
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesigning a bioactive scaffold from coassembled collagen–laminin short peptide hydrogels for controlling cell behaviour†
Rashmi Jain and
Sangita Roy *
*
Institute of Nano Science and Technology, Mohali, Punjab, India. E-mail: sangita@inst.ac.in
First published on 26th November 2019
Abstract
Synthetic bioactive hydrogels have been widely recognized as key elements of emerging strategies for tissue engineering. The complex hierarchical structure and chemical composition of the natural ECM inspires us to design multi-component ECM mimics that have potential applications in biomedicine. Taking inspiration from natural proteins, we hypothesized that designing a multi-component synthetic matrix based on short ECM derived peptides will be highly beneficial for providing a functional scaffold, which still remains a challenge in the field of biomaterials. For the proof of concept, we designed a composite hydrogel scaffold inspired by the two essential components of native ECM, i.e. collagen and laminin, which play diverse roles in supporting cell growth. To the best of our knowledge, we have designed the shortest collagen inspired peptide sequence i.e. Nap-FFGSO, which has propensity to self-assemble in the presence of short laminin mimetic peptides. Interestingly, only 10% w/w of laminin peptides was sufficient to provide nucleation and growth of the nanostructures in the collagen inspired peptide and induces further self-assembly to create higher order structures. Such a nucleation and growth mechanism can trigger gelation in the collagen inspired peptides, which otherwise failed to form a gel under physiological conditions. We could achieve similar complexity in the designed matrix through utilization of simple non-covalent interactions, rather than covalent synthetic methodologies to create a dual functional matrix from the non-gelator collagen inspired peptide. The nanofibrous morphology generated through co-assembly can essentially mimic the structure and function of natural ECM, which enables the scaffold to communicate with cells through biochemical signals and promote cell growth, adhesion, proliferation and migration. We envisage that this structural mimicry of a native collagen fibrillar network using such a short peptide sequence can lead to new opportunities for developing next generation functional materials. The strategy of supramolecular assembly using multiple components could develop a plethora of viable biomaterials under physiological conditions.
Introduction
The extracellular matrix (ECM) is a complex extracellular environment that provides structural support to the cells and regulates several important functions like cell growth, cell–cell interactions and tissue organization.1,2 Self-assembling hydrogels possess great potential to be developed as substitutes for native ECM owing to their capability of providing a desirable environment for cell survival and biodegradation.3,4 Hydrogels can be tailored to closely mimic the physical properties, stiffness and topography to match that of natural ECM, but inducing complex cell–ECM interactions still remains a challenge.5,6 This cell–ECM interaction encodes a huge amount of information which determines the cell fate.7 Incorporation of bioactive cues majorly in the form of proteins derived from native ECM is the most versatile and facile strategy to promote cell–matrix interactions.8In nature, bio-molecules have remarkable ability to self-assemble into highly ordered well-defined complex structures.9 For example, collagen, the most abundant member of ECM protein family, has attracted considerable attention due to its functional significance, structural diversity and its excellent self-assembling properties.10,11 But, difficulties like, thermal instability, relatively difficult isolation and specific modifications and possibility of contamination of collagen obtained from natural sources or by microbial expression have made synthetic collagen models a popular target of biomimetic design.12 Inspite of having wide variety of structural organizations, all collagens share a common primary structure. They are composed of distinct repetitive units of Gly–X–Y, where X and Y are usually proline and hydroxyproline.13,14 The characteristic triple helical structure of collagen is stabilized by the close packing of glycine and hydrogen bond formed between the amide proton of glycine and carbonyl group of nearby X-residue.15 A classical report by Ramachandran and co-workers explained the role of hydroxyproline residues in stabilizing the collagen triple helix via additional H-bonding between hydroxyl group of hydroxyproline and water molecules.16 Further, self-assembly of triple helices provides mechanical strength and structural integrity to the body.15
Crucial advancements have been made in replicating the multi-hierarchical self-assembly of collagen and towards understanding the fundamentals of design of native collagen. To this direction, collagen mimetic peptides (CMP) have been evolved as the potential biomaterial for biomedical applications, like, tissue engineering, drug delivery, diagnostics and therapy, etc.17–21 Many approaches have been adapted to elucidate the driving forces of self-assembly in synthetic systems mimicking the similar steps as that of natural collagen, but it still remains a challenge.11 To overcome such challenge, researchers explored wide range of intermolecular interactions like electrostatic interactions,11,22,23 π–π stacking,24 hydrophobic interactions,25 π–cation interactions,26 metal–ligand interactions27–31 and triple helical nucleation32 and accessed diverse higher order structures.33,34 Hartgerink and co-workers designed a 36-mer CMP which can mimic each step of multi-hierarchical assembly of collagen. However, their smart design included classical intermolecular interactions like H-bonding complemented with salt bridges between lysine and aspartate residues. The electrostatic interactions stabilized the triple helix in a sticky end assembly.11,12
Based on basic repeating sequence Gly–X–Y, extensive efforts have been made to overcome the synthetic challenge in designing a collagen inspired peptide (CIP). However, only a few short collagen inspired peptide amphiphiles have been developed that posses the ability to form supramolecular hydrogels.35,36 For example, the octapeptide library of collagen inspired supramolecular hydrogelator was developed by Z. Yang et al., having random coil conformation. The gel strength was found to match that of the matrix of the fibroblast cells and hence they were suitable for the growth of 3T3 cells.37 Another recent report by Teckinay's group demonstrated the collagen presenting scaffold based on octapeptide amphiphile having Pro–Hyp–Gly (POG) functional sequence, which forms β-sheets. These scaffolds were further explored for the prevention of progressive IVD (intra vertebral disc) degeneration.38 However, both these reports demonstrated self-assembling octapeptide sequences, which were the smallest length collagen inspired peptides, reported so far. Hence, it would be extremely interesting to design even shorter peptides based on the native collagen structure (GXO), which would be easy to synthesize and can have the potential to achieve the similar structural complexity as well as functions of collagen. It was evident that attempts were directed to modify –X or –O positions with charged amino acids, inducing additional electrostatic interactions for stabilizing collagen mimetic oligopeptide sequences.13 As mentioned in earlier reports, the cis–trans isomerisation of prolines can have destabilizing effects on collagen assembly, so substitution of proline residues offers a target site for CIP modification and explore sequences, keeping the position of glycine and hydroxyproline residues constant.39
Moreover, the previous reports in the literature utilized single functional peptide sequence (collagen inspired) to design a bioactive scaffold.10–14 However, the complex hierarchical structure and composition of native ECM provides clues to incorporate multiple signals in a single homogenous scaffold, which would provide synergistic cellular response.40–43 Laminin is another important class of extracellular matrix protein. Laminin is a heterotrimeric protein, mainly present in the basement membrane. Two most popular short peptide sequences, like, IKVAV which is present on the A-chain and YIGSR, present on the B-chain of native laminin protein, are identified to mimic the biological activity of the laminin protein.44 Laminin has diverse biological roles including cell adhesion, migration, differentiation and growth.44 However, a very few IKVAV and YIGSR based short laminin peptides were reported, showing classical role in controlling cell behavior.44,45 A report by Nisbet et al., illustrated the minimalist Fmoc self-assembling peptide design using laminin derived IKVAV and YIGSR, along with fibronectin based RGD peptide for delivering cortical neural progenitor cells in mouse brain.46,47 In particular, the role of laminin peptides along with collagen IV has been widely studied for their nerve regeneration properties.48–50 The role of multicomponent gels composed of isolated collagen, laminin, fibronectin and hyaluronic acid in neurite extension was shown by Schmidt et al.40 In the similar line, H. Iwata's group showed the development of hydrogels composed of collagen and laminin derived cell adhesive peptides isolated from microbial genetic engineering for improved survival of neural cell.51 To the best of our knowledge, no synthetic co-assembled multi-component oligopeptide gels inspired from collagen and laminin peptides have been reported in the literature.52 The previous studies clearly explained that synthetic oligopeptide sequences derived from collagen and laminin individually show the promising biological applications, and therefore, in combination, can present an exclusive bioactive scaffold, which can provide a more realistic model of ECM mimetic biomaterials for tissue engineering. The combined matrix can provide the necessary biochemical and biomechanical cues to different cell types.
In this paper, we have illustrated the design of collagen inspired peptide (CIP), the shortest CIP sequence, containing only a single –GXO unit, which makes it difficult to attain α-helical structures. However, this CIP structural mimic can self-assemble to form spherical aggregates, but fails to induce higher order fibrillar network to form gel like structure. Interestingly, the incorporation of a very less quantity of laminin mimetic peptide (10% w/w) serves dual purpose. Initially, it acts as a nucleation center and induces gelation to the non-gelating self-assembling CIP through growth of organized nanostructures, in aqueous solvent. And secondly, it provides additional functionality to the scaffold for cell adhesion. The nanofibrillar structure of the co-assembled gels exhibited homogenous morphology, which indicates that the peptides formed non self-sorted fibrous network showing the β-turn like secondary structures. Further investigations were made to study the cell–matrix interactions, which revealed that supramolecular co-assembled hydrogels supported the adhesion, survival and proliferation of different types of cells. We aim to achieve the similar complexity of natural ECM within our designer self-assembled hydrogel domain, though they are the shortest CIP and LMP sequences designed so far which can mimic the functional attributes of large proteins. We envisage that, these multicomponent co-assembled collagen and laminin mimetic peptide hydrogels based on molecular self-assembly can be developed as potential biocompatible platforms for presentation of biochemical functionality at nanoscale.
Results and discussions
Peptide design
The scaffolds designed with combination of multiple signals offer a synergistic effect on cellular response. To mimic the co-existence of collagen and laminin in native extracellular matrix, we designed a multi-component hydrogel scaffolds composed of collagen and laminin derived functional short peptide sequences.54,55 To the best of our knowledge, it is the first time we demonstrate the minimalist approach for co-assembly of two different bioactive short peptides resulting into supramolecular gels, without any polymeric network utilization. Most interestingly, these are the shortest peptide sequence designed so far, attempting minimal modification to the known functional sequence. The design of collagen inspired peptide sequence i.e. Nap-FFGSO was based on incorporation of the basic collagen motif, Gly–X–O, with aromatic dipeptide gelator, Nap-FF (napthoxy-diphenylalanine) (Fig. 1a). In our design, 2nd position ‘proline’ was mutated with aliphatic amino acid, serine, with an additional hydroxyl group, which can facilitate the extensive H-bonding interactions in the aqueous solvent. The aromatic dipeptide, Nap FF is an excellent gelator and its gelation properties were well explored in the literature.56–58 The evidences for the presence of aromatic amino acid residues in the native collagen can be found in the literature, which catalyze the formation of stable fibril formation. It was proposed that such type of short non-helical peptides flanked at the terminals of long chain triple helix confers rigidity and stability to the more flexible collagen fibrils.59 In this context, only a few reports on short IKVAV and YIGSR based peptides forming gels are available in the literature, in conjugation with an aliphatic chain, or polymeric network or additional amino acid sequences.44–47 However, in combination to collagen inspired peptide, the pentapeptide sequences i.e. IKVAV (A) and YIGSR (B), derived from native laminin protein, were also synthesized with 2-nathoxyacetic-acid attached at their N-terminal (Fig. 1a). The variable side chains in the peptide backbone gives rise to variable intermolecular interactions (aromatic π–π interactions, hydrophobic interactions and H-bonding) resulting in differential co-assembly, forming different types of self-assembled structures.60 The designed peptide sequences and their representative assembly mechanism is shown in Fig. 1a and b. The peptides were synthesized using standard solid phase synthesis protocols. The purity of synthesized peptides was checked through HPLC and LC-MS. The compound was found to be >99% pure (Table S1 and Fig. S1†).61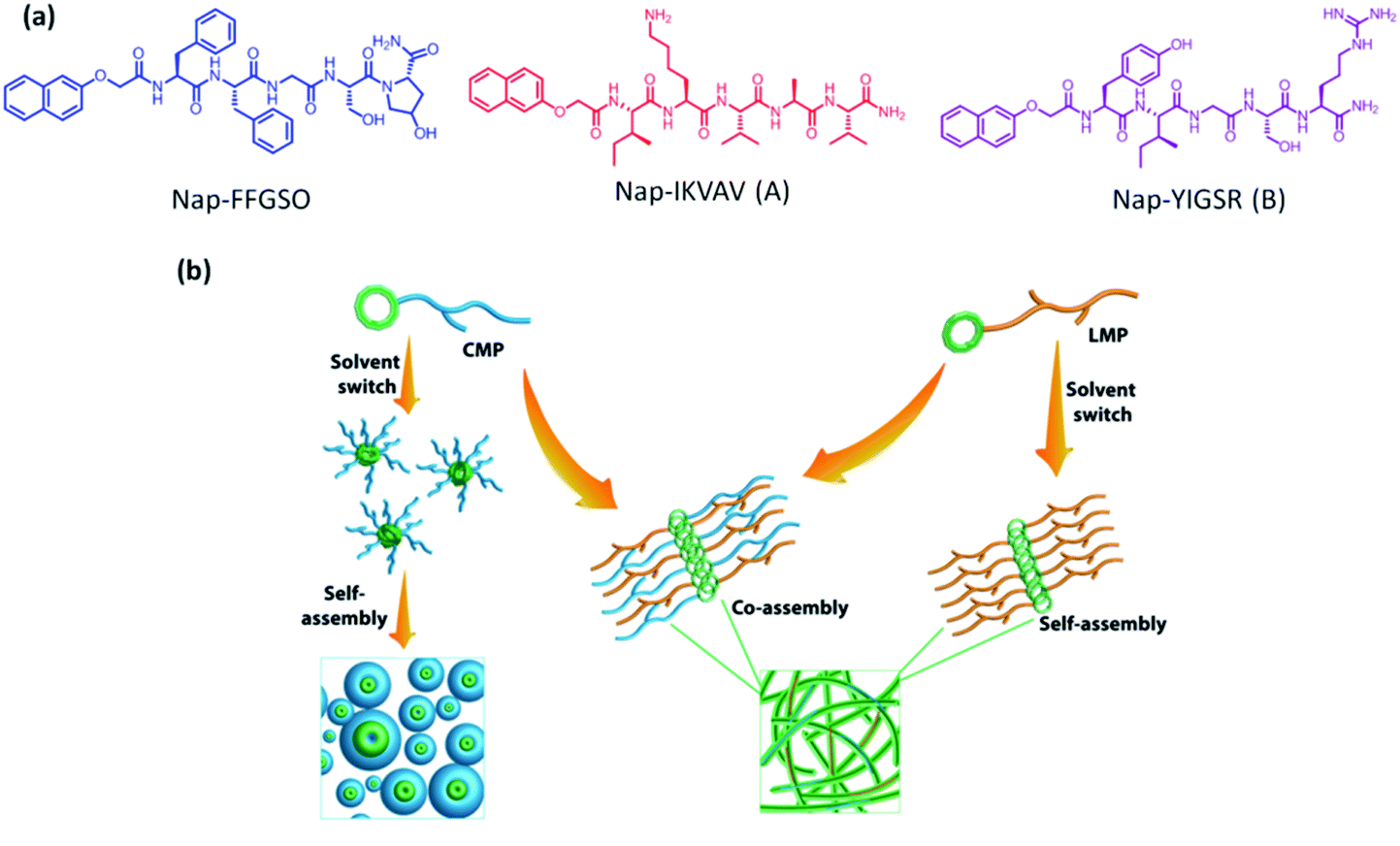 | ||
| Fig. 1 (a) Molecular structure of CIP (collagen inspired peptide) as Nap-FFGSO and LMP (laminin mimetic peptides) as Nap IKVAV and Nap YIGSR. (b) Schematic representation of co-assembly of CIP and LMP using 10% DMSO/water as a solvent switch. | ||
Gelation studies
It is expected that variable amino acid sequences in hydrophobic IKVAV i.e. A as well as hydrophilic YIGSR i.e. B, will induce differential intermolecular interactions (π–π, hydrophobic and H-bonding) with Nap-FFGSO, leading to differential co-assembly. These peptides were found to be insoluble in aqueous media owing to their extensive hydrophobicity which prevents them forming an organized network structure in water. Therefore, we employed a unique strategy of solvent switch to induce organized network in the peptide solution. This strategy involves the dilution of DMSO stock of peptides with water and the solution was vortexed until a uniform mixture was obtained. To investigate the differential self-assembling properties of collagen inspired peptide individually as well as their co-assembly with LMP, their critical aggregation concentration (cac) was determined using thioflavin T (Th T).62 Th T is a fluorescent dye that binds to hydrophobic patches of amyloid fibrils. The intensity of Th T fluorescence is dependent on the concentration of aggregates. As the aggregation starts, more hydrophobic patches are exposed to the Th T dye and fluorescence emission intensity of Th T tends to increase. Addition of increasing concentration of peptides to a fixed 10 μM concentration of Th T results in drastic increase in the fluorescence intensity, after a certain concentration, and that can be considered as cac. For individual peptides, the cac for Nap-FFGSO, Nap-IKVAV and Nap-YIGSR was found to be ∼10 μM, ∼2.5 μM and ∼2.5 μM respectively (Fig. S2a–c†). This indicates that LMPs have higher aggregation propensity than Nap-FFGSO, as they aggregate at the lower concentrations. The induction of co-assembly after addition of LMP was assessed by adding the varying concentrations of LMP peptide to sub-aggregation concentration of Nap-FFGSO i.e. 5 μM. It was observed that 1 μM of Nap-IKVAV and 1.8 μM of Nap-YIGSR was able to induce aggregation in 5 μM of Nap-FFGSO (Fig. S2d and e†). The cac results suggest that the addition of LMP reduces the cac of Nap-FFGSO because multiple interaction points were introduced by adding LMP in the solution which helped in nucleation and growth of the co-assembled structures. Later, gelation was tried at higher concentrations of CIP and LMPs to achieve macroscopic self-assembly which is expected to result from higher order assembly of aggregated spherical structures.To prepare co-assembled gels, the peptides in different proportions were dissolved in DMSO/water and results for gelation studies are summarized in Table 1. The role of DMSO fraction in the system was well explained in a report by Adams and his group.63 The report clearly states that lower DMSO fractions result in highly turbid solutions, initially, which clears out over the course of time. The increased turbidity was caused due to the formation of dispersed spherical structures, leading to phase separation. With time, the spheres acts as a nucleation sites for the growth of fibrillar network, which can be correlated with the formation of gels. On the other hand, the higher concentrations of DMSO results in higher solubility with the clear transparent solutions, which sometimes may fail to form gel. Our observations were consistent with the reported phenomenon, at 2% of DMSO fraction, a lump of solubilised peptide was formed which takes longer time to get miscible with water, with the aid of vigorous agitation. As the peptide (with DMSO) dissolution proceeds in water, the solution becomes more turbid and resulted into gelation after complete dissolution. The images of gels with 2% DMSO were shown in Fig. S3a and c†.63 While with 10% DMSO, the stable translucent gels were obtained (Fig. S4†). The microscopic evaluation of the gels prepared in 2% DMSO/water were consistent with the previous studies that reports the formation of turbid gels with lesser amount of DMSO and forms mixed structures with spherical aggregates and discrete fibrillar structures in both Nap-FFGSO + A and Nap-FFGSO + B (Fig. S3b and d†). In addition to this, longer vigorous agitations and lesser organised structures account for the lower mechanical strength of co-assembled CIP-LMP gels in 2% DMSO/water (Fig. S3e†). Therefore, we used 10% DMSO/water as a solvent for gelation of CIP peptides in further studies.
| Peptide | Concentration (mM) | Gelation status |
|---|---|---|
| Nap-FFGSO | 30 | Sol |
| Nap-IKVAV (A) | 3 | Sol |
| 5 | Gel | |
| Nap-YIGSR (B) | 3 | Sol |
| 5 | Gel | |
| Nap-FFGSO + A | 30 + 3 | Gel |
| Nap-FFGSO + B | 30 + 3 | Gel |
| Nap-FFGSO + A + B | 30 + 1.5 + 1.5 | Gel |
| Nap-FFGSO + A + B | 20 + 1.5 + 1.5 | Sol |
| Nap-FFGSO + A + B | 20 + 2.25 + 2.25 | Gel |
| Nap-FFGSO + A | 20 + 3 | Sol |
| Nap-FFGSO + B | 20 + 3 | Sol |
| Nap-FFGSO + A | 20 + 4.5 | Gel |
| Nap-FFGSO + B | 20 + 4.5 | Gel |
The self-assembly of Nap-FFGSO alone was checked over a wide range of concentrations (up to 100 mM), but it failed to form gel even at higher concentrations. The probable reason for non-gelating properties of Nap-FFGSO could be the improper hydrophilic–lipophilic balance in aqueous solvent, which plays an extremely important role in determining the intermolecular interactions between the peptide molecules and thus affects the self-assembling behaviour of short amyloid like peptides. Probably, the additional hydroxyl group of serine participates in preferential H-bonding with water molecules, which disturbs the gelator–gelator interactions and thus interrupts in the formation of higher ordered structures. Interestingly, when 30 mM concentration of Nap-FFGSO was mixed with 10% of either of the LMP's (Nap IKVAV or Nap YIGSR), it resulted into the formation of self-supporting translucent to opaque gels. We also observed that the combination of 5% of each LMP when mixed with 30 mM of Nap-FFGSO, the peptides showed the hierarchical structure formation leading to supramolecular gels (Table 1). In order to develop a superior surrogate of natural ECM, such co-assembled supramolecular gels with short bioactive peptide sequences will be highly beneficial, which remain unexplored, so far in the field of tissue engineering. It was also interesting to know that the concentration of the laminin peptides used in the co-assembled gels was just 10%. Surprisingly, at this low concentration, both Nap IKVAV and Nap YIGSR individually can induce well organized network structure, though none of these two peptides could lead to supramolecular gelation. The minimum gelation concentration for Nap IKVAV and Nap YIGSR peptides separately was found to be 5 mM in 10% DMSO/water system (Table 1). The results indicated that addition of only a small quantity (∼10%) of LMP, at their sub-gelation concentration, was able to induce gelation with non-gelator CIP scaffold via co-assembly. To check the gelation ability of CIP's in the presence of LMP's, different ratios of both the peptides were tried, as a control. On decreasing the CIP concentration up to 20 mM and increasing LMP conc. up to 4.5 mM (i.e. 15% of 30 mM CIP), results into gelation (Fig. S5a, b, e and f†). The probable reason for gelation at lower concentration of CIP is that the concentration of LMP's reached near to their gelation concentration, which is 5 mM. However, 20 mM CIP added with 3 mM of either LMP or 1.5 mM of both LMPs (i.e. 10% of 30 mM), failed to form gel (Fig. S5c and g†). However, the Nap-FFGSO (20 mM) + A (2.25 mM) + B (2.25 mM) formsed table gel, showing the presence of fibrous network (Fig. S5d and h†). The optical images of all the gels with 30 mM of Nap-FFGSO with 3 mM of LMPs are shown in Fig. S3.† We hypothesize that laminin peptides act as the nucleating site for self-assembly and thus triggers the growth of higher ordered structures. The nucleating centers further support the association of the peptides through extensive aromatic π–π interactions among CIP molecules. We expect that in addition to aromatic π–π interactions, the stronger hydrophobic interaction induced by IKVAV and additional intermolecular H-bonding domain in YIGSR facilitates the growth of nucleated nanostructures. Thus, addition of LMPs probably improves the hydrophilic–lipophilic balance of the system in aqueous environment by enhancing the intermolecular interactions, which could trigger supramolecular gels formation.32
Morphological characterization
It is highly crucial to know the morphology of these co-assembled gels at nanoscale, as the variable topology is expected to induce differential cell–matrix interactions. Atomic force microscopy (AFM) was used to investigate the morphology of the nanostructures formed due to co-assembly (Fig. 2a–d). As anticipated, the non-gelator Nap-FFGSO at 30 mM concentration showed the random aggregate like morphology. As discussed earlier, at 3 mM concentration, which is the sub-gelation concentration, both the LMPs i.e. A and B were unable to form gel but showed the presence of soluble fibrous morphologies. The LMP, A showed short fibrous structures with an average fiber diameter of 40 ± 4.5 nm, while LMP B formed thin long fibers with an average diameter of 28.7 ± 4.4 nm as evident from AFM studies (Fig. S6 and Table S2†). The induction of gel formation in Nap-FFGSO upon addition of LMPs at their sub gelation concentration was monitored using AFM. In the presence of A and B, the spherical aggregates formed by Nap-FFGSO completely disappeared and the appearance of well ordered fibrous network in the co-assembled gels was observed, which led to the formation of supramolecular gels. Interestingly, the dimensions of the conjugated gels were found to be quite different. The CIP with hydrophobic LMP derivative, A showed the existence of short length twisted nanofibers of diameter ∼62 ± 4.8 nm (Fig. 2a–d and Table S2†), whereas CIP with hydrophilic LMP B formed fibers of much longer length, however, the fiber appears to be thin with a diameter of ∼17 ± 2.3 nm. In the tri-component gels with both LMPs, i.e. Nap-FFGSO + A + B, the presence of fibers of variable lengths were observed with uniform diameter in the range of ∼26 ± 3.1 nm. The morphological investigation clearly indicated that there were no distinct subpopulations of fibrils in co-assembled gels that might indicate the self-sorted assembly of mixed LMP peptides into mutually exclusive fibrils.64 Therefore, it was assumed that the triconjugate peptides co-assembled to form homogenous fibrils with similar diameter. However the length of the fibers are dependent on the laminin additive, as more hydrophobic IKVAV derivative induces shorter length and wider fibers while the hydrophilic YIGSR derivative yield thin but longer fibrous network due to involvement of H-bonding interactions in aqueous solvents. The control gels, at 20 mM concentration of CIP and 4.5 mM of LMPs showed the presence of uniform fibers of shorter lengths, while 20 mM of CIP with 1.5 mM of each LMP showed aggregates like structure and 2.25 mM of each LMP (equivalent to 4.5 mM of total LMP concentration) showed the formation of entangled fibrous network. The probable reason for the failure of gelation in Nap-FFGSO (20 mM) + A (1.5 mM) + B (1.5 mM) was the lack of sufficient interactions which could support the growth of aggregate like nanostructures into fibres (Fig. S5†). | ||
| Fig. 2 AFM and TEM images of collagen and laminin co-assembled peptide hydrogels of (a and e) Nap-FFGSO (30 mM), (b and f) Nap-FFGSO (30 mM) + A (3 mM), (c and g) Nap-FFGSO (30 mM) + B (3 mM) and (d and h) Nap-FFGSO (30 mM) + A (1.5 mM) + B (1.5 mM). | ||
The presence of fibrous structures confirms the formation of co-assembled gels. To further confirm the morphology of the co-assembled peptides, transmission electron microscopy was performed (Fig. 2e–h and Table S2†). Similar morphologies were also evident from TEM studies. As evident from AFM, the Nap-FFGSO formed spherical structures, which are incapable of entrapping water and thus cannot form gels. In corroboration, to AFM, Nap-FFGSO + A showed short fibers of diameter in the range ∼38 ± 5.6 nm and Nap-FFGSO + B forms long fibrous network with ∼8 ± 2.7 nm diameter. Also, Nap-FFGSO + A + B showed mixed length fiber population, but interestingly their diameter was in the similar range of ∼10 ± 2.3 nm. TEM and AFM confirmed the formation of nanofibrous structures after co-assembly which supports the three dimensional gel network for further applications.
Mechanical strength evaluation
Matrix stiffness of the gels shows prominent effect on cell processes such as adhesion, proliferation and differentiation. Hence, it is necessary to assess stiffness of hydrogel matrix to determine the behavior of developed biomaterials in tissue engineering application. The mechanical strength of all the three co-assembled supramolecular gels was measured using oscillatory rheology. Higher storage modulus (G′) in comparison to its corresponding loss modulus (G′′) for all the co-assembled systems confirmed the gelation. During amplitude sweep, the equilibrium storage moduli of all the gels remained in linear viscoelastic region up to 0.1% with minimal variation up to 10% strain and decreased significantly above 10%. This means that the gels retained elastic properties up to 10% and above that loses its elastic nature. Mechanical strength of co-assembled gels was further assessed using frequency sweep mode, keeping the strain constant as 0.1% in the linear viscoelastic region. However, no considerable difference in the gel strength was noted for Nap-FFGSO + A and Nap-FFGSO + A + B, which showed G′ in the range of 1.3 ± 0.2 kPa and 0.7 ± 0.1 kPa respectively (Fig. 3a and S7†). But, Nap-FFGSO + B showed relatively higher storage modulus i.e. 3.7 ± 0.3 kPa, in comparison to other two systems. The probable reason for relatively higher strength of Nap-FFGSO + B could be the favorable H-bonding interactions between the hydroxyl groups of Nap-FFGSO and Nap YIGSR in aqueous system.65 Very interestingly, the higher mechanical strength of the gels can be further correlated with the thinner fiber diameters of Nap-FFGSO + B with more entanglements leading to strongest gels of the co-assembled CIP and LMP gel family.66 It was noted that the mechanical strength of these co-assembled gels was much higher than the short collagen inspired hydrogels reported by Z. Yang et al., which reports storage modulus in the range of <1 Pa to <50 Pa.37 Similarly, the storage modulus of gels reported in our work was also comparable or greater than short collagen peptide amphiphile gels reported by Teckinay and his group. They showed the storage modulus of Col-PA/E-PA in the range of 1 kPa while we showed the storage modulus in the range of ∼1 kPa to ∼4 kPa for the CIP-LMP co-assembled gels.38 Thus, the co-assembled systems offer an advantage to adjust the mechanical stiffness of the nanofibrous hydrogels by modulating the nature of the dopant peptide as well as the concentration of the individual peptide components.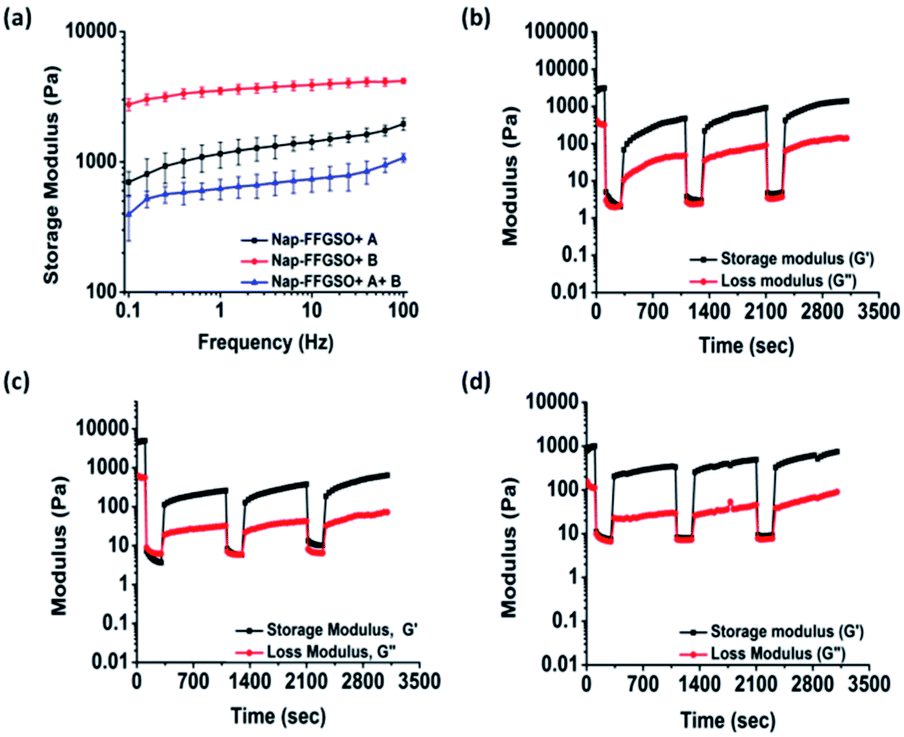 | ||
| Fig. 3 Mechanical strength measurements of co-assembled peptide hydrogels (a) frequency sweep graph of Nap-FFGSO with LMP A and B (b–d) thixotropic behaviour of (b) Nap-FFGSO (30 mM) + A (3 mM), (c) Nap-FFGSO (30 mM) + B (3 mM) and (d) Nap-FFGSO (30 mM) + A (1.5 mM) + B (1.5 mm) at 50% strain for 100 s followed by recovery at 0.1% strain for 200 s. | ||
Mechanoresponsiveness studies
To get an insight into another important aspect, i.e. injectable behaviour of these gels, the hydrogels were subjected to different extent of mechanical stress, which is crucial for their biomedical applications.67 The thixotropic studies were performed to study reversible gel–sol–gel transitions by applying external mechanical stress in a cyclic manner.68 The gels were deformed by applying the strain of 50% for 100 s followed by decreasing the strain up to 0.1% (LVE range) for 200 s which allows the recovery of the structure of gels.69 We observed significant difference in the percentage recovery of co-assembled gels, depending upon the type of laminin peptide added. The weak gels formed by Nap-FFGSO + A showed the highest recovery of ∼50%. However, under similar experimental conditions, Nap-FFGSO + B and Nap-FFGSO + A + B showed comparatively lesser recovery ∼10% and 25% respectively (Fig. 3 and S7†). We hypothesize that the lesser recovery of Nap-FFGSO with hydrophilic laminin peptide may be attributed to compromised H-bonding interactions due to loss of water during deformation. However in case of Nap-FFGSO + A, the hydrophobic interactions were restored faster.70 Therefore, the thixotropic studies revealed that these co-assembled systems can be developed as potential injectable biomaterial for cell culture and drug delivery applications.Secondary structure investigation
To assess the intermolecular interactions involved in the formation of secondary structures, several spectroscopic techniques were employed (Fig. 4). Circular dichroism (CD) is a powerful tool to elucidate the molecular arrangements of peptides and provides information regarding self-assembled supramolecular structures in the gel phase.71 For a non-gelator, Nap-FFGSO, no characteristic CD signal was observed which also complies with its morphological analysis indicating the absence of any highly ordered hierarchical organization (Fig. 4a). The CD spectrum of all the three co-assembled gels showed a positive band near 195 nm, a negative peak in between 205–210 nm, a broad positive band near 217 nm with an additional negative peak or broad band near 230 nm (n–π* transition) indicating the existence of β-turns as the major secondary structures (Fig. 4a).71 The negative cotton effect in co-assembled gels induces helical orientation of the naphthalene groups, as evidenced by the distinct peak maxima at 288 nm.72 The peak corresponds to the π–π* transition of naphthalene moiety. The single type of characteristic signal in Nap-FFGSO + A + B also indicates that homogenous structures were formed via co-assembly and no self-sorting of the different peptide nanofibers were visible, which also complies with morphological analysis.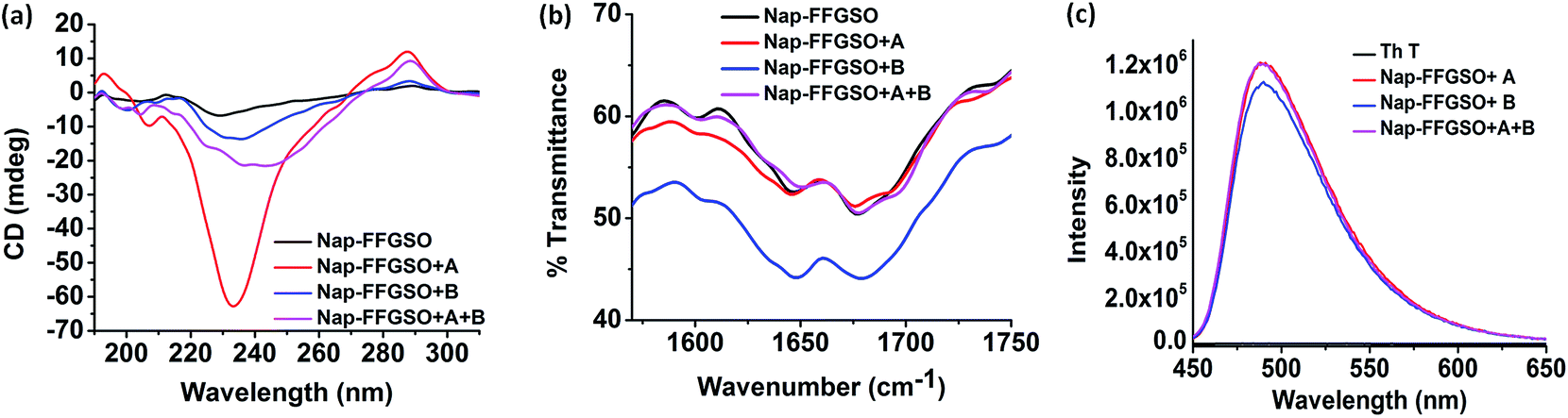 | ||
| Fig. 4 Secondary structure investigation of co-assembled hydrogels prepared 30 mM concentration of CIP and 10% LMP (3 mm) using (a) CD spectroscopy and (b) FTIR spectroscopy and (c) Th T binding assay through fluorescence spectroscopy. | ||
To further investigate the H-bonding interactions in the peptide secondary structures, FTIR spectroscopy was employed (Fig. 4b).73,74 The coupling of H-bonding between carbonyl and NH group of peptide backbone results in enhancement of signals in amide I region (1600–1700 cm−1). For all the peptide systems in our studies, a transmittance at ∼1645 cm−1 and ∼1680 cm−1 was observed which is indicative of ‘β-turns’ conformation in the structures (Fig. 4b).73,74 The FTIR spectra of LMP's (Nap IKVAV and Nap YIGSR) alone showed the presence of β-sheet structure, supporting the fibrous morphology (Fig. S6c†). The FTIR results support the CD analysis, as both the techniques indicated the formation of β-turns in the co-assembled gels.
Further, to authenticate the secondary structures within these gels, thioflavin T (Th T) binding assay was carried out. Th T is a widely used tool for the detection of cross β-structures as formed by the amyloid fibrils. Interestingly, the Th T binding is mediated by contacts through aromatic residues in the hydrophobic pockets.75 As analyzed by CD and FTIR, the β-turn secondary structures were formed by the co-assembly of CIPs and LMPs which also exhibits Th T binding (Fig. 4c). The Th T binding with the aromatic residues of these peptides is marked by the enhancement in fluorescence intensity of emission peak at ∼480 nm.75 The relative enhancement with Nap-FFGSO + A and Nap-FFGSO + A + B were found to be comparatively higher than Nap-FFGSO + B. The more hydrophobic nature of IKVAV in comparison to YIGSR might create more hydrophobic pockets and thus relatively higher increment in the Th T fluorescence intensity was observed (Fig. 4c). The Th T bound amyloid like fibrils can be examined using fluorescence microscope (Fig. S8†). In all the three co-assembled gels, the fluorescent fibrous patches can be observed, which confirms the amyloid like behavior of supramolecular assemblies formed from short peptide sequences derived from collagen and laminin.76,77
Evaluation of co-assembly
To get an insight of the molecular arrangement of two different peptides during co-assembly, the fluorescent tagging approach was used. One of the two co-assembling peptide was tagged with fluorophore i.e. FITC. In the absence of free fluorophore binding site (i.e. −NH2 or –COOH group), Nap-FFGSO could not be fluorescently coupled and therefore Nap-IKVAV was selected to be coupled with FITC. Both the peptides were dissolved in 10% DMSO/water and co-assembled structures were observed under fluorescence microscope. Initially, non-fluorescent aggregate like structures were observed with embedded fluorescent peptides within it. With time, the peptides align in the form of fibers (Fig. S9†). The presence of fluorescent patches throughout the length of the fibers was observed. This clearly reveals that the two peptides are supramolecularly arranged to form a single type of fiber, thus avoiding self-sorted fiber formation.78 We hypothesize that due to less hydrophobicity of LMP peptides, their faster gelation kinetics in aqueous solvent can be observed, leading to the formation of nucleation centers (short fibrous structures) for the further self-assembly of Nap-FFGSO.Solvent exchange
It is expected that 10% DMSO in gels can be detrimental to biological applications like cell culture. A report by T. Eljezi, showed the effect of different concentrations on the proliferation of cells. This report suggests that DMSO concentrations above 1% can reduce the cell proliferation significantly.79 However, the tunability achieved using solvent switch is highly appreciable. Therefore, to eliminate the deleterious effects of solvents, we considered solvent exchange method post-gelation, which was earlier shown by Dave Adams and co-workers.80 The CIP-LMP gels (30 + 3 mM) were initially prepared in 10% DMSO/water according to the procedure described. Post-gelation, 1 ml of Milli-Q water was added gently at the top of the gel and allowed to exchange with the solvent entrapped in gel network for 2 h. The supernatant aqueous layer was removed without disturbing the gel. Subsequently, the similar exchange was done three more times. The gels remain intact after the three cycles of solvent exchange. The removal of DMSO was confirmed by FTIR which was indicated by the diminished sulfoxide peak at 1020 cm−1, in comparison to sharp peak present in 10% DMSO as control (Fig. S10a†).80 It was important to evaluate the effect of solvent exchange on the mechanical strength of the gels, so we carried out rheology of solvent exchanged gels (Fig. S10b†). A reduction in mechanical strength was observed after solvent exchange, but gels retain their structural integrity. For Nap-FFGSO + A and Nap-FFGSO + B gels, the mechanical strength reduces to 0.8 ± 0.1 kPa from 1.3 ± 0.3 kPa and 2 ± 0.3 kPa from 3.7 ± 0.3 kPa, respectively. However, for Nap-FFGSO + A + B gels does not show very significant reduction in storage modulus and showed 0.5 ± 0.06 kPa after exchange in comparison to 0.7 ± 0.1 kPa without exchange. Interestingly, the mechanical strength of the gels remained higher, even after solvent exchange, in comparison to the previously reported short collagen mimetic peptide hydrogels. Similar exchange can be done with cell culture media in order to make these gels more suitable for cell culture applications.Biocompatibility studies
Biocompatibility studies are pre-requisite criteria to assess the suitability of these CIP/LMP co-assembled peptide gels as biomaterials.81 The short duration treatment (4 h) was performed to assess the initial contact cytotoxicity of peptides with the cells.82 The colorimetric MTT assay was performed to quantitatively determine the number of metabolically active cells after treatment with respect to control cells (Fig. 5). The mouse fibroblast L929 cells and rat glioma C6 cells were seeded in 96 well plate and allowed to adhere for 24 h. The cells were then treated with different concentrations i.e. 100 μg ml−1 (∼0.13 mM) and 1000 μg ml−1 (∼1.3 mM) of soluble peptides for 4 h to check the cytotoxicity of these peptides.83 The reason for using sub-gelation concentrations for cytotoxicity studies was that the previous reports suggested that oligomeric lower ordered self-assembled structures of amyloid peptides are more toxic to the cells than the higher ordered self-assembled fibrous network.84 Moreover, the dilution of peptide stock (5 mg ml−1 or 6.5 mM) also reduces the DMSO content to 0.2% in 100 μg ml−1 and 2% in 1000 μg ml−1, which further reduces the chances of toxicity due to DMSO. The presence of soluble self-assembled structures in the diluted peptide solutions at concentration of 100 μg ml−1 (0.13 mM) and 1000 μg ml−1 (1.3 mM) were confirmed by using AFM (Fig. S13†). The control cells were treated with 10% DMSO containing media with similar dilution (0.2% and 2%). The results suggest that peptides were highly biocompatible; however the percentage of viable cells was relatively higher with L929 cells in comparison to C6 cells. With both the cell types, CIP alone (Nap-FFGSO) showed more than 100% cell viability, which might be due to promoted proliferation (Fig. 5). The biocompatibility of the co-assembled gels was found to be inversely proportional to the hydrophobic nature of the resulting gels. Among all the three co-assembled gel systems, the most hydrophobic sequence, Nap-FFGSO + A resulted in comparatively less biocompatible matrix, which showed nearly 80% viability with L929 cells and nearly 60% for C6 cells at 1000 μg ml−1 (1.3 mm) concentration (Fig. 5a and b). The greater hydrophobicity of IKVAV which leads to the formation of shorter length fibers may not providing uniform network structure for adherence of cells, which is likely to affect its interaction with cells and made it comparatively less biocompatible. In contrast, the hydrophilic Nap-FFGSO + B induces less toxicity to the cells with ∼90% viability of C6 cells and 93% viability of L929 cells, at 1000 μg ml−1 (1.3 mM) concentration of peptide. However, doping of hydrophobic A (5%) with Nap-FFGSO + B, in a triconjugate gels, biocompatibility was found to be reduced than Nap-FFGSO + B, which becomes ∼70% for C6 cells and ∼84% for L929 cells. Further, to check the long term cytotoxicity of the peptides, the biocompatibility assay after the treatment of 24 h, 48 h and 72 h was also performed with both C6 and L929 cells (Fig. S14†). The concentration used for the treatment was 1000 μg ml−1 (1.3 mM). The cell viability of more than 80% was observed even after 72 h for all the peptides except for Nap-FFGSO + A, which showed ∼70% cell viability up to 72 h. The biocompatibility results suggested that the peptides were relatively more compatible with L929 cells than C6 cells. We also checked the morphology of cells after the treatment of peptides and it was observed that cells retain their healthy morphology and remain unaffected by the treatment even at higher concentration i.e. 1000 μg ml−1 (1.3 mM) (Fig. S11 and S12†).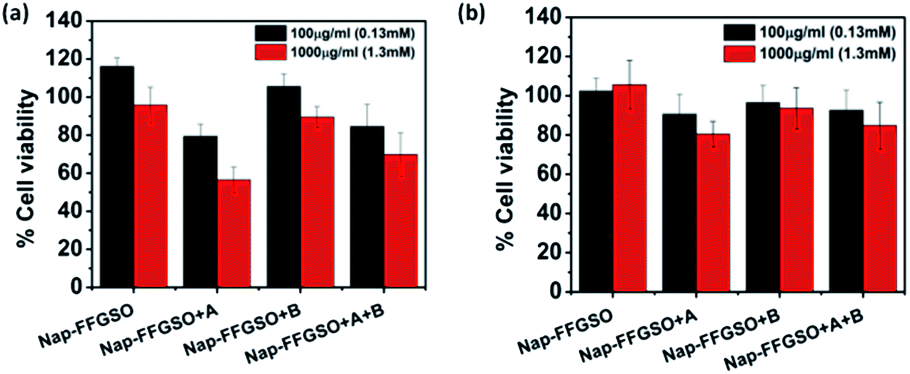 | ||
| Fig. 5 Biocompatibility studies of collagen inspired peptide and its co-assemblies with laminin mimetic peptides at the concentration of 100 μg ml−1 (0.13 mM) and 1000 μg ml−1 (1.3 mM) with (a) C6 cells and (b) L929 cell lines, using MTT. | ||
2D cell culture
Prior to assessment of these co-assembled gels as substrate for cell culture applications, we checked the integrity of these gels by exposing them to cell culture media. The perfusion of self-assembled gels with culture media also replaces the organic co-solvent (DMSO) used during gelation procedure.85 The pre-formed hydrogels prepared in 10% DMSO/water were spreaded uniformly over a coverslip and allowed to set for 24 h. The cell culture media was added on the top of the gels and kept for perfusion for next 24 h, after which the media was replaced. After several washes with culture media, all the three co-assembled gels retained their integrity and remain adhered on the surface of coverslips. It was also expected that the washings with media would reduce the DMSO content of gels and results in better cytocompatability.86 The gels showed excellent stability which can offer a biocompatible bioactive scaffold for cell culture applications.Further, these co-assembled hydrogels containing the functionality similar to both collagen as well as laminin proteins were employed for the 2D cell culture applications.87 Mouse fibroblast L929 and rat glioma C6 cells were seeded onto the surface of each co-assembled gels (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per well in a 6-well plate). For control, the cells were seeded on untreated coverslip. After 24 h of seeding, cell adhesion was observed with cells adapting the polyhedral or spindal shapes (Fig. S15†). Live/dead staining was performed after 24, 48 and 72 h after seeding (Fig. 6, 7 and S16†). The cell density on the co-assembled hydrogels was found to be comparatively less in comparison to control cell culture surface, but the healthy morphology was retained, similar to that of control. The probable reason for this difference in cell density may arise due to unfamiliar microenvironment, on which they are grown. Initially, cells may take time to adapt to its microenvironment and prolonged cell survival can be observed.85 Interestingly, after 24 h, no dead cells were found (Fig. S16†). The cultured cells were checked for viability after 48 h and 72 h using confocal laser microscopy. All the co-assembled gels were highly biocompatible up to 72 h. In corroboration with MTT results, the CIP in combination with hydrophilic laminin derivative, i.e. Nap-FFGSO + B, which showed thin-entangled long fibrous network exhibited excellent biocompatibility, with negligible number of dead cells, for both the cell lines (C6 and L929) even after 72 h (Fig. 6 and 7), whereas Nap-FFGSO + A showed very few dead cells which might probably due to the hydrophobic nature or IKVAV and also shorter fiber lengths might not provide proper support to the growing cells.88 For both the cell types, the Nap-FFGSO + A + B gels resulted in enhancement of cell density after 72 h along with the distinguished cell morphology. However in case of Nap-FFGSO + A and Nap-FFGSO + B, the clusters of C6 cells were observed which indicates the dominant proliferation phase of the cells, after 72 h40 In contrast to this, L929 cell density has been relatively increased after 72 h, in comparison to 48 h, indicating growth phase. The above difference in cell growth and proliferation of different cell types with different peptides clearly showed the differential interaction of each co-assembled gels with the cells. The probable reason for such differential behavior is variable mechanical stiffness and morphology of the hydrogels, along with its biofunctional moieties. We believe that the positive charge of lysine and arginine residues in IKVAV and YIGSR on the co-assembled peptide gels surface is likely to interact with the negative surface of cells and provides suitable cell–matrix interactions.89,90 Thus the 2D culture observations clearly indicated that these collagen and laminin mimetic peptide co-assembled gels promotes cell adhesion and proliferation and might result in prolonged cell viability.
000 cells per well in a 6-well plate). For control, the cells were seeded on untreated coverslip. After 24 h of seeding, cell adhesion was observed with cells adapting the polyhedral or spindal shapes (Fig. S15†). Live/dead staining was performed after 24, 48 and 72 h after seeding (Fig. 6, 7 and S16†). The cell density on the co-assembled hydrogels was found to be comparatively less in comparison to control cell culture surface, but the healthy morphology was retained, similar to that of control. The probable reason for this difference in cell density may arise due to unfamiliar microenvironment, on which they are grown. Initially, cells may take time to adapt to its microenvironment and prolonged cell survival can be observed.85 Interestingly, after 24 h, no dead cells were found (Fig. S16†). The cultured cells were checked for viability after 48 h and 72 h using confocal laser microscopy. All the co-assembled gels were highly biocompatible up to 72 h. In corroboration with MTT results, the CIP in combination with hydrophilic laminin derivative, i.e. Nap-FFGSO + B, which showed thin-entangled long fibrous network exhibited excellent biocompatibility, with negligible number of dead cells, for both the cell lines (C6 and L929) even after 72 h (Fig. 6 and 7), whereas Nap-FFGSO + A showed very few dead cells which might probably due to the hydrophobic nature or IKVAV and also shorter fiber lengths might not provide proper support to the growing cells.88 For both the cell types, the Nap-FFGSO + A + B gels resulted in enhancement of cell density after 72 h along with the distinguished cell morphology. However in case of Nap-FFGSO + A and Nap-FFGSO + B, the clusters of C6 cells were observed which indicates the dominant proliferation phase of the cells, after 72 h40 In contrast to this, L929 cell density has been relatively increased after 72 h, in comparison to 48 h, indicating growth phase. The above difference in cell growth and proliferation of different cell types with different peptides clearly showed the differential interaction of each co-assembled gels with the cells. The probable reason for such differential behavior is variable mechanical stiffness and morphology of the hydrogels, along with its biofunctional moieties. We believe that the positive charge of lysine and arginine residues in IKVAV and YIGSR on the co-assembled peptide gels surface is likely to interact with the negative surface of cells and provides suitable cell–matrix interactions.89,90 Thus the 2D culture observations clearly indicated that these collagen and laminin mimetic peptide co-assembled gels promotes cell adhesion and proliferation and might result in prolonged cell viability.
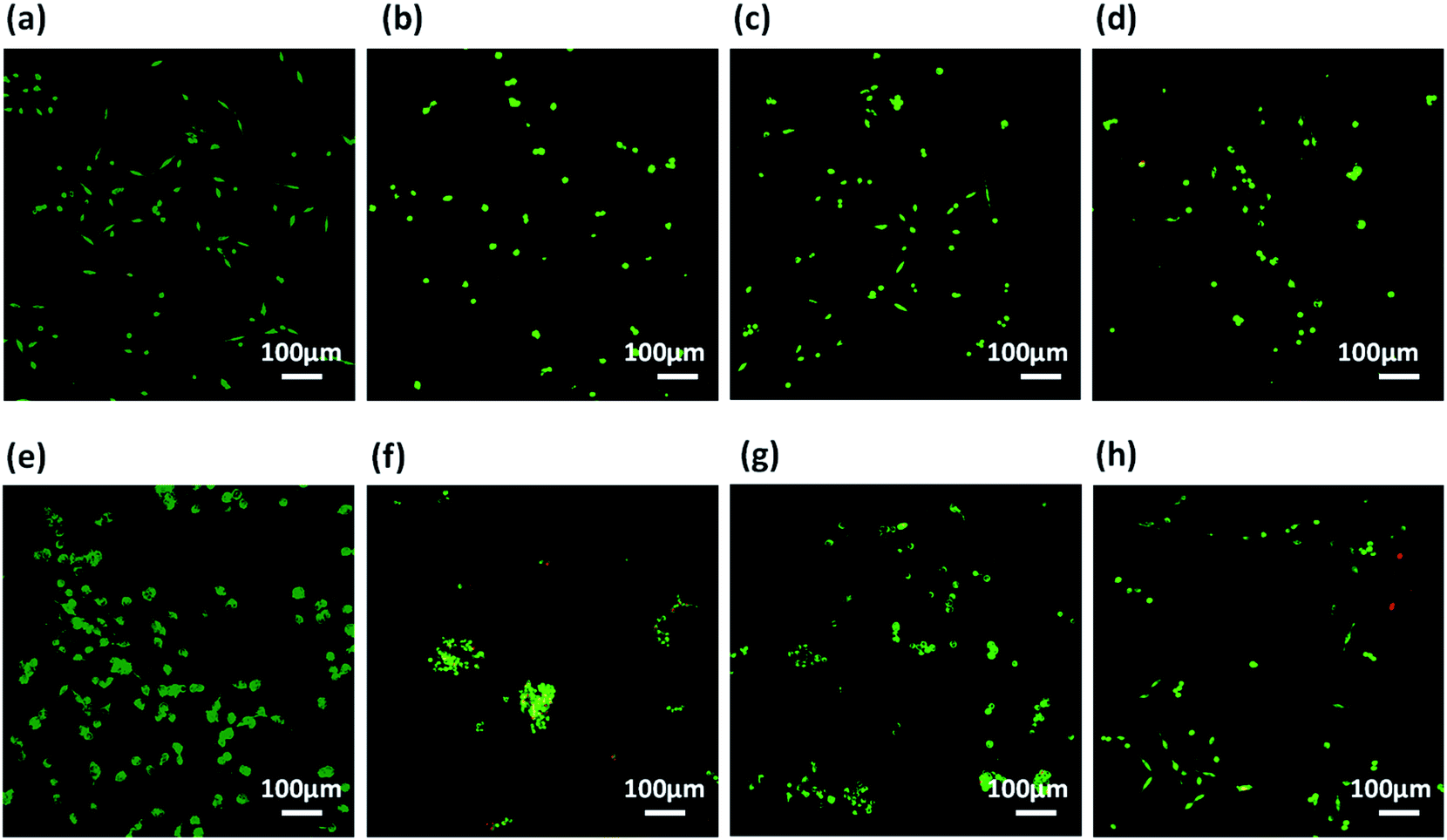 | ||
| Fig. 6 Confocal laser scanning microscopic images using C6 cells showing live (green) and dead (red) cells cultured over (a and e) plastic (control), (b and f) Nap-FFGSO + A gels (c and g) Nap-FFGSO + B gels and (d and h) Nap-FFGSO + A + B gels after 48 h and 72 h respectively. | ||
 | ||
| Fig. 7 Confocal laser scanning microscopic images using L929 cells showing live (green) and dead (red) cells cultured over (a and e) plastic (control), (b and f) Nap-FFGSO + A gels (c and g) Nap-FFGSO + B gels and (d and h) Nap-FFGSO + A + B gels after 48 h and 72 h respectively. | ||
Cell proliferation and migration studies
The cell proliferation behaviour of C6 cells and L929 cells in the presence of co-assembled CIP/LMP peptides was quantified by using Alamar blue reduction at different time points.91 Alamar blue is a resazurin based colorimetric reagent, widely used for quantification of metabolically active cells, without interfering with the viability of the cells, over the period of time.92 The matrigel was used as positive control to compare the bioactivity of our peptide hydrogels. The results suggested that there was a slight difference in the cell viability of all the three co-assembled gel systems after 24 h, 48 h, and 72 h as indicated by nearly similar fluorescence intensity of reduced Alamar blue (Fig. 8a and b). On comparing the proliferation of cells treated with matrigel with the CIP treated cells, it was found that Nap-FFGSO showed higher proliferation rates than matrigel. While other CIP/LMP combined peptides showed relatively similar proliferation rates to that of matrigel, which indicates the equivalent bioactivity of our developed peptides. The results clearly reveal that our peptide gels showed comparatively better bioactivity than commercial matrigel in promoting proliferation. In comparison to control, all the peptide treated cells showed relatively higher intensity, at all the three time points. It was also observed that the Nap-FFGSO + B showed statistically significant proliferation from 24 h to 72 h, while matrigel treated cells does not show statistically significant difference in the proliferation rates of the cells. This difference was more prominent with L929 cells. This indicates that our designed peptides were non-cytotoxic and promoted proliferation of both types of cells owing to its dual functionality. The results were in agreement with live/dead assay, which indicated increased live cell density in the confocal images of treated cells (Fig. 6 and 7).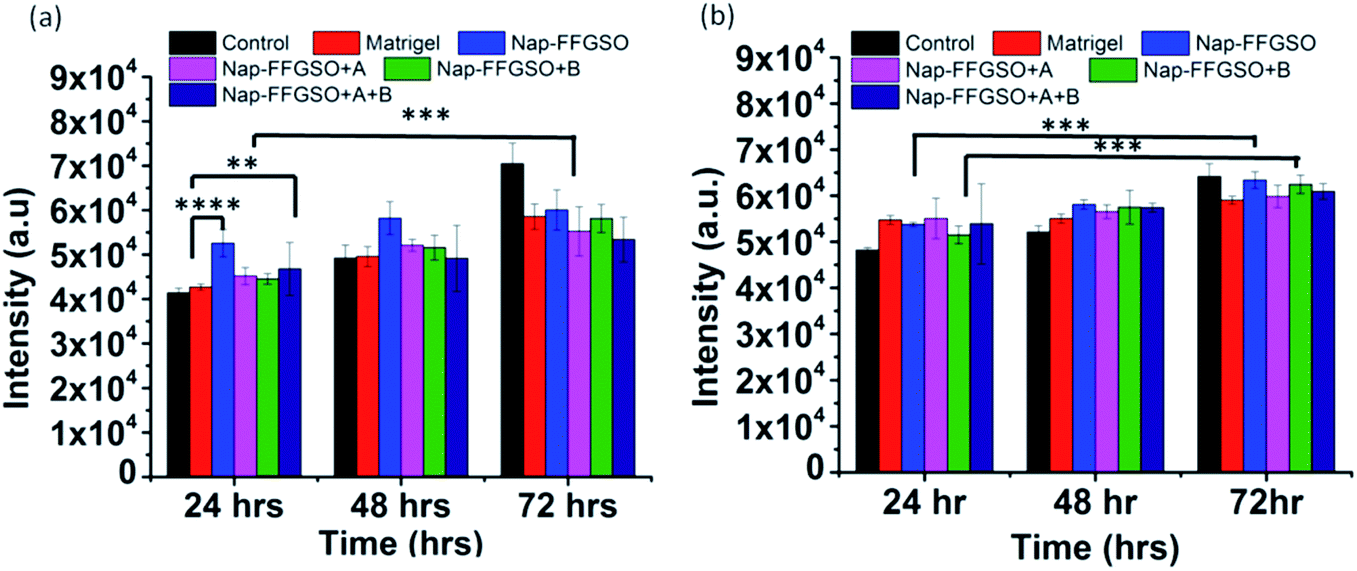 | ||
| Fig. 8 Proliferation studies of (a) C6 cells and (b) L929 cells after treatment with positive control as matrigel and different co-assembled CIP-LMP gels diluted to concentration of 1000 μg ml−1 (1.3 mM), after 24 h, 48 h and 72 h, using Alamar blue reagent. Data is represented as mean ± SD with N = 3 indicating p ≤ 0.0059 for **, p ≤ 0.003 for *** and p ≤ 0.0001 for ****. | ||
The proliferation was visually studied by migration studies. Proliferation followed by migration is an important phenomenon for wound healing and this wound healing capability of co-assembled nanostructures was assessed by in vitro scratch assay.93 This assay is based on creation of artificial gap in the monolayer of cells which was filled by the migration of proliferating cells present at the edges of the gap.94 The cell suspension with soluble peptide nanostructures were seeded in 48 well plate. When the 60–70% confluency was reached, a scratch was made using a sterile micropipette tip. The movement of proliferated cells towards scratched region was visualized at different time points, as indicated by the region covered by box in the images in Fig. S17 and S18.† After 24 h, very few numbers of cells invaginates into the marked region. However, in comparison to control (without treatment), the treated wells showed no significant difference in migration rates (Fig. S17 and S18†). The treated cells remain adhered and retain their healthy morphology, even after 48 h. In corroboration with cell viability assay results and live/dead microscopic assessment, the hydrophilic Nap-YIGSR along with CIP showed higher rate of proliferations, as shown by dense network of cells, after 48 h, for C6 cells (Fig. S17†). Similarly, for L929 cells, relatively more number of cells migrates towards the scratched region within 24 h and nearly 50% confluency was reached after 48 h (Fig. S18†). As expected from 2D culture, the hydrophilic laminin derivative in combination with CIP, i.e. Nap-FFGSO, was found to show maximum potency for development of a tissue engineering scaffold. The results demonstrated that collagen–laminin co-assembled peptides supported proliferation and migrations, which are important steps of wound healing and can be promising materials for wound healing applications.
Experimental section
Materials
Fmoc (9-fluorenylmethoxycarbonyl) protected amino acids, DIC (diisopropyl carbodiimide), piperazine, EDT (1,2-ethanedithiol), anisole, thioanisole were purchased from Sigma-Aldrich. Fmoc-Rinkamide MBHA resin (200–400 mesh), TFA (trifluoro acetic acid), oxyma, 2-napthoxy-acetic acid, DMF (dimethyl formamide) were purchased from Merck.Peptide synthesis
Designed collagen and laminin mimetic peptides were synthesized using standard Fmoc solid phase peptide chemistry by using Microwave Automated Solid Phase Peptide Synthesizer (Liberty Blue CEM, Matthews, NC, USA). Fmoc-protected amino acids were coupled on solid support Rinkamide resin at 0.1 mmol scale. 2 equivalents of Fmoc-protected amino acids were coupled in the presence of 5 equivalents of DIC and 10 equivalents of oxyma. 20% w/v piperazine/DMF solution was used for Fmoc-group deprotection after each subsequent coupling. A cocktail mixture composed of TFA:anisole:EDT:thioanisole in the ratio of 95:2:3:5 was added for 3 h to cleave peptides from resin. The purity of cleaved peptides was checked using HPLC and found to be >99% pure and molecular weights were confirmed by LC-MS analysis.
Critical aggregation concentration determination
Formation of self-assembled aggregates of CIP and LMP peptides were determined by the thioflavin T (Th T) fluorescence assay. Th T stock of 2.5 mM was prepared by dissolving 8 mg of Th T in 10 ml water. 200 μl of stock was further diluted up to 50 ml to produce 10 μM working solution of Th T. The peptide stocks were separately prepared at the concentration of 0.1 mM and 0.5 mM, which were added in different volumes, to 100 μl of Th T working solution, to attain different peptide concentrations for studying aggregation. Similarly, for studying co-assembly, the Nap-FFGSO concentration was fixed at 5 μM, to which different volumes of LMP stock solutions were added. The peptides were incubated for an hour with Th T, before the emission scan was recorded in the range of 450–600 nm, using Tecan microplate reader with an excitation wavelengths of 440 nm.Hydrogel formation
Peptides were weighed individually and mixed in solid form and then dissolved in 10% DMSO of the total volume. After complete dissolution of solid, the final volume was made up, by addition of MQ water. The mixture was vortexed and sonicated intermittently to form a uniform solution. The peptide solution was allowed to form gel and left undisturbed for 24 h, to ensure complete gelation. Throughout the studies, the collagen inspired peptide (Nap-FFGSO) was used at the concentration of 30 mM along with 10% (3 mM) of laminin mimetic peptides i.e. Nap-IKVAV and Nap-YIGSR. The minimum gelation concentration (MGC) of the co-assembled gels was assessed by inverted-vial method.Morphological analysis
Rheological measurements
Viscoelastic properties of the nanofibrous co-assembled hydrogels were measured using oscillatory rheology with 50 mm parallel plate geometry (Anton Parr MCR302). The co-assembled hydrogel samples with 30 mM collagen inspired peptide and 3 mM (in biconjugate system) and 1.5 mM (in tri-conjugate system) of each laminin mimetic peptide were prepared and incubated for 24 h before measurements. The optimum strain values were obtained in the linear viscoelastic region by performing the amplitude sweep. Further the storage (G′) and loss modulus (G′′) were recorded as a function of frequency sweep in the range of 0.1 to 100 Hz. The experiments were performed using a solvent trap to prevent any drying affect, at 20 °C and temperature controlled with an integrated electrical heater. The measurements were repeated 3 times to ensure reproducibility, with the average data shown.Thixotropic studies
To explore the mechano-responsiveness of these co-assembled hydrogels, step-strain rheology was done. Before performing thixotropic measurements, we checked the recovery properties by mechanical shaking, which transforms the gel material into solution (sol) and its recovery into the self-supporting gel state upon resting, however the time of reformation may vary from sample to sample. The initial strain values were fixed as 0.1% within linear viscoelastic (LVE) region followed by 50% strain which deforms the gel and transforms it to sol, as indicated by G′ < G′′. As soon as the stress is removed (strain 0.1%), the gel like character is recovered. Both, during deformation and recovery cycles the frequency was fixed at 1 Hz. Three consecutive cycles each having 200 s deformation phase and 800 s recovery phase were carried out. The temperature was maintained at 20 °C throughout the measurement, through automated temperature controller.Secondary structure investigation
Fluorescence microscopy of Th T binding
The diluted peptide gels were mixed with Th T working solution (as prepared for Th T fluorescence spectroscopy) in 2:1 ratio and allowed to incubate for 5 min. 50 μl of mixture was added on a glass slide and covered with coverslip. The image was observed by the Olympus BX53 fluorescence microscope and captured using Q-Capture Pro 7 software. The 40× lens was used to capture bright field images and the fluorescence images with the help of FITC emission filters (an excitation wavelength of 490 nm).
Evaluation of co-assembly
The Nap IKVAV peptide was fluorescently tagged with FITC (fluorescein isothiocyanate) using standard coupling protocol. In the absence of free N and C terminal, the FITC was coupled with lysine side chain of IKVAV, under basic conditions, in the presence of excess DIPEA.53 The tagged Nap IKVAV peptide was mixed with untagged Nap-FFGSO to form co-assembled gel. The peptide solution was drop casted on a slide a co-assembly was visualized using fluorescence microscope.Solvent exchange
CIP/LMP co-assembled gels prepared at 30 mM + 3 mM concentration in DMSO/water. After 24 h of gelation, 1 ml of Milli-Q water was added at the top of the gel and allowed to perfuse for 2 h. After 2 h, the supernatant water is replaced by fresh water. Similar procedure was repeated up to 3 cycles. After this the DMSO gel were used for FTIR analysis using Vertex 70v FTIR spectrometer (Bruker). Similarly, the solvent exchanged gels were analysed for mechanical strength using rheometer.In vitro cell culture experiments
Cell culture experiments were performed with C6 glioma cells and L929 fibroblast cells. Cells were cultured using DMEM media with 10% foetal bovine serum (FBS), and 1% penicillin–streptomycin antibiotic solution and were incubated at humidified chamber with 5% CO2 at 37 °C.Biocompatibility studies
To assess the initial contact cytotoxicity of these peptides and quantify cell survival in the presence of peptides, an MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay was performed. For biocompatibility studies, 5000 cells were seeded in each well in a 96 well plate and allowed to adhere for 24 h. The peptide stock solutions were prepared of 5 mg ml−1 (6.5 mM) concentration in 10% DMSO/water. The peptide solutions were sterilized before treatment. Peptide stock solution was diluted up to 100 μg ml−1 (0.13 mM) and 1000 μg ml−1 (1.3 mM) with DMEM media, resulting in final DMSO concentration to 0.2% and 2% respectively, and added to the cells. The experiments were done in triplicates. For control, the cells were treated with 10% DMSO/water diluted with DMEM media up to 0.2% and 2% DMSO. After 4 h of treatment, the media was changed and MTT was added to the cells to check the viability of cells. MTT is a colorimetric assay reagent which converts into purple colored insoluble formazan upon activity with metabolically active cells. After 4 h of MTT addition, the supernatant media was removed and formazan crystals were dissolved in DMSO (100 μl). The absorbance of each well was recorded using plate reader at 495 nm wavelength. The % viability of treated cells was calculated with respect to control cells. To check the toxicity after long term treatment, the MTT assay was carried out after 24 h, 48 h and 72 h, after the treatment of 1000 μg ml−1 (1.3 mM) of peptide solution.2D culture
Prior to 2D cultures, the stability of gels was assessed for media exchange and long duration media perfusion. The gels were placed on coverslips and UV sterilized, after which the media was perfused to provide cells, a uniform environment. The sub-confluent monolayer cells were trypsinized using trypsinase (0.25%)–EDTA (0.02%) solution and resuspended in complete media. The cell density was counted using haemocytometer. Then 1 ml of cell suspension, having 10000 cells per ml was added in each well of 6 well-plate. The cells were maintained at 37 °C with 5% CO2 and allowed to adhere.
Live/dead staining
The standard protocol according to live/dead cell mediated cytotoxicity kit (Invitrogen) was used for differential fluorescent staining of live and dead cells. 4 μl of DiOC18 (3) was diluted with 1 ml of complete media (1:250 dilution) while 2 μl of PI (propidium iodide) was diluted with 1 ml of PBS buffer and filtered using 0.2μ filters. The cells were incubated with DiOC18 (3) for 45 min in dark and then washed three times with PBS. Further, PI solution was added to wells and left for 20 min. At last three washes with PBS were performed and plates were visualized under confocal microscope under the green (480 nm) and red (530 nm) channels.
Cell proliferation assay
Proliferation of C6 cells and L929 cells were assessed by Alamar blue assay after 24 h and 48 h of treatment. To perform this assay, both the types of cells were seeded in 96 well plates at the cell density of 5 × 103. The gels were prepared according to the described procedure and diluted up to the concentration of 1000 μg ml−1 (1.3 mM) with culture media. The dispersed nanostructures were added to each well and incubated in 95% humidified incubator maintained at 37 °C, with 5% CO2, for different time durations, with triplicates for each sample. The cells treated with blank 2% DMSO in media were used as negative control and the cells treated with Matrigel as positive control. The media of each well was replaced with 100 μl of fresh media with 10% Alamar blue reagent, after different time points (24 h, 48 and 72 h). The Alamar containing plates were further incubated for 4 h. At the end of the incubation, the 96-well plate was read by using micro plate reader (Tecan Infinite M Plex). The fluorescence intensity of Alamar blue was determined by excitation/emission of 530 nm/600 nm. The diluted solutions of peptide gel were used as controls and their fluorescence intensity values were subtracted from the corresponding cell embedded peptides. The data was statistically analysed by using GraphPad Prism 8.0 software. Two way-ANOVA test was applied on data obtained in triplicates (N = 3) with 95% confidence level. The data was analysed using Bonferroni statistical test. The data having P values >0.01 were considered as non-significant.In vitro scratch wound assay
Induction of wound healing ability of the bio-inspired peptide conjugates was tested by scratch wound assay, in vitro. L929 fibroblast cells and C6 glioma cells were passaged and diluted to the density of 1 × 104 and were seeded in a 48 well plate. The cells were allowed to adhere and become 60–70% confluent. After that, a scratch was made and each well is treated with diluted peptide gels with 1000 μg ml−1 concentration. The plates were placed in a microscope incubator (37 °C/5% CO2). The plates were imaged using an upright Olympus microscope at 10× magnification, at different time points.Conclusions
In summary, we showed the sophisticated design of the self-assembling peptide hydrogel scaffolds which could play dual functionality. The minimalist design approach was used to develop the shortest structural mimic of collagen. By simply exploring non-covalent interactions, we coupled short laminin derived peptides with CIP's without relying on the covalent modification of the collagen peptide monomer. We, for the first time, reported the co-assembled supramolecular hydrogels solely based on bioactive peptides, without any polymeric scaffold or long chain hydrophobic domain. Although, these self-assembled scaffolds formed β-turns but they shared similarity with structure and nanofibrous morphology of the collagen, which have the potential to adapt functional attributes of the native collagen protein. The in vitro studies demonstrated that these scaffolds are highly biocompatible and its dual functionality supports both fibroblast as well as neuronal cells. The mechanical stiffness along with the chemical functionality determines the cellular response like adhesion, proliferation and growth. Studies are underway to understand the differential role of both structural protein and functional protein mimics of natural ECM for controlling cell behavior for their prolonged use. In future, these hydrogels can be explored for the encapsulation of cells for cell delivery for tissue repair applications. We envisage that these collagen–laminin derived biomimetic systems provide an excellent biocompatible platform to be explored for stem cell differentiation and tissue regeneration.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors acknowledge Department of Biotechnology, India (BT/PR22067/NNT/28/1163/2016) for providing the financial support. The authors thank Department of Science and Technology, India and Institute of Nano Science and Technology, Mohali, for providing infrastructural support and Doctoral fellowship. The authors would like to thank Mr Vijay Kumar Pal, PhD scholar at INST Mohali, for providing support in designing schemes.References
- H. Geckil, F. Xu, H. Zhang, S. Moon and U. Demirci, Nanomedicine, 2010, 5, 469–484 CrossRef CAS PubMed.
- Y. Loo, M. Goktas, A. B. Tekinay, M. O. Guler, C. A. E. Hauser and A. Mitraki, Adv. Healthcare Mater., 2015, 4, 2557–2586 CrossRef CAS PubMed.
- H. Chang, C. Li, R. Huang, R. Su, W. Qi and Z. He, J. Mater. Chem. B, 2019, 7, 2899–2910 RSC.
- N. Singh, M. Kumar, J. F. Miravet, R. V. Ulijn and B. Escuder, Chem.–Eur. J., 2017, 23, 981–993 CrossRef CAS PubMed.
- K. Liang, K. Hyun Bae and M. Kurisawa, J. Mater. Chem. B, 2019, 7, 3775–3791 RSC.
- S. Wan, S. Borland, S. M. Richardson, C. L. R. Merry, A. Saiani and J. E. Gough, Acta Biomater., 2016, 46, 29–40 CrossRef CAS PubMed.
- N. D. Evans and E. Gentleman, J. Mater. Chem. B, 2014, 2, 2345–2356 RSC.
- H. Green, G. Ochbaum, A. Gitelman-Povimonsky, R. Bitton and H. Rapaport, RSC Adv., 2018, 8, 10072–10080 RSC.
- J. Luo and Y. W. Tong, ACS Nano, 2011, 5, 7739–7747 CrossRef CAS PubMed.
- M. M. Islam, R. Ravichandran, D. Olsen, M. K. Ljunggren, P. Fagerholm, C. J. Lee, M. Griffith and J. Phopase, RSC Adv., 2016, 6, 55745–55749 RSC.
- L. E. R. O'Leary, J. A. Fallas, E. L. Bakota, M. K. Kang and J. D. Hartgerink, Nat. Chem., 2011, 3, 821–828 CrossRef PubMed.
- V. A. Kumar, N. L. Taylor, A. A. Jalan, L. K. Hwang, B. K. Wang and J. D. Hartgerink, Biomacromolecules, 2014, 15, 1484–1490 CrossRef CAS PubMed.
- S. M. Yu, Y. Li and F. Kim, Soft Matter, 2011, 7, 7927–7938 RSC.
- O. D. Krishna and K. L. Kiick, Biomacromolecules, 2009, 10, 2626–2631 CrossRef CAS PubMed.
- L. Yao, M. He, D. Li, H. Liu, J. Wua and J. Xiao, New J. Chem., 2018, 42, 7439–7444 RSC.
- M. Bansal, C. Ramakrishnan and G. N. Ramachandran, Proc. Indian Acad. Sci., 1975, 82, 152–164 CrossRef CAS.
- W. Wang, G. Li, W. Zhang, J. Gao, J. Zhang, C. Li, D. Ding and D. Kong, RSC Adv., 2014, 4, 30168–30171 RSC.
- T. Luo and K. L. Kiick, Bioconjugate Chem., 2017, 28, 816–827 CrossRef CAS PubMed.
- B. Brodsky, G. Thiagarajan, B. Madhan and K. Kar, Biopolymers, 2008, 89, 345–353 CrossRef CAS PubMed.
- J. Sun, Y. Guo, R. Xing, T. Jiao, Q. Zou and X. Yan, Colloids Surf., A, 2017, 514, 155–160 CrossRef CAS.
- R. Xing, L. Liu, T. Jiao, N. Zhang, K. Ma, R. Zhang, Q. Zou, G. Ma and X. Yan, Adv. Mater., 2016, 28, 3669–3676 CrossRef CAS PubMed.
- S. Rele, Y. Song, R. P. Apkarian, Z. Qu, V. P. Conticello and E. L. Chaikof, J. Am. Chem. Soc., 2007, 129, 14780–14787 CrossRef CAS PubMed.
- T. Jiang, C. Xu, Y. Liu, Z. Liu, J. S. Wall, X. Zuo, T. Lian, K. Salaita, C. Ni, D. Pochan and V. P. Conticello, J. Am. Chem. Soc., 2014, 136, 4300–4308 CrossRef CAS PubMed.
- M. A. Cejas, W. A. Kinney, C. Chen, G. C. Leo, B. A. Tounge, J. G. Vinter, P. P. Joshi and B. E Maryanoff, J. Am. Chem. Soc., 2007, 129, 2202–2203 CrossRef CAS PubMed.
- M. A. Cejas, W. A. Kinney, C. Chen, J. G. Vinter, H. R. Almond, K. M. Balss, C. A. Maryanoff, U. Schmidt, M. Breslav, A. Mahan, E. Lacy and B. E. Maryanoff, Proc. Natl. Acad. Sci., 2008, 105, 8513–8518 CrossRef CAS PubMed.
- C. C. Chen, W. Hsu, T. C. Kao and J. C. Horng, Biochemistry, 2011, 50, 2381–2383 CrossRef CAS PubMed.
- D. E. Przybyla, C. M. R. Perez, J. Gleaton, V. Nandwana and J. Chmielewski, J. Am. Chem. Soc., 2013, 135, 3418–3422 CrossRef CAS PubMed.
- M. M. Pires and J. Chmielewski, J. Am. Chem. Soc., 2009, 131, 2706–2712 CrossRef CAS PubMed.
- M. He, L. Wang, J. Wu and J. Xiao, Chem.–Eur. J., 2016, 22, 1914–1917 CrossRef CAS PubMed.
- M. M. Pires, J. Lee, D. Ernenwein and J. Chmielewski, Langmuir, 2012, 28, 1993–1997 CrossRef CAS PubMed.
- W. Hsu, Y. L. Chen and J. C. Horng, Langmuir, 2012, 28, 3194–3199 CrossRef CAS PubMed.
- B. Sarkar, L. E. O'Leary and J. D. Hartgerink, J. Am. Chem. Soc., 2014, 136, 14417–14424 CrossRef CAS PubMed.
- K. McGuinness, I. J. Khan and V. Nanda, ACS Nano, 2014, 8, 12514–12523 CrossRef CAS PubMed.
- T. Luo, M. A. David, L. C. Dunshee, R. A. Scott, M. A. Urello, C. Price and K. L. Kiick, Biomacromolecules, 2017, 18, 2539–2551 CrossRef CAS PubMed.
- C. M. Yamazaki, S. Asada, K. Kitagawa and T. Koide, Biopolymers, 2008, 90, 816–823 CrossRef CAS PubMed.
- P. J. Skrzeszewska, F. A. de Wolf, M. W. T. Werten, A. P. H. A. Moers, M. A. C. Stuart and J. van der Gucht, Soft Matter, 2009, 5, 2057–2062 RSC.
- Y. Hu, H. Wang, J. Wang, S. Wang, W. Liao, Y. Yang, Y. Zhang, D. Kong and Z. Yang, Org. Biomol. Chem., 2010, 8, 3267–3271 RSC.
- O. Uysal, E. Arslan, G. Gulseren, M. C. Kilinc, I. Dogan, H. Ozalp, Y. S. Caglar, M. O. Guler and A. B. Tekinay, ACS Appl. Bio Mater., 2019, 2, 1686–1695 CrossRef CAS.
- M. D. Shoulders and R. T. Raines, Annu. Rev. Biochem., 2009, 78, 929–958 CrossRef CAS PubMed.
- C. Deister, S. Aljabari and S. E. Schmidt, J. Biomater. Sci., Polym. Ed., 2007, 18, 983–997 CrossRef CAS PubMed.
- S. P. Baldwin, C. E. Krewson and W. M. Saltzman, Int. J. Dev. Neurosci., 1996, 14, 351–3641 CrossRef CAS PubMed.
- R. J. Linnola, M. Sund, R. Ylönen and T. Pihlajaniemi, J. Cataract Refractive Surg., 1999, 25, 1486–1491 CrossRef CAS PubMed.
- J. P. Jung, A. K. Nagaraj, E. K. Fox, J. S. Rudra, J. M. Devgun and J. H. Collier, Biomaterials, 2009, 30, 2400–2410 CrossRef CAS PubMed.
- K. Tashiro, G. C. Sephel, B. Weeks, M. Sasaki, G. R. Martin, H. K. Kleinman and Y. Yamada, J. Biol. Chem., 1989, 264, 16174–16182 CAS.
- B. Mammadov, M. Sever, M. Gecer, F. Zor, S. Ozturk, H. Akgun, U. H. Ulas, Z. Orhan, M. O. Guler and A. B. Tekinay, RSC Adv., 2016, 6, 110535–110547 RSC.
- A. L. Rodriguez, T. Y. Wang, K. F. Bruggeman, C. C. Horgan, E. Li, R. J. Williams, C. L. Parish and D. R. Nisbet, J. Mater. Chem. B, 2014, 2, 7771–7778 RSC.
- C. C. Horgan, A. L. Rodriguez, R. Li, K. F. Bruggeman, N. Stupka, J. K. Raynes, L. Day, J. W. White, R. J. Williams and D. R. Nisbet, Acta Biomater., 2016, 38, 11–22 CrossRef CAS PubMed.
- R. O. Labrador, M. Butí and X. Navarro, Exp. Neurol., 1998, 149, 243–252 CrossRef CAS PubMed.
- K. D. Newman, C. R. Mclaughlin, D. Carlsson, F. Li, Y. Liu and M. Griffith, Int. J. Artif. Organs, 2006, 29, 1082–1091 CrossRef CAS PubMed.
- S. Sur, M. O. Guler, M. J. Webber, E. T. Pashuck, M. Ito, S. I. Stupp and T. Launey, Biomater. Sci., 2014, 2, 903 RSC.
- M. Hiraoka, K. Kato, T. Nakaji-Hirabayashi and H. Iwata, Bioconjugate Chem., 2009, 20, 976–983 CrossRef CAS PubMed.
- J. H. Collier, J. S. Rudra, J. Z. Gasiorowski and J. P. Jung, Chem. Soc. Rev., 2010, 39, 3413–3424 RSC.
- Y. Zheng, S. Ji, A. Czerwinski, F. Valenzuela, M. Pennington and S. Liu, Bioconjugate Chem., 2014, 25, 1925–1941 CrossRef CAS PubMed.
- C. B. Berkholtz, B. E. Lai, T. K. Woodruff and L. D. Shea, Histochem. Cell Biol., 2006, 126, 583–592 CrossRef CAS PubMed.
- M. J. Horacek, J. C. Thompson, M. O. Dada and L. Terracio, Acta Anat., 1993, 147, 69–74 CrossRef CAS PubMed.
- R. Jain, G. Khandelwal and S. Roy, Langmuir, 2019, 35, 5878–5889 CrossRef CAS PubMed.
- X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165–13307 CrossRef CAS PubMed.
- Y. Zhang, Y. Kuang, Y. Gao and B. Xu, Langmuir, 2011, 27, 529–537 CrossRef CAS PubMed.
- K. Kar, S. Ibrar, V. Nanda, T. M. Getz, S. P. Kunapuli and B. Brodsky, Biochemistry, 2009, 48, 7959–7968 CrossRef CAS PubMed.
- C. J. Bowerman, W. Liyanage, A. J. Federation and B. L. Nilsson, Biomacromolecules, 2011, 12, 2735–2745 CrossRef CAS PubMed.
- C. Chen, Y. Zhang, Z. Hou, X. Cui, Y. Zhao and H. Xu, Biomacromolecules, 2017, 18, 3563–3571 CrossRef CAS PubMed.
- A. Kroes-Nijboer, P. Venema, J. Bouman and E. van der Linden, Food Biophysics, 2009, 4, 59–63 CrossRef.
- L. Chen, J. Raeburn, S. Sutton, D. G. Spiller, J. Williams, J. S. Sharp, P. C. Griffiths, R. K. Heenan, S. M. King, A. Paul, S. Furzeland, D. Atkins and D. J. Adams, Soft Matter, 2011, 7, 9721–9727 RSC.
- C. Colquhoun, E. R. Draper, E. G. B. Eden, B. N. Cattoz, K. L. Morris, L. Chen, T. O. McDonald, A. E. Terry, P. C. Griffiths, L. C. Serpell and D. J. Adams, Nanoscale, 2014, 6, 13719–13725 RSC.
- C.-L. Shen and R. M. Murphy, Biophys. J., 1995, 69, 640–651 CrossRef CAS PubMed.
- J. B. Guilbaud, C. Rochas, A. F. Miller and A. Saiani, Biomacromolecules, 2013, 14, 1403–1411 CrossRef CAS PubMed.
- N. Nandi, K. Gayen, S. Ghosh, D. Bhunia, S. Kirkham, S. K. Sen, S. Ghosh, I. W. Hamley and A. Banerjee, Biomacromolecules, 2017, 18, 3621–3629 CrossRef CAS PubMed.
- A. Baral, S. Roy, S. Ghosh, D. Hermida-Merino, I. W. Hamley and A. Banerjee, Langmuir, 2016, 32, 1836–1845 CrossRef CAS PubMed.
- C. K. Thota, N. Yadav and V. S. Chauhan, Sci. Rep., 2016, 6, 1–12 CrossRef PubMed.
- C. Yan, A. Altunbas, T. Yucel, R. P. Nagarkar, J. P. Schneider and D. J. Pochan, Soft Matter, 2010, 6, 5143–5156 RSC.
- C. Shu, T. Li, W. Yang, D. Li, S. Ji and L. Ding, R. Soc. Open Sci., 2018, 5, 171814 CrossRef PubMed.
- Z. Yang, G. Liang, M. Ma, Y. Gaoa and B. Xu, J. Mater. Chem., 2007, 17, 850–854 RSC.
- E. Vass, M. Hollósi, F. Besson and R. Buchet, Chem. Rev., 2003, 103, 1917–1954 CrossRef CAS PubMed.
- J. Kong and S. Yu, Acta Biochim. Biophys. Sin., 2007, 39, 549–559 CrossRef CAS PubMed.
- M. Biancalana and S. Koide, Biochim. Biophys. Acta, 2010, 1804, 1405–1412 CrossRef CAS PubMed.
- M. J. Krysmann, V. Castelletto and I. W. Hamley, Soft Matter, 2007, 3, 1401–1406 RSC.
- P. Sharma, H. Kaur and S. Roy, Biomacromolecules, 2019, 207, 2610–2624 CrossRef PubMed.
- S. Onogi, H. Shigemitsu, T. Yoshii, T. Tanida, M. Ikeda, R. Kubota and I. Hamachi, Nat. Chem., 2016, 8, 743–752 CrossRef CAS PubMed.
- T. Eljezi, P. Pinta, D. Richard, J. Pinguet, J.-M. Chezal, M.-C. Chagnon, V. Sautou, G. Grimandi and E. Moreau, Chemosphere, 2017, 173, 452–459 CrossRef CAS PubMed.
- J. Raeburn, C. Mendoza-Cuenca, B. N. Cattoz, M. A. Little, A. E. Terry, A. Z. Cardoso, P. C. Griffiths and D. J. Adams, Soft Matter, 2015, 11, 927–935 RSC.
- A. D. Martin, A. B. Robinson and P. Thordarson, J. Mater. Chem. B, 2015, 3, 2277–2280 RSC.
- P. Gavel, D. Dev, H. S. Parmar, S. Bhasin and A. K. Das, ACS Appl. Mater. Interfaces, 2018, 10, 10729–10740 CrossRef CAS PubMed.
- Z. Tu, M. Volk, K. Shah, K. Clerkin and J. F. Liang, Peptides, 2009, 30, 1523–1528 CrossRef CAS PubMed.
- N. Habibi, N. Kamaly, A. Memic and H. Shafiee, Nano Today, 2016, 11, 41–60 CrossRef CAS PubMed.
- W. Liyanage, K. Vats, A. Rajbhandary, D. S. W. Benoit and B. L. Nilsson, Chem. Commun., 2015, 51, 11260–11263 RSC.
- H. Choi, M. Go, Y. Cha, Y. Choi, K.-Y. Kwon and J. H. Jung, New J. Chem., 2017, 41, 4793–4796 RSC.
- K. M. Hainline, F. Gu, J. F. Handley, Y. F. Tian, Y. Wu, L. de Wet, D. J. Vander Griend and J. H. Collier, Macromol. Biosci., 2019, 19, 1800249 CrossRef PubMed.
- A. T. Wood, D. Everett, S. Kumar, M. K. Mishra and V. Thomas, J. Biomed. Mater. Res., Part B, 2019, 107, 332–341 CrossRef CAS PubMed.
- S. A. Makohliso, R. F. Valentini and P. Aebischer, J. Biomed. Mater. Res., 1993, 27, 1075–1085 CrossRef CAS PubMed.
- G. B. Schneider, A. English, M. Abraham, R. Zaharias, C. Stanford and J. Keller, Biomaterials, 2004, 25, 3023–3028 CrossRef CAS PubMed.
- W. Sun, T. Incitti, C. Migliaresi, A. Quattrone, S. Casarosa and A. Motta, J. Tissue Eng. Regener. Med., 2017, 11, 1532–1541 CrossRef CAS PubMed.
- G. Damodaran, R. Collighan, M. Griffin, H. Navsaria and A. Pandit, Acta Biomater., 2009, 5, 2441–2450 CrossRef CAS PubMed.
- X. Zhang, X. Kang, L. Jin, J. Bai, W. Liu and Z. Wang, Int. J. Nanomedicine, 2018, 13, 3897–3906 CrossRef CAS PubMed.
- C.-C. Liang, A. Y. Park and J.-L. Guan, Nat. Protoc., 2007, 2, 329–333 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: ESI includes analytical HPL chromatograms, optical images of gels, AFM images LMP solutions, Th t binding fluorescence microscopic images, quantitative data of rheology and thixotropy, cell images after treatment and phase contrast and fluorescence microscopy images for 2D culture, microscopic images for cell migration studies. See DOI: 10.1039/c9ra07454f |
| This journal is © The Royal Society of Chemistry 2019 |