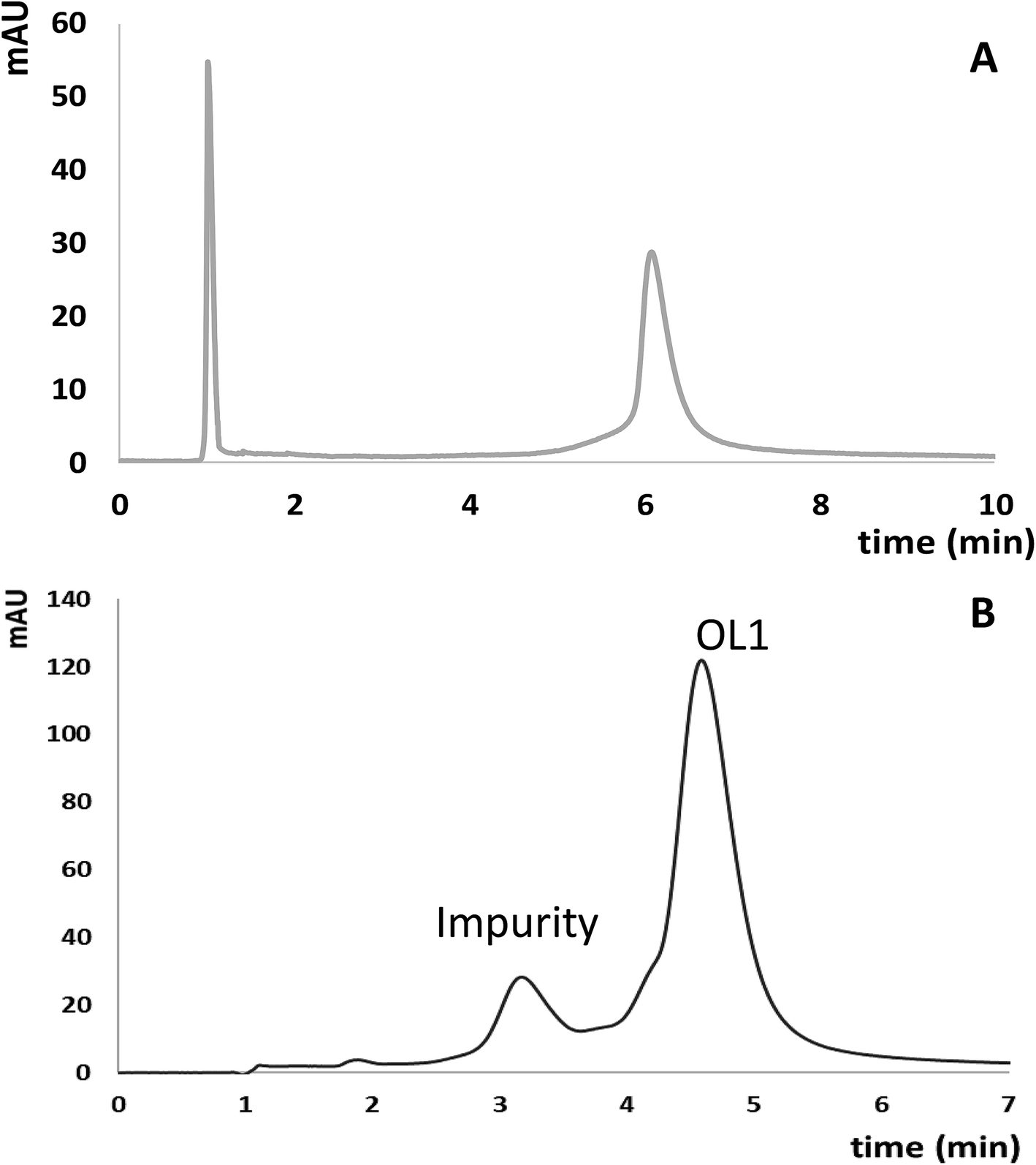 | ||
| Fig. 5 Chromatograms obtained for: (A) OL4 extracted from enriched serum sample, (B) separation of OL1 and its potential sequence related impurity. Chromatographic conditions: (A) Kinetex F5 stationary phase, gradient elution program: 45–60% v/v of MeOH in 10 minutes, (B) syncronis aQ stationary phase, gradient elution program: 48–55% v/v of MeOH in 10 minutes. UV detection at λ = 260 nm, autosampler and column temperature: 30 °C, mobile phase flow rate 0.3 ml min−1. Injection volume: 2 μl. | ||
Liquid–liquid extraction with the phenol:chloroform:isoamyl alcohol at the ratio 25:24:1 was used for ASO isolation from serum samples since it allows reducing of specific ASO affinity to plasma proteins. In order to remove phenol residue it was necessary to carry out an additional extraction step with the use of chloroform at the ratio of 3:1 to the supernatant.
Linearity of the method was established by calculating the determination coefficient (R2) for the calibration curve in the range of 8–20 μM (five different concentrations). Calibration curve was prepared by enriching of post-extracted diluted serum samples by an appropriate concentration of OL4. The obtained calibration curve (y = 2.3063x − 6.4301) was linear with R2 equal 0.9959; thus the developed method is characterized by acceptable linearity within the test range of concentrations. LOD of the method has been calculated using the following equation: 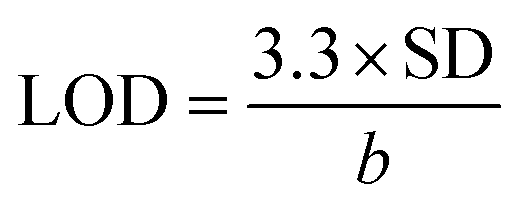 , where SD is standard deviation of linear regression and b is a slope of the calibration line, while LOQ has been determined by triplicating of LOD value. LOD of the method equaled 1.74 μM, while LOQ value was 5.23 μM. The matrix effect was determined by comparison of the ASO peak area for a standard sample with the peak area of the same concentration of ASO in a serum sample enriched post-extraction and its equaled 14.8%. Recovery was also evaluated by comparison of the ASO concentration determined in enriched serum samples with concentration of ASO added to serum sample and its equaled 62.18% ± 0.37. It has to be pointed out that the developed method may be used for real ASO samples analysis; however, recovery value needs to be further improved.
, where SD is standard deviation of linear regression and b is a slope of the calibration line, while LOQ has been determined by triplicating of LOD value. LOD of the method equaled 1.74 μM, while LOQ value was 5.23 μM. The matrix effect was determined by comparison of the ASO peak area for a standard sample with the peak area of the same concentration of ASO in a serum sample enriched post-extraction and its equaled 14.8%. Recovery was also evaluated by comparison of the ASO concentration determined in enriched serum samples with concentration of ASO added to serum sample and its equaled 62.18% ± 0.37. It has to be pointed out that the developed method may be used for real ASO samples analysis; however, recovery value needs to be further improved.
The developed method was also applied for the determination of potential impurities formed during synthesis process of oligonucleotides. aQ stationary phase was used for this purpose. Application of gradient elution allowed for complete separation of OL1 from its impurity (Fig. 5B). Calibration curves were prepared by the subsequent dilution of the OL1 and OL2 mixture. The concentration range of OL2 was 2–15 μM, while for OL1 5–25 μM (five different concentrations). Linearity of the method was determined by determination of R2 coefficient, which equaled 0.9977 for OL1 and 0.9967 for OL2. Based on the calibration curves equations for OL1 (y = 1.4848x + 4.016) and OL2 (y = 0.724x + 0.918) concentration of these compounds was determined and it equaled 24.17 μM for OL1 and 8.89 μM for OL2. The developed method is suitable for separation and determination of sequence related impurities of oligonucleotides.
4. Conclusions
To the best of our knowledge, it was the first time an analysis of ASOs was performed with the use of ILs as mobile phase additives. The results obtained during this investigation proved that an important factor which influences ASO retention was IL concentration: regardless of the IL type, increase in their concentrations resulted in greater ASO retention. It may be supposed that for the lowest [BMIM][Cl] concentration (0.1 mM) only the activity of free silanols is suppressed because ASOs are not retained on C18 stationary phase. When concentration increases, ILs cations probably adsorb on the stationary phase ligands and interact with ASOs by electrostatic interactions as well as they form ion pairs with ASOs, which consequently leads to their increased retention. Tendencies noted during this investigation are similar to those observed when alkylamines are used as IPRs. Therefore, the probable retention mechanism of ASOs includes hydrophobic interactions, formation of ASO-IL ion pairs as well as hydrogen bonding. ILs may be used as an alternative to the standard IPRs used in IPC analysis of ASOs.Another important factor which has an impact on the retention of ASOs is ILs cation size. The greatest ASO k values were obtained for [HMIM][Cl] with the longer alkyl ligand embedded in the imidazolium cation. This cation probably more effectively adsorbs on the stationary phase ligands, compared to the [BMIM] and [EMIM] ones, which leads to the enhancement of ASO retention. Moreover, for the larger imidazolium cation ([HMIM]), the improvement in ASO peak shapes was also noted.
The method of separation of synthetic metabolites was developed during the study involving mobile phases containing [HMIM][Cl] and methanol. However, it has to be pointed out that addition of [HMIM][Cl] to the mobile phase gives sufficient selectivity only for ASO with different lengths of the phosphate backbone, because separation of sequential isomers failed. Finally, ILs as the mobile phase additives were used in determination of ASOs in fortified serum extracts. The LOD and LOQ values of the developed method were as follows: 1.74 μM and 5.23 μM. Moreover, method was characterized by acceptable linearity at the concentration range 8–20 μM. However, the obtained recovery value needs to be increased further.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by the National Science Center (Cracow, Poland) under Sonata Bis project (2016/22/E/ST4/00478).References
- W. Zhang, L. He, Y. Gu, X. Liu and S. Jiang, Anal. Lett., 2003, 36, 827–838 CrossRef CAS.
- L. J. He, W. Z. Zhang, B. Wen, X. Liu and S. X. Jiang, Chin. Chem. Lett., 2003, 14, 39–45 Search PubMed.
- M. Buszewska-Forajta, M. J. Markuszewski and R. Kaliszan, J. Chromatogr. A, 2018, 1559, 17–43 CrossRef CAS PubMed.
- X. Xiaohua, Z. Liang, L. Xia and J. Shengxiang, Anal. Chim. Acta, 2004, 519, 207–211 CrossRef.
- C. F. Poole, J. Chromatogr. A, 2004, 1037, 49–82 CrossRef CAS PubMed.
- S. Studzińska, K. Krzemińska, M. Szumski and B. Buszewski, Talanta, 2016, 154, 270–277 CrossRef PubMed.
- T. Cecchi, Ion-Pair Chromatography and Related Techniques, Google eBook, 2009 Search PubMed.
- E. K. Mustonen, T. Palomäki and M. Pasanen, Regul. Toxicol. Pharmacol., 2017, 90, 328–341 CrossRef CAS PubMed.
- A. Kaczmarkiewicz, Ł. Nuckowski and S. Studzińska, Talanta, 2019, 196, 54–63 CrossRef CAS PubMed.
- A. Kaczmarkiewicz, Ł. Nuckowski, S. Studzińska and B. Buszewski, Crit. Rev. Anal. Chem., 2019, 15 Search PubMed.
- S. Studzińska, Talanta, 2018, 176, 329–343 CrossRef PubMed.
- A. C. McGinnis, E. C. Grubb and M. G. Bartlett, Rapid Commun. Mass Spectrom., 2013, 27, 2655–2664 CrossRef CAS PubMed.
- B. Bothner, K. Chatman, M. Sarkisian and G. Siuzdak, Bioorg. Med. Chem. Lett., 1995, 5, 2863–2868 CrossRef CAS.
- A. Apffel, J. A. Chakel, S. Fischer, K. Lichtenwalter and W. S. Hancock, J. Chromatogr. A, 1997, 777, 3–21 CrossRef CAS.
- C. G. Huber and A. Krajete, Anal. Chem., 1999, 71, 3730–3739 CrossRef CAS PubMed.
- A. Premstaller, H. Oberacher and C. G. Huber, Anal. Chem., 2000, 72, 4386–4393 CrossRef CAS.
- H. Oberacher, W. Parson, R. Muhlmann and C. G. Huber, Anal. Chem., 2001, 73, 5109–5115 CrossRef CAS PubMed.
- L. Gong and J. S. O. McCullagh, J. Chromatogr. A, 2011, 1218, 5480–5486 CrossRef CAS PubMed.
- S. Studzińska, F. Łobodziński and B. Buszewski, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2017, 1040, 282–288 CrossRef PubMed.
- A. V. Herrera-Herrera, J. Hernández-Borges and M. Á. Rodríguez-Delgado, Anal. Bioanal. Chem., 2008, 392, 1439–1446 CrossRef CAS PubMed.
- M. Caban and P. Stepnowski, Anal. Chim. Acta, 2017, 967, 102–110 CrossRef CAS PubMed.
- A. Berthod, M. J. Ruiz-Angel and S. Huguet, Anal. Chem., 2005, 77, 4071–4080 CrossRef CAS PubMed.
- R. Kaliszan, M. P. Marszałł, M. J. Markuszewski, T. Bączek and J. Pernak, J. Chromatogr. A, 2004, 1030, 263–271 CrossRef CAS PubMed.
- A. C. McGinnis, B. Chen and M. G. Bartlett, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2012, 883–884, 76–94 CrossRef CAS PubMed.
- M. Gilar, K. J. Fountain and Y. Budman, J. Chromatogr. A, 2014, 1245, 65–74 Search PubMed.
- S. Studzińska, R. Rola and B. Buszewski, Anal. Bioanal. Chem., 2016, 408, 1585–1595 CrossRef PubMed.
- M. Biba, E. Jiang, B. Mao, D. Zewge, J. P. Foley and C. J. Welch, J. Chromatogr. A, 2013, 1304, 69–77 CrossRef CAS PubMed.
- A. Berthod, M. J. Ruiz-Ángel and S. Carda-Broch, J. Chromatogr. A, 2018, 1559, 2–16 CrossRef CAS PubMed.
- M. Biba, J. P. Foley and C. J. Welch, Liquid chromatographic separation of oligonucleotides, Elsevier Inc., second edn, 2017, vol. 2 Search PubMed.
- S. Studzińska, R. Rola and B. Buszewski, J. Pharm. Biomed. Anal., 2017, 138, 146–152 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra06483d |
| This journal is © The Royal Society of Chemistry 2019 |