
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBrCl+ elimination from Coulomb explosion of 1,2-bromochloroethane induced by intense femtosecond laser fields†
Hua
Wu
 *,
Yuanxin
Xue
,
Junqing
Wen
,
Hui
Wang
,
Lihua
Bai
,
Wanlin
He
,
Ruijuan
Sun
and
Wenli
Zheng
*,
Yuanxin
Xue
,
Junqing
Wen
,
Hui
Wang
,
Lihua
Bai
,
Wanlin
He
,
Ruijuan
Sun
and
Wenli
Zheng
School of Sciences, Xi'an Shiyou University, Shanxi 710065, P. R. China. E-mail: whua@xsyu.edu.cn
First published on 7th October 2019
Abstract
By using a dc-slice imaging technique, photodissociation of 1,2-C2H4BrCl was investigated at 800 nm looking for heteronuclear unimolecular ion elimination of BrCl+ in an 80 fs laser field. The occurrence of fragment ion BrCl+ in the mass spectrum verified the existence of a unimolecular decomposition channel of BrCl+ in this experiment. The relative quantum yield of the BrCl+ channel was measured to be 0.8%. By processing and analyzing the velocity and angular distributions obtained from the corresponding sliced images of BrCl+ and its partner ion C2H4+, we concluded that BrCl+ came from Coulomb explosion of the 1,2-bromochloroethane dication 1,2-C2H4BrCl2+. With the aid of quantum chemical calculations at the M06-2X/def2-TZVP level, the potential energy surface for BrCl+ detachment from 1,2-C2H4BrCl2+ has been examined in detail. According to the ab initio calculations, two transition state structures tended to correlate with the reactant 1,2-C2H4BrCl2+ and the products BrCl+ + C2H4+. In this entire dissociation process, the C–Br and C–Cl bond lengths were observed to elongate asymmetrically, that is, the C–Br chemical bond broke firstly, and subsequently a new Br–Cl chemical bond started to emerge while the C–Cl bond continued to exist for a while. Hence, an asynchronous concerted elimination mechanism was favored for BrCl+ detachment.
1. Introduction
The photodissociation of polyhalogenalkanes induced by intense laser fields has been extensively studied and is the subject of recent reviews.1–14 Two major reactions are possible for dissociation of polyhalogenalkanes: the halogen atom release reaction channel and the halogen molecular elimination reaction channel. Thus far, most studies have been focused on the halogen atomic product channel, and the halogen molecular elimination channel as a research topic has received less attention in earlier papers.Rather than breaking one carbon-halogen chemical bond, a halogen molecular detachment process involves cleavage of two carbon-halogen chemical bonds and formation of one halogen–halogen bond. Generally, two types of reaction mechanism (sequential and concerted) are associated with halogen molecular elimination from polyhaloalkanes. Sequential dissociation mechanism refers to that two halogen atoms detach from polyhaloalkanes in turn and then combine by a collisional process. A concerted reaction is defined as one for which breaking of the two carbon–halogen bond and formation of the one halogen–halogen bond occur in a single kinetic step.15–17 Asynchronous and synchronous mechanisms are included for concerted mechanism. In an asynchronous concerted reaction, the first carbon–halogen bond breaks and halogen–halogen bond is formed while the second carbon-halogen is still part of the molecule, the reaction ends when the second carbon-halogen chemical bond breaks; synchronous concerted mechanism refers to that the two carbon–halogen chemical bonds start to break at the same time and break at the same rate, and the cleavage processes are accompanied by the formation of a halogen–halogen bond.9,12 Many authors have tried to explain the detachment mechanism of halogen molecular fragments from polyhalogenalkanes.4–14 Lin and co-workers4–7 investigated photodissocation of CH2Br2, C2H4Br2, CH2I2, and CH2BrI to molecular products of Br2, I2, and BrI by cavity ring-down absorption spectroscopy at 248 nm. Their theoretical calculations on potential energy surface of the polyhaloalkanes revealed that these halogen molecular dissociation channels were anticipated to proceed via an asynchronous concerted mechanism. By using femtosecond pump-probe technique, Dantus and co-workers8–12 identified the molecular detachment channel of YZ and X2 from polyhaloalkanes CX2YZ (X represents for Cl, H, or F, and Y, Z represent for Cl, Br, or I) under an ultraviolet laser field, and a ∼50 fs ultrafast process was observed for their elimination processes. They proposed that synchronous concerted process was observed for heteronuclear YZ elimination while asynchronous concerted process was detected for homonuclear X2 ejection. Janssen and co-workers13 studied the I2 elimination from C2F4I2 at 396 nm, and demonstrated that I2 was ejected through an asynchronous concerted mechanism. They used the ion pair model to elucidate the I2 formation. From the theoretical aspects, Bozzelli and co-workers14 demonstrated that C2H4Cl2 could give rise to Cl2 by ab initio calculations.
Although there is a great amount of studies on the halogen molecular elimination reactions, these studies were performed under ultraviolet excitation in a relatively weak laser field, and halogen molecular elimination reactions induced by near-infrared (800 nm) intense femtosecond laser field have been rarely studied. When polyhaloalkanes interact with an intense femtosecond laser field, several electrons could be rapidly removed from polyhaloalkanes, resulting in departure of parent molecular ion into cationic fragments. This is the so-called Coulomb explosion (CE) processes. Liu and co-workers18 observed molecular ions I2+ and ICl+ under intense femtosecond laser irradiation, and they stated that their elimination processes were concerted elimination mechanism. However, whether asynchronous or synchronous elimination mechanism was not distinguished for I2+ and ICl+ in this experiment. Our group have demonstrated that homonuclear Br2+ elimination from 1,2-dibromoethane under 800 nm femtosecond laser field was a synchronous concerted mechanism.19 In this paper, photodissociation of 1,2-bromochloroethane was investigated in search of unimolecular ion elimination of heteronuclear BrCl+ and its elimination mechanism via primary channel by dc-slice ion imaging technique at 800 nm under 80 fs laser field. The occurrence of BrCl+ in mass spectrum verified the existence of unimolecular ion elimination channel of BrCl+ in this experiment. The relative quantum yield of BrCl+ was measured to be 0.8%. By analyzing the corresponding velocity and angular distributions of BrCl+ and its partner ion C2H4+, we concluded that BrCl+ came from two-body CE of 1,2-C2H4BrCl2+. With the aid of quantum chemical calculations at M06-2X/def2-TZVP level, the potential energy surface for BrCl+ detachment from 1,2-C2H4BrCl2+ have been examined in detail. According to our theoretical calculations, two transition state structures tended to correlate with the reactant 1,2-C2H4BrCl2+ and the products BrCl+ + C2H4+, and an asynchronous concerted elimination mechanism was favored for BrCl+ detachment.
2. Experimental section
The detailed description of our DC slice ion-imaging system was explained elsewhere.19–21 Briefly, the experimental apparatus is mainly composed of a supersonic molecular beam system and a time-of-flight (TOF) mass spectrometer system. A pulsed valve was operated synchronously with the laser pulses to generate the pulsed molecular beam. The sample beam entered into the ionization region through a skimmer. Usually the sample mixture was prepared with 1,2-bromochloroethane (Sinopharm Chemical Reagent Co., Ltd, 98+%) seeded in He at 1 atm without further purification. The 800 nm linearly polarized laser pulses (80 fs, 1 kHz) were then focused onto the sample beam by a 40 cm focal-length lens and perpendicularly intersected the sample beam. The ions were accelerated and focused by ion lens to generate an image onto a 2D position sensitive detector. The detector was composed of a pair of microchannel plates (MCPs), a P47 phosphor screen (PS), an intensified charge coupled device (ICCD) camera (512 × 512 pixels) with time gate of 4 ns, which were mounted at the end of the drift tube. The output signals from PS could also be fed into a photomultiplier tube (PMT) to get TOF mass spectra. The laser intensities were calibrated according to the idea of Guo et al.,22 and the calibrated intensities were 5.0 × 1013–1.6 × 1014 W cm−2. All the time delays were provided by a pulse generator (DG535).3. Computational details
The BrCl+ elimination channel on the singlet ground-state potential energy surface of 1,2-bromochloroethane dication 1,2-C2H4BrCl2+ was characterized. M06-2X/def2-TZVP level of theory23,24 has been used to calculate the geometries, the vibrational frequencies, and the energies of reactants (1,2-BCE2+ and S1), transition states (TS1 and TS2) and fragmentation products (BrCl+ and C2H4+). Here, the structure S1 represents the geometry of the intermediate in BrCl+ elimination process. The calculated vibrational frequencies could be used to get zero-point vibrational corrections and to determine whether the structure was a transition state or a local minimum. The geometries of 1,2-BCE2+, S1, BrCl+, and C2H4+ were ascertained when the number of negative frequencies were zero. However, the structures of TS1 and TS2 were confirmed with the number of imaginary frequency being one. All of the above calculated energies contained zero-point energy corrections. The intrinsic reaction coordinate (IRC) calculations at M06-2X/def2-TZVP level were done on TS1 and TS2 geometries to make sure that the transition states were connected to the specified reactants and products. The Gaussian 16 program25 was employed for these mentioned quantum chemical calculations.4. Results and discussions
Fig. 1 shows photoionization mass spectrum of 1,2-C2H4BrCl (1,2-BCE) induced by 80 fs laser pulse at 1.2 × 1014 W cm−2 intensity. Because of the existence of Br and Cl in 1,2-BCE, the Br- and Cl-containing ion signals exhibit intensity patterns. This can be well understood by the natural abundance of bromine (79Br:81Br = 1.02)26 and chlorine (35Cl:37Cl = 3.08).26 The fragment ions stemming from the single C–Br, C–Cl and C–C bond rupture, Cl2+, Br2+, Cl+, Br+, CH2Cl+, CH2Br+, C2H4Cl+, and C2H4Br+, are the major products in the spectrum. The generation of doubly-charged fragment ions C2+, Cl2+ and Br2+ implies the presence of Coulomb explosion (CE). The ion BrCl+ with m/q = 114, 116 and 118, resulting from breakage of C–Br and C–Cl bonds and formation of Br–Cl bond, can be clearly identified in the inset spectrum of Fig. 1, which indicates the occurrence of BrCl+ unimolecular ion elimination in this experiment. As illustrated in this figure, a single C–Br and C–Cl bond-breaking channel leading to Br+, Br2+ and Cl+, Cl2+ are the major channel for dissociation of 1,2-BCE, and their corresponding relative quantum yields are measured to be 30% and 21% for Br+, Br2+ and Cl+, Cl2+ respectively. In contrast, the relative quantum yield of BrCl+ is calculated to be 0.8%. These relative yields are calculated by the ratio of the corresponding ion signal to the total ion signals in TOF mass spectrum. This conclusion of 1,2-BCE is in agreement with the previous literature on polyhalogenalkanes that halogen product pathway is regarded as the primary fragmentation pathway for polyhaloalkanes.1–12 The aim of the present work has been to explore the mechanism on unimolecular ion elimination of BrCl+, so we would concentrate on BrCl+ and its partner C2H4+ in the sections below.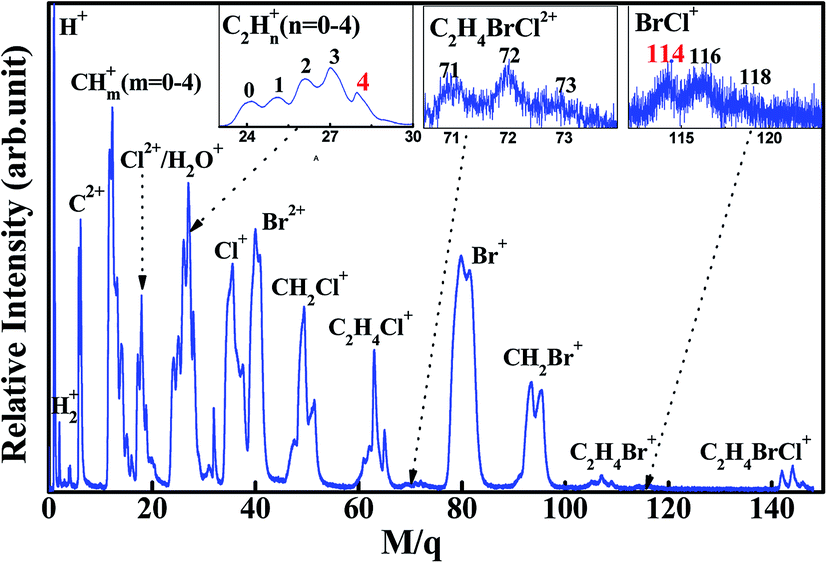 | ||
| Fig. 1 Mass spectrum of 1,2-bromochloroethane induced by 80 fs laser pulses at 1.2 × 1014 W cm−2 intensity. The inset is the enlarged BrCl+, C2H4BrCl2+ and C2H4+ spectrum. | ||
The DC slice ion imaging technique can provide velocity distribution and angular distribution of fragment ions simultaneously. The images and the corresponding velocity distributions of BrCl+ and C2H4+ obtained from photodissociation of 1,2-BCE are shown in Fig. 2. It should be noted that the velocity distribution of BrCl+ can be simulated by a single Gaussian function with velocity and kinetic energy release (KER) values peaked at 773 m s−1 and 0.35 eV, indicating that one dissociation channel is involved. Whereas for fragment ion C2H4+, two curves are needed (the peak values of velocity and KER are 1014 m s−1, 0.15 eV and 3183 m s−1, 1.47 eV respectively), which means more than one pathway participate in the photodissociation process.
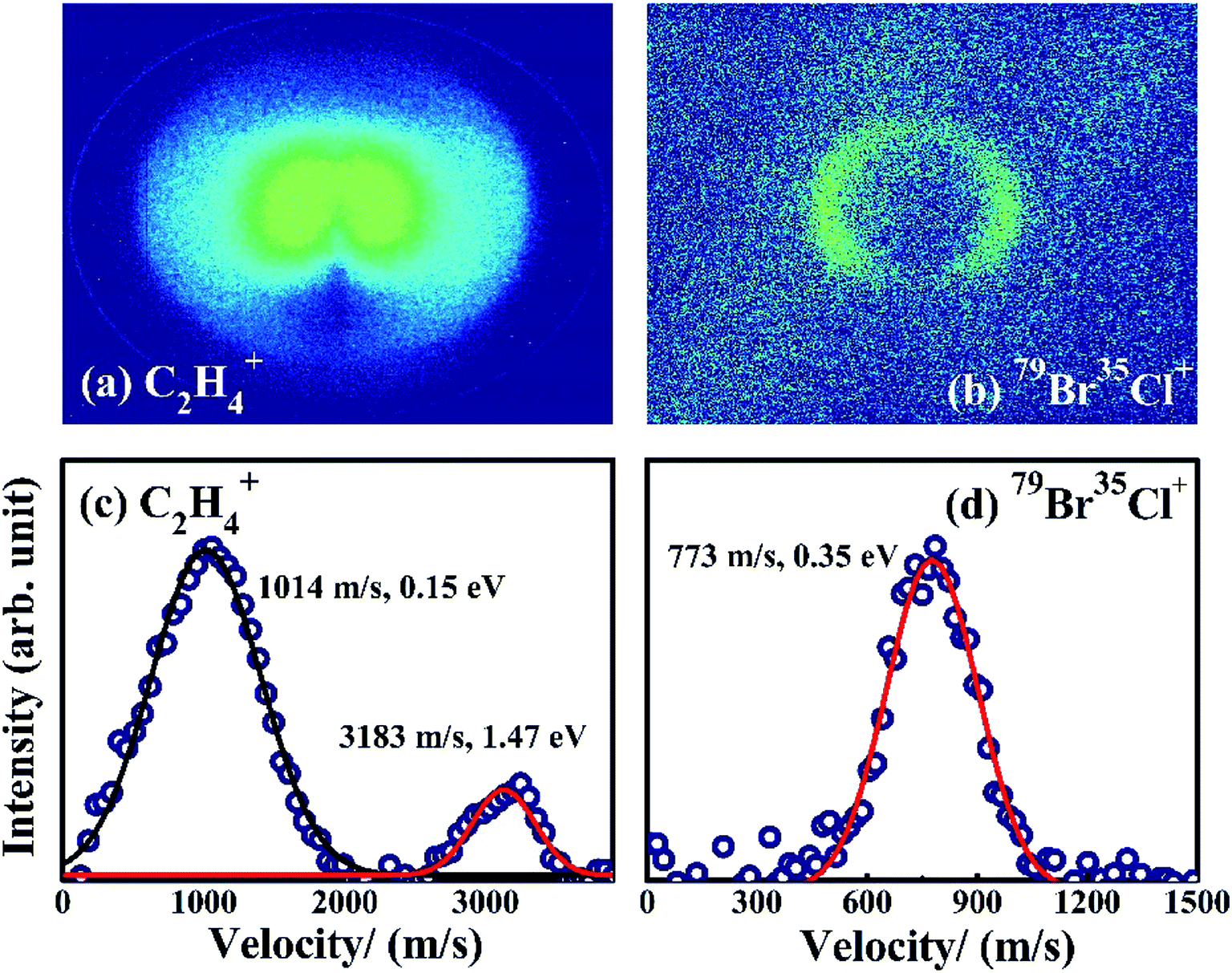 | ||
Fig. 2 Images and velocity distributions of C2H4+ and 79Br35Cl+ at 1.2 × 1014 W cm−2 intensity. The navy circles ( ) refer to experimental data, and the red ( ) refer to experimental data, and the red ( ) solid lines represent for the fitted curves for CE pathways. ) solid lines represent for the fitted curves for CE pathways. | ||
In our experiment, 1,2-BCE is the main component of the samples, although trace BrCl molecule might exist because of the decomposition of 1,2-BCE. If BrCl+ stems from direct photoionization of BrCl in the sample, an isotropic distribution peaked at 0 eV should be detected in the image and velocity distribution of BrCl+. Since no such distribution is observed, it is evident that BrCl+ is from the dissociation of 1,2-BCE. We know that the motion of the two ions produced by a two-body CE channel follow the law of conservation of momentum. In other words, an inverse relationship exists between the energy ratio and the mass ratio of the two ions.20,27,28 In this paper, the high-KER ratio of C2H4+ and BrCl+ is measured to be 4.20, and the inverse mass ratio of BrCl+ and C2H4+ is calculated to be 4.07. These two values are quite close to each other, so we believe 79Br35Cl+ and C2H4+ come from two-body CE of 1,2-C2H4BrCl2+
| 1,2-C2H4BrCl2+ → C2H4+ + BrCl+ |
One can see another low-KER component of C2H4+ (0.15 eV) in Fig. 2, and it is generally thought to arise from dissociative ionization of 1,2-BCE+ by experimental analysis and theoretical calculations.1,21,27,29 However, this dissociative ionization channel is our next work, so we won't elaborate here. From the viewpoint of momentum conservation, two ions generated by the same two-body CE channel must have the same angular distribution. Fig. 3 demonstrates that fragment ions 79Br35Cl+ and C2H4+ have a similar distribution, which is a good proof for our CE channel identification. Janssen et al.13 have studied the concerted reaction of C2F4I2 to I2 and C2F4, and they used the ion pair model to elucidate the I2 formation. On the basis of the 80 fs laser pulse we used and the isolated samples within our supersonic molecular beam, we tend to employ this argument to understand the BrCl+ generation process in this experiment.
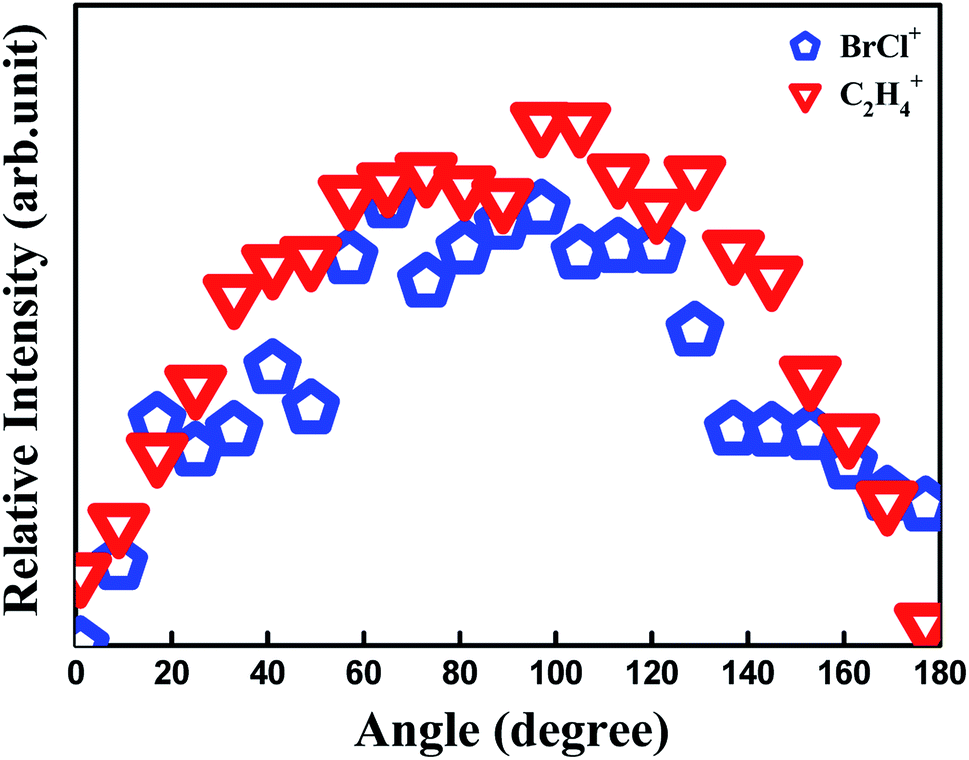 | ||
| Fig. 3 Angular distributions for BrCl+ and C2H4+ at 1.2 × 1014 W cm−2 intensity. | ||
The appearance energy from 1,2-C2H4BrCl2+ to C2H4+ and BrCl+ is calculated to be 2.92 eV by Gaussian 09 software packages at M06-2X/def2-TZVP theoretical level. The KER values of C2H4+ and BrCl+ are 1.47 eV and 0.35 eV from Fig. 2, respectively. That is to say that at least 4.74 eV should be required in this dissociation process, and thus at least four photons are needed and a maximum of 1.46 eV available energy can be remained for this process in our experiment when the process proceeds under multiphoton conditions. The angular distributions of fragment ions BrCl+ and C2H4+ in Fig. 3 can be fitted by Legendre expansion:30,31

![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ) is the Legendre polynomial, and βL represents the expansion coefficient. The leading expansion coefficient β2 is an important parameter that reflects the lifetime of the precursor species state, and a smaller β2 represents a longer lifetime of precursor species state.30,31 In our experiment, the β2 value of BrCl+ and C2H4+ is determined to be β2 = −0.28. Therefore, a relatively long life-time can be inferred for the precursor ion C2H4BrCl2+, and the observation of C2H4BrCl2+ in our mass spectrum is consistent with this conjecture.
θ) is the Legendre polynomial, and βL represents the expansion coefficient. The leading expansion coefficient β2 is an important parameter that reflects the lifetime of the precursor species state, and a smaller β2 represents a longer lifetime of precursor species state.30,31 In our experiment, the β2 value of BrCl+ and C2H4+ is determined to be β2 = −0.28. Therefore, a relatively long life-time can be inferred for the precursor ion C2H4BrCl2+, and the observation of C2H4BrCl2+ in our mass spectrum is consistent with this conjecture.
As we shall know, there are two mechanisms for halogen molecule detachment: stepwise and concerted mechanism.15–17 In our experiment, the supersonic molecular beam has a very low temperature (with rotation temperature below ten Kelvin and vibration temperature a little more than ten Kelvin to tens of Kelvin). Therefore, the stepwise mechanism (collisional process of Br and Cl is involved) for the BrCl+ formation can be ruled out. Moreover, Dantus et al. have addressed that the concerted elimination of halogen molecules (X2) from RCHX2 is an ultrafast process about 50 fs.8–12 Therefore, on the basis of the 80 fs laser pulse and the isolated samples within our supersonic molecular beam, we prefer to use the concerted mechanism to understand the generation process. In order to explain the formation of BrCl+ from dication 1,2-BCE2+, dissociation process calculations of 1,2-BCE2+ to BrCl+ + C2H4+ was conducted at M06-2X/def2-TZVP level.23–25 The geometries of reactants (1,2-BCE2+ and S1), the transition states (TS1 and TS2), and products (BrCl+ and C2H4+) were fully optimized. The geometries of 1,2-BCE2+, S1, BrCl+, and C2H4+ were ascertained when the number of negative frequencies were zero. However, the structures of TS1 and TS2 were confirmed with the number of imaginary frequency being one. The intrinsic reaction coordinate (IRC) calculations were done on TS1 and TS2 geometries to make sure that the transition states were connected to the specified reactants and products. The optimized structures involved in this unimolecular ion reaction process are shown in Fig. 4.
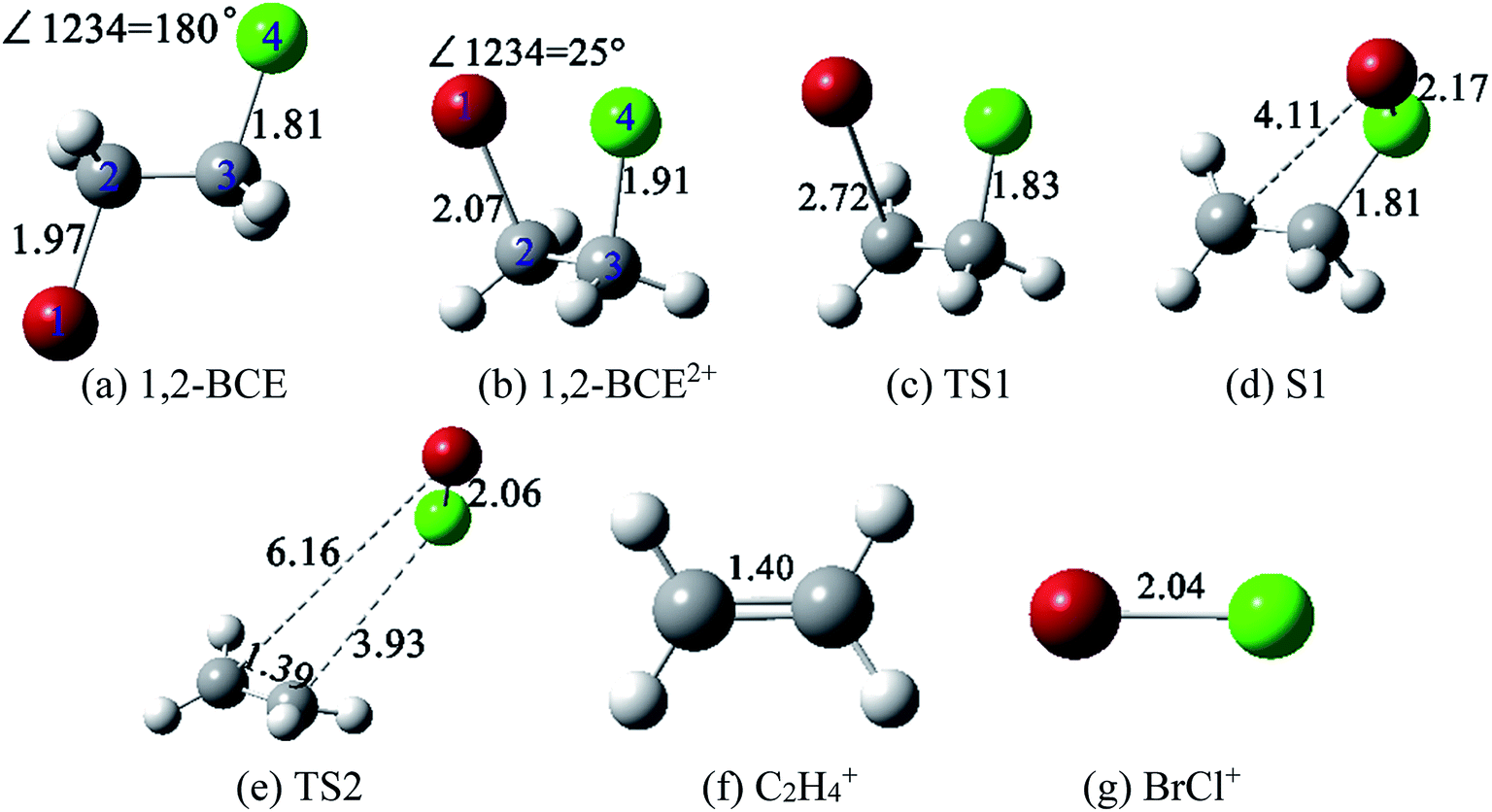 | ||
| Fig. 4 Structures of 1,2-BCE, 1,2-BCE2+, TS1, S1, TS2, C2H4+, and BrCl+ ((a)–(g)) at M06-2X/def2-TZVP level. The unit of the decimals is in Å. | ||
There are some noticeable differences in stable structure between ground-state 1,2-BCE and 1,2-BCE2+: ∠BrCCCl of 180° and 25°, C–Br bond distance of 1.97 Å and 2.07 Å, and the C–Cl bond distance of 1.81 Å and 1.91 Å, respectively. Weitzel et al.'s and Mebel et al.32,33 studied the structures of parent ions C2H62+ and C6H123+, and they reached a close conclusion for the markedly differences in molecular structure between neutral C2H6 and C6H12 respectively. After 1,2-BCE2+ is formed, how is the BrCl+ ion brought out? Fig. 5 presents a complete reaction path from the stable structure 1,2-BCE2+ through two designed TSs toward the products C2H4+ + BrCl+. This reaction starts with the elongation of C–Br bond length of 2.07 Å in 1,2-BCE2+ to 2.72 Å in TS1 by surpassing an energy barrier of 0.26 eV. This leads to the S1 structure with the continued elongation of C–Br bond length to 4.11 Å, and the slightly shortening of the C–Cl bond length at the same time. After that, the bond distances of C–Br and C–Cl in TS2 are asynchronously lengthened to 6.16 Å and 3.93 Å as compared with S1 where the bond distances of C–Br and C–Cl are 4.11 Å and 1.81 Å, respectively. The barrier calculated for this process from S1 to TS2 is 1.80 eV. In addition, the Br–Cl bond distance is shortened to 2.06 Å in TS2. After crossing the barrier TS2, the dication 1,2-BCE2+ decompose to BrCl+ and C2H4+via CE process. Our computed results of IRC definitely demonstrate that TS1 and TS2 geometries are connected with their designated reactants and products.
 | ||
| Fig. 5 Dissociation of 1,2-BCE2+ to fragments C2H4+ and BrCl+ at M06-2X/def2-TZVP level of theory. | ||
Three reaction coordinates are chosen to describe this dissociation reaction: RCBr, RCCl, and RBrCl. RCBr and RCCl are the C–Br and C–Cl bond lengths, respectively; RBrCl is the distance between Br and Cl atoms. One can see in Table 1 that RCBr and RCCl increase asymmetrically, and RBrCl decreases during the entire dissociation process. In other words, in this dissociation process, the C–Br and C–Cl bond are not broken synchronously, C–Br chemical bond break apart firstly, and subsequently a new Br–Cl chemical bond starts to emerge while C–Cl bond continues to exist for a while. According to these results, we can deduce that 1,2-BCE2+ decomposes into BrCl+ and C2H4+ by an asynchronous concerted mechanism.
| R CBr (Å) | R CCl (Å) | R BrCl (Å) |
|---|---|---|
| 2.07 | 1.91 | 2.19 (BCE2+) |
| 2.13 | 1.88 | 2.19 |
| 2.72 | 1.83 | 2.19 (TS1) |
| 3.20 | 1.81 | 2.17 |
| 3.63 | 1.81 | 2.17 |
| 4.11 | 1.81 | 2.17 (S1) |
| 4.82 | 2.04 | 2.18 |
| 5.29 | 2.78 | 2.09 |
| 5.65 | 3.24 | 2.07 |
| 6.16 | 3.93 | 2.06 (TS2) |
| 6.87 | 4.99 | 2.06 |
| 7.10 | 5.32 | 2.06 |
So far, unimolecular ion elimination of BrCl+ from CE of 1,2-BCE2+ is securely demonstrated in this experiment. Using femtosecond pump-probe technique, Dantus and co-workers8–12 studied concerted elimination of CX2YZ to halogen molecules YZ (X represents for Cl, H, or F, and Y, Z represent for Cl, Br, or I). They pointed out that these halogen molecular elimination reactions from CX2YZ were ultrafast processes around 50 fs. On the basis of their conclusions and the 80 fs laser pulse we used, it is reasonable to assume that unimolecular ion elimination process of BrCl+ from 1,2-BCE2+ proceeds during the presence of laser pulse. Hence, we can understand this whole process in this way: femtosecond lasers cause vertical ionization of 1,2-BCE into a repulsive region of the potential energy hyper surface of 1,2-BCE dication, followed by a relaxation of unstable 1,2-BCE dication (the previous step) to the local minimum of 1,2-BCE dication. As displayed in Fig. 4, the relaxation involves rotation of ∠BrCCCl and elongation of both C–Br and C–Cl bond. Afterwards, fragmentation of 1,2-BCE2+ into C2H4+ and BrCl+ take place on ground PES of 1,2-BCE2+ with presence of the laser field. Eventually, the BrCl+ is produced through an asynchronous concerted mechanism.
5. Conclusions
We have used dc-slice ion imaging technique to study unimolecular ion elimination of BrCl+ from photodissociation of 1,2-BCE at 800 nm in an 80 fs laser field. The observation of fragment ion BrCl+ in TOF spectrum confirmed the existence of unimolecular ion elimination channel of BrCl+ in this experiment, and the relative quantum yield of BrCl+ was measured to be 0.8%. Through analysis of the velocity and angular distributions of BrCl+ and C2H4+, BrCl+ was demonstrated to stem from CE of 1,2-bromochloroethane diaction 1,2-BCE2+. Theoretically, the pathway of 1,2-BCE2+ to BrCl+ + C2H4+ was simulated at M06-2X/def2-TZVP level. The calculation results showed that 1,2-C2H4BrCl2+ went over a small barrier of 0.26 eV to form S1 in which C–Br and C–Cl bond lengths were 4.11 Å and 1.81 Å respectively, and then S1 overcame a 1.80 eV barrier to dissociate into BrCl+ + C2H4+ through an asynchronous concerted elimination mechanism.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by University Students' Innovation and Entrepreneurship Training Program of Xi'an Shiyou University (201810705019), National Natural Science Foundation of China (Grant No. 11747111, 11847138), Natural Science Basic Research Plan in Shanxi Province of China (Grant No. 2016JQ1027, 2019JM296), and Scientific Research Program Funded by Shannxi Provincial Education Department (Grant No. 19JK0667). The authors wish to acknowledge Prof. Zhenrong Sun and Dr Yan Yang of East China Normal University for their help on experimental design.References
- Y. M. Wang, S. Zhang, Z. G. Wei and B. Zhang, Velocity map imaging of dissociative ionization and Coulomb explosion of CH3I induced by a femtosecond laser, J. Phys. Chem. A, 2008, 112, 3846 CrossRef CAS PubMed.
- Y. R. Lee, C. C. Chen and S. M. Lin, A concerted three-body formation X + Y + C2H4 in the photodissociation of CH2XCH2Y (X, Y=Br, Cl) at 193 nm, J. Chem. Phys., 2004, 120, 1223 CrossRef CAS PubMed.
- W. G. Roeterdink and M. H. M. Janssen, Femtosecond velocity map imaging of dissociative ionization dynamics in CF3I, Phys. Chem. Chem. Phys., 2002, 4, 601 RSC.
- P. Y. Wei, Y. P. Chang, W. B. Lee, Z. F. Hu, H. Y. Huang, K. C. Lin, K. T. Chen and A. H. H. Chang, 248 nm photolysis of CH2Br2 by using cavity ring-down absorption spectroscopy: Br2 molecular elimination at room temperature, J. Chem. Phys., 2006, 125, 133319 CrossRef.
- H. L. Lee, P. C. Lee, P. Y. Tsai, K. C. Lin, H. H. Kuo, P. H. Chen and A. H. H. Chang, Photodissociation of dibromoethanes at 248 nm: an ignored channel of Br2 elimination, J. Chem. Phys., 2009, 130, 184308 CrossRef PubMed.
- S. Y. Chen, P. Y. Tsai, H. C. Lin, C. C. Wu, K. C. Lin, B. J. Sun and A. H. H. Chang, I2 molecular elimination in single-photon dissociation of CH2I2 at 248 nm by using cavity ring-down absorption spectroscopy, J. Chem. Phys., 2011, 134, 034315 CrossRef.
- M. Balaganesh, D. P. Roibás, T. kasai and K. C. Lin, Photodissociation of CH2BrI using cavity ring-down spectroscopy: in search of a BrI elimination channel, Phys. Chem. Chem. Phys., 2019, 21, 13943 RSC.
- U. Marvet, Q. G. Zhang, E. J. Brown and M. Dantus, Femtosecond dynamics of photoinduced molecular detachment from halogenated alkanes. I. Transition state dynamics and product channel coherence, J. Chem. Phys., 1998, 109, 4415 CrossRef CAS.
- U. Marvet, E. J. Brown and M. Dantus, Femtosecond concerted elimination of halogen molecules from halogenated alkanes, Phys. Chem. Chem. Phys., 2000, 2, 885 RSC.
- Q. G. Zhang, U. Marvet and M. Dantus, Concerted elimination dynamics from highly excited states, Faraday Discuss., 1997, 108, 63 RSC.
- Q. G. Zhang, U. Marvet and M. Dantus, Femtosecond dynamics of photoinduced molecular detachment from halogenated alkanes. II. Asynchronous concerted elimination of I2 from CH2I2, J. Chem. Phys., 1998, 109, 4428 CrossRef CAS.
- U. Marvet and M. Dantus, Femtosecond observation of a concerted chemical reaction, Chem. Phys. Lett., 1996, 256, 57 CrossRef CAS.
- W. G. Roeterdink, A. M. Rijs and M. H. M. Janssen, Imaging of ultrafast molecular elimination reactions, J. Am. Chem. Soc., 2006, 128, 576 CrossRef CAS.
- L. Zhu and J. W. Bozzelli, Cl2 molecular elimination reaction from 1,2-dichloroethane, Chem. Phys. Lett., 2002, 357, 65 CrossRef CAS.
- W. T. Borden, R. J. Loncharich and K. N. Houk, Synchronicity in multibond reactions, Annu. Rev. Phys. Chem., 1988, 39, 213 CrossRef CAS.
- M. J. S. Dewar, Multibond reactions cannot normally be synchronous, J. Am. Chem. Soc., 1984, 106, 209 CrossRef CAS.
- C. E. M. Strauss and P. L. Houston, Correlations without coincidence measurements: deciding between stepwise and concerted dissociation mechanisms for ABC.-+. A+ B+ C, J. Phys. Chem., 1990, 94, 8751 CrossRef CAS.
- Z. H. Liu, Y. Q. Wang, J. J. Ma, L. Wang and G. Z. He, Concerted elimination of CH2I2 and CH2ICl under intense femtosecond laser excitation, Chem. Phys. Lett., 2004, 383, 198 CrossRef CAS.
- H. Wu, Y. Yang, S. Z. Sun, J. Zhang, L. Deng, S. A. Zhang, T. Q. Jia, Z. G. Wang and Z. R. Sun, Concerted elimination of Br2+ resulting from the Coulomb explosion of 1,2-dibromoethane in an intense femtosecond laser field, Chem. Phys. Lett., 2014, 607, 70 CrossRef CAS.
- Y. Yang, L. L. Fan, S. Z. Sun, J. Zhang, Y. T. Chen, S. A. Zhang, T. Q. Jia and Z. R. Sun, Dissociative double ionization of 1-bromo-2-chloroethane irradiated by an intense femtosecond laser field, J. Chem. Phys., 2011, 135, 064303 CrossRef PubMed.
- H. Wu, Y. X. Xue, J. Q. Wen, H. Wang, Q. F. Fan, G. X. Chen, J. Zhu, F. H. Qu and J. Guo, Theoretical and experimental studies on hydrogen migration in dissociative ionization of the methanol monocation to molecular ions H3+ and H2O+, RSC Adv., 2019, 9, 16683 RSC.
- C. L. Guo, M. Li, J. P. Nibarger and G. N. Gibson, Single and double ionization of diatomic molecules in strong laser fields, Phys. Rev. A, 1998, 58, R2471 CrossRef.
- L. Goerigk, A. Hansen, C. Bauer, S. Ehrlich, A. Najibi and S. Grimme, A look at the density functional theory zoo with the advanced GMTKN55 database for general main group thermochemistry, kinetics and noncovalent interactions, Phys. Chem. Chem. Phys., 2017, 19, 32184 RSC.
- M. Walker, A. J. A. Harvey, A. Sen and C. E. H. Dessent, Performance of M06, M06-2X, and M06-HF density functionals for conformationally flexible anionic clusters: M06 functionals perform better than B3LYP for a model system with dispersion and ionic hydrogen-bonding interactions, J. Phys. Chem. A, 2013, 47, 12590 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, et al.Gaussian 16, Revision B.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- K. E. Singleton, R. G. Cooks and K. V. Wood, Utilization of natural isotopic abundance ratios in tandem mass spectrometry, Anal. Chem., 1983, 55, 762 CrossRef CAS.
- Y. M. Wang, S. Zhang, Z. R. Wei and B. Zhang, Velocity map imaging of dissociative ionization of ICl in femtosecond laser field, Chem. Phys. Lett., 2009, 468, 14 CrossRef CAS.
- M. E. Corrales, G. Gitzinger, J. G. Vazquez, V. Loriot, R. de Nalda and L. Banares, Velocity map imaging and theoretical study of the Coulomb explosion of CH3I under intense femtosecond IR pulses, J. Phys. Chem. A, 2012, 116, 2669 CrossRef CAS.
- H. Wu, S. A. Zhang, Y. Yang, S. Z. Sun, J. Zhang, L. Deng, T. Q. Jia, Z. G. Wang and Z. R. Sun, Coulomb explosion and dissociative ionization of 1,2-dibromoethane under an intense femtosecond laser field, RSC Adv., 2014, 4, 45300 RSC.
- G. E. Busch and K. R. Wilson, Triatomic photofragment spectra. II. Angular distributions from NO2 photodissociation, J. Chem. Phys., 1972, 56, 3638 CrossRef CAS.
- T. Okino, Y. Furukawa, P. Liu, T. Ichikawa, R. Itakura, K. Hoshina and H. Nakano, Coincidence momentum imaging of ejection of hydrogen molecular ions from methanol in intense laser fields, Chem. Phys. Lett., 2006, 419, 223 CrossRef CAS.
- P. M. Kraus, M. C. Schwarzer, N. Schirmel, G. Urbasch, G. Frenking and K. M. Weitzel, Unusual mechanism for H3+ formation from ethane as obtained by femtosecond laser pulse ionization and quantum chemical calculations, J. Chem. Phys., 2011, 134, 114302 CrossRef.
- T. S. Zyubina, S. H. Lin, A. D. Bandrauk and A. M. Mebel, Dissociation pathways of cyclohexane trication, Chem. Phys. Lett., 2004, 393, 470 CrossRef CAS.
Footnote |
| † PACS numbers: 82.53.St, 33.80.Rv, 31.15.A-, 33.80.Eh. |
| This journal is © The Royal Society of Chemistry 2019 |