
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCO2 reduction using paper-derived carbon electrodes modified with copper nanoparticles†
Federico J. V. Gomez a,
George Chumanovb,
Maria Fernanda Silvab and
Carlos D. Garcia*b
a,
George Chumanovb,
Maria Fernanda Silvab and
Carlos D. Garcia*b
aInstituto de Biología Agrícola de Mendoza (IBAM-CONICET), Facultad de Ciencias Agrarias, Universidad Nacional de Cuyo, Mendoza, Argentina
bDepartment of Chemistry, Clemson University, 211 S. Palmetto Blvd, Clemson, SC 29634, USA. E-mail: cdgarci@clemson.edu
First published on 18th October 2019
Abstract
The conversion of CO2 into useful chemicals can lead to the production of carbon neutral fuels and reduce greenhouse gas emissions. A key technological challenge necessary to enable such a process is the development of substrates that are active, cost effective, and selective for this reaction. In this regard, the reduction of CO2 via electrochemical means is one of the most attractive alternatives but still requires rather unique electrodes. Considering the potential of this approach, this report describes a one-step methodology for the synthesis of carbon electrodes derived from simple paper and modified with various metallic nanoparticles. Upon a preliminary selection based on the catalytic activity towards CO2 reduction, the electrodes containing CuNPs were further characterized by Raman spectroscopy, and electrical/electrochemical techniques. These electrodes were then applied for the electrochemical reduction of CO2, leading to the formation of compounds with one carbon atom (formic acid), two carbon atoms (ethenone), three carbon atoms (propanoic acid) and four carbon atoms (butanol and butanoic acid).
1. Introduction
The development of efficient methods for the conversion of CO2 into other chemicals can lead to the production of carbon neutral fuels and simultaneously reducing greenhouse gas emissions. The conversion of CO2 into feedstock chemicals is not only important for selected industrial processes (synthesis of salicylic acid, urea, methanol, cyclic carbonates, polycarbonates, and polyether carbonates),1 but also critically important for future space exploration missions because it can provide access to local resources freeing up the cargo space for other mission-critical supplies.2–5 Because CO2 is one of the most abundant molecules in many alien atmospheres, its activation can promote local agriculture and manufacturing.6Currently, the activation of CO2 is performed in batch reactors based on either the Sabatier7 or Bosch reactions,8 in which high pressure and high temperature are required to overcome the high activation energy of CO2. In other words, and despite recent advances,1,9 most current processes still require large energy inputs to transform CO2 into useful chemicals.10 Alternatively, the electrochemical reduction of CO2 can proceed in ambient environments and is capable of producing a variety of hydrocarbons with high selectivity.3,11–13 Various electrocatalysts, including composites containing transition metals14 such as Au,15 Ag,16,17 Fe,18 Co,19,20 Sn,21 Pd,22,23 and Ni24 have been proposed to improve the yield of the electrochemical reduction of CO2. In addition to those, a variety of Cu-based nanocatalysts can also enhance the electrochemical formation of hydrocarbons from CO2-saturated aqueous solutions and do so at ambient temperature and pressure.12,25–27 More importantly, the unique configuration and density of uncoordinated sites has positioned copper as the most suitable heterogeneous catalyst to produce valuable hydrocarbons and alcohols from CO2.28 The surface sites facilitate the initial formation of CO via breaking one of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds in the CO2 molecule, that can progress (chemically or electrochemically)29 to yield hydrocarbons. In addition to its binding capacity, the surface of Cu-based nanocatalyst can be also tuned to minimize the binding energy of the reaction products, so the surface can be effectively regenerated for the next catalytic cycle. Despite these advantages, the application of Cu-based electrodes is still limited by costs, specific fabrication procedures, and activity.
O bonds in the CO2 molecule, that can progress (chemically or electrochemically)29 to yield hydrocarbons. In addition to its binding capacity, the surface of Cu-based nanocatalyst can be also tuned to minimize the binding energy of the reaction products, so the surface can be effectively regenerated for the next catalytic cycle. Despite these advantages, the application of Cu-based electrodes is still limited by costs, specific fabrication procedures, and activity.
To address these limitations, this report describes the possibility of using paper-derived carbon electrodes, modified with metallic NPs towards the catalytic reduction of CO2. The electrodes were fabricated by pyrolysis (under reductive conditions) of paper strips presoaked with solutions containing various metallic salts. The method led to the in situ formation of abundant NPs, dispersed throughout the structure of the substrates (without the need of surfactants, commonly used to avoid the NP aggregation). Because they exhibited higher catalytic activity towards the reduction of CO2, the electrodes containing CuNP were selected and further characterized by Raman spectroscopy and electrical/electrochemical techniques. When applied for the reduction reaction, the electrodes allowed the synthesis of compounds with one carbon atom (formic acid), two carbon atoms (ethenone), three carbon atoms (propanoic acid) and four carbon atoms (butanol and butanoic acid). These results show the potential impact of the proposed electrodes for the up-conversion of CO2.
2. Materials and methods
2.1 Reagents¶
Sulfuric acid (ACS/FCC, BDH Aristar, 95.0–98.0%) was acquired from VWR (Pittsburgh, PA, USA). Sodium bicarbonate and hexa-ammine ruthenium(III) chloride from Sigma-Aldrich (St. Louis, MO, USA). Copper(II) sulfate, copper(II) chloride, copper(II) acetate, cobalt(II) acetate, and silver nitrate were purchased from Fisher Scientific (Fair Lawn, NJ, USA). All aqueous solutions were prepared using 18 MΩ cm water (NANOpure Diamond, Barnstead; Dubuque, IA) and analytical reagent grade chemicals. Phosphate buffer solution was prepared by dissolving anhydrous Na2HPO4 (Fisher Scientific; Fair Lawn, NJ, USA) in ultrapure water. The pH of the solutions was measured using a glass electrode connected to a digital pH meter (Orion 420A+, Thermo; Waltham, MA, USA) and adjusted with 1 mol L−1 solutions of either NaOH or HCl.2.2 Fabrication of NPs-modified carbon electrodes
Following previous publications from our group,30–32 electrodes were obtained by pyrolysis of paper strips (4.5 cm × 1.5 cm; Whatman 3MM chromatography paper; GE Health Care; Pittsburgh, PA) pre-soaked in 1 M solutions of the following metallic salts: CuSO4, AgNO3, CuCl2, Cu(CH3COO)2 or Co(CH3COO)2. Upon immersion in the selected solution for 5 min, the paper strips were dried in a convection oven (at 50 °C for 30 min), placed between two silicon wafers, and then transferred to a tube furnace (Type F21100, Barnstead–Thermolyne; Dubuque, IA, USA) for pyrolysis. The quartz tube was first flushed with forming gas (5% H2/95% Ar, 1 L min−1) for 5 min (to remove the O2 and avoid oxidation reactions) and then allowed to reach a temperature of 1000 °C, at a rate of 20 °C min−1. After 1 h, the tube furnace was turned off and allowed to cool-down to room temperature while maintaining the flow of forming gas. Finally, the pyrolyzed samples were removed from the furnace and stored in a Petri dish until use. The resulting carbon electrodes will be referred to as MeNPs@CE. In order to obtain electrodes with uniform dimensions, MeNPs@CE were fixed to a Plexiglas substrate using double-sided tape and cut using a commercial 30 W CO2 laser engraver (Mini 24, Epilog Laser Systems; Golden, CO, USA). The resulting electrodes featured a circular pad (0.50 cm2) and a stem, similar to those previously used as sensors.30 Then, stripped 20 gauge tinned copper wires were attached to the stem of the electrodes and silver paint (SPI Supplies; West Chester, PA, USA) was applied to secure the electrical connection. In order to prevent water from wicking up the stem of the electrode (and therefore increasing the electrode area), acrylic paint was spotted on base of the stem, between the circular pad and the contact area. Plain carbon electrodes (without MeNPs) were also prepared using the conditions described above (but soaking the paper strips in DI water) and used as control.2.3 Electron microscopy
Micrographs of bare pyrolyzed paper samples and NPs modified carbonized paper were obtained using a scanning electron microscope (SEM, Hitachi S3400) with the acceleration voltage of 15.0 kV. Micrographs of single fiber CuSO4 modified pyrolized paper were obtained using a dual-beam FIB-SEM (Hitachi NB5000) with the acceleration voltage 5.0 kV.2.4 Electrochemical techniques
Cyclic voltammetry (CV) was performed to investigate the electrochemical performance of the MeNPs@CE. The experiments were performed using 100 mM H2SO4, as supporting electrolyte, containing 1 mM K3Fe(CN)6/K4Fe(CN)6, as the redox couple. In all cases, a standard three-electrode cell comprised of CuNPs@CE, a silver/silver chloride (Ag|AgCl|KClsat), and a platinum wire were used as working, reference, and counter electrode, respectively. CV experiments were carried out using a CHI660A Electrochemical Analyzer (CH Instruments, Inc.; Austin, TX).2.5 Electrolysis
CO2 reduction experiments were performed in a hermetically sealed three-electrode cell comprised of the CuNPs@CE, a silver/silver chloride (Ag|AgCl|KClsat), and a platinum wire; used as working, reference, and counter electrode, respectively. The electrolyte was 0.5 M NaHCO3 saturated with CO2 (pH 7.2) by purging the gas for 30 min at room temperature. The electrolyte was stirred during the electrolysis to assist the surface/bulk exchange of reactants and products. Electrolysis was performed at −1.5 V (vs. Ag|AgCl|KClsat).2.6 Electrical and physical characterization of the electrochemical device
In order to gain preliminary information related to the macroscopic properties of the obtained material, both contact angle and resistivity were measured. The contact angle was studied to gain insight into the wettability of the electrode by placing a drop (5 μL) of ultrapure water (18 MΩ cm) onto the CuNPs@CE and taking a lateral black & white photo of the droplet (using an Iphone SE camera). The contact angle was then measured using Low Bond Axismmetric Drop Shape Analysis (LB-ADSA) plugin for ImageJ software. Resistivity measurements were taken with a Keithley 2636A SourceMeter coupled to a Jandel cylindrical four-point probe with a 1 mm probe spacing. The thickness of the material was then measured using scanning electron microscopy (SEM).2.7 Raman spectroscopy
Raman spectra were excited with 514.5 nm light from an Ar+ ion laser (Innova 200, Coherent, 500 mW). Scattered light was collected by a f/1.2 camera lens (Nikon) in a 45° backscattering geometry and analyzed by triple spectrometer (Triplemate 1877, Spex) equipped with CCD camera (iDUS 420, Andor). The spectral resolution at the excitation wavelength was 0.2 nm. Raman spectrum of indene was used for the spectral calibration.2.8 Product identification
Although a number of techniques,33,34 can be used to determine the reduction products in both the liquid and gas phase, a simple GC-MS methodology was developed for this project. Solid phase micro extraction (SPME) was performed to extract the products from the head space and also from the electrolyte. The selected SPME fiber was a StableFlex™ polydimethylsiloxane/divinylbenezene (PDMS/DVB, 65 μm). SPME support and holder manual for SPME were obtained from Supelco (Bellefonte, PA, USA). Before use, the fiber was conditioned following the instructions from the manufacturer. The fiber was exposed during the 30 min of the electrolysis in both head space and liquid face. Identification of compounds was carried out by GC using a PerkinElmer Elite-5MS and an Agilent DB-WAX (30 m length, 32 mm diameter and 0.25 μm film thickness) fitted in a capillary gas chromatograph-electron impact mass spectrometer (GC-EIMS; Clarus 500, PerkinElmer, Shelton, CT). The GC column was eluted with He (0.7 ml min−1). The GC temperature program was from 35 to 200 °C at 20 °C min−1, then raised to 240 °C at 4 °C min−1 and held for 15 min. The mass spectrometer was operated with electron impact ionization energy of 70 eV. The injector temperature was 240 °C, ion source temperature was 120 °C and the interface temperature was 150 °C.3. Results and discussion
3.1 Surface topography
Scanning electron micrographs were obtained to determine the size and morphology of the NPs formed in situ during the pyrolysis process. In agreement with previous reports,35 a significant reduction of the mass and dimensions of the substrates was observed but in all cases, the initial structure of the cellulose was preserved after the pyrolysis step (Fig. 1A–F). Also, a large number of metallic NPs ranging in size between 500 nm and 1800 nm (with varying shape) were formed on the surface of the carbonized cellulose fibers (Fig. 1B–F). As it can be observed, the shape and the size appeared to be dependent on the metallic salt used to soak the paper and reduced in the presence of the forming gas (containing 5% H2). These conditions provide a clean and simple way to promote the reduction of the metallic salts to the corresponding nanoparticles.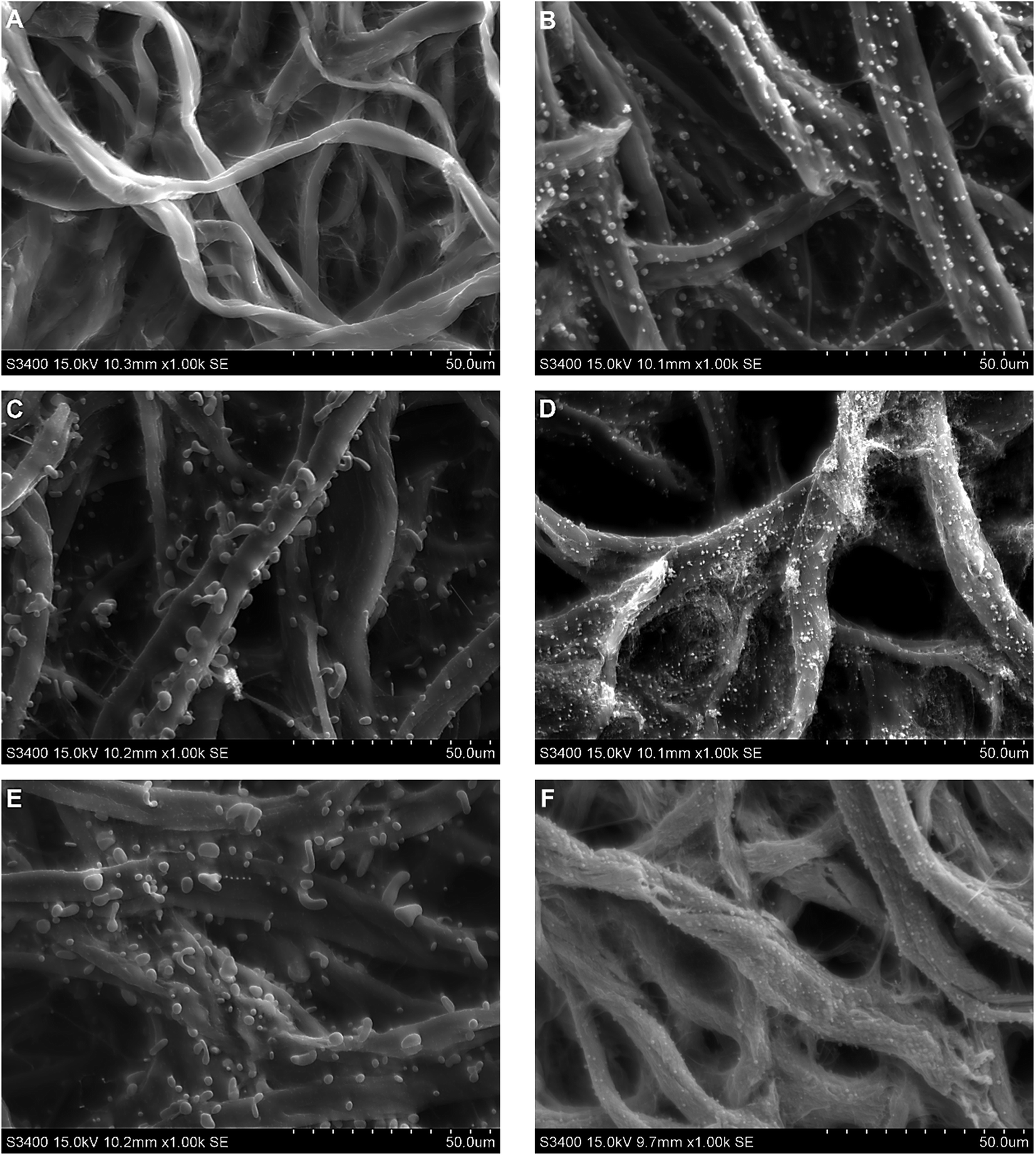 | ||
| Fig. 1 SEM micrographs for (A) control (plain paper), (B) CuSO4, (C) Co(CH3COO)2, (D) Cu(CH3COO)2, (E) CuCl2, (F) AgNO3. | ||
Table 1 summarizes the diameter and shape of the obtained NPs. It should be also pointed out that the NPs synthesized during the pyrolysis are not only observed on the surface of the material but also distributed within the individual carbon fibers, as can be observed in the cross-sections of a pyrolyzed paper modified with CuSO4 (Fig. S1 and S2†). This is important because the particles on the surface of the material can provide enhancements in the catalytic activity of the material while those within the fibers can bridge clusters of graphitic carbon and decrease the resistivity of the material.
3.2 Electrochemical characterization
Cyclic voltammograms using 0.010 M K3[Fe(CN)6]/K4[Fe(CN)6] were recorded varying the scan rate, between 50 mV s−1 and 400 mV s−1 to establish the electrochemical performance of MeNPs@CE. As can be observed in Fig. 2A, the peak current (ip) corresponding to both the reduction and oxidation processes increased linearly with the square root of the scan rate in all tested electrodes, suggesting that the response is (partially) controlled by the diffusion of the electroactive species. However, the peak potential separation between the reduction and oxidation waves also increased with the scan rate and was significantly larger than 0.059 V/n (Fig. 2B), the value expected for fully reversible systems. In fact, the peak potential differences observed (>200 mV) indicate that the electrochemical process should be characterized as irreversible and that such behavior could be attributed to the intrinsic resistance of the material.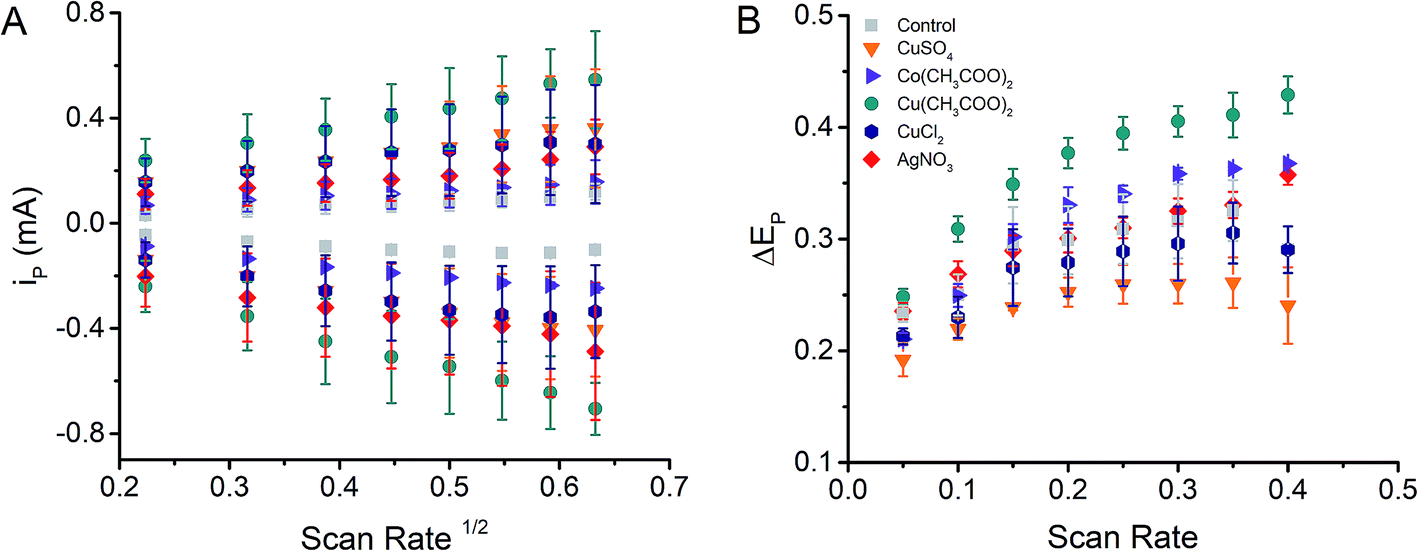 | ||
| Fig. 2 Dependence of the peak current (ip) on the square root of the scan rate (A) and dependence of the peak potential difference (ΔEp) on the scan rate (B) for the electrodes produced. | ||
3.3 Electrochemical reduction of CO2
To investigate the performance of the produced MeNPs@CE towards the CO2 reduction, cyclic voltammetry experiments were performed. Fig. 3 shows the voltammograms obtained with the produced electrodes using an electrolyte composed of 0.5 M NaHCO3 saturated with CO2. As it can be observed, all MeNPs@CE featured well-defined cathodic waves with onset potentials ranging from −0.5 to −0.8 V (vs. Ag|AgCl|KClsat). Although these values are significantly larger than the equilibrium potentials for the most commonly reported electrochemical products, they do fall within commonly reported overpotentials for the production of hydrocarbons.29,36–38 It is also important to note that while the current densities obtained are slightly lower than those reported with other electrodes (2–20 mA cm−2 (ref. 29, 37, 39 and 40)), the simplicity of the proposed fabrication strategy could enable their utilization in low-income settings.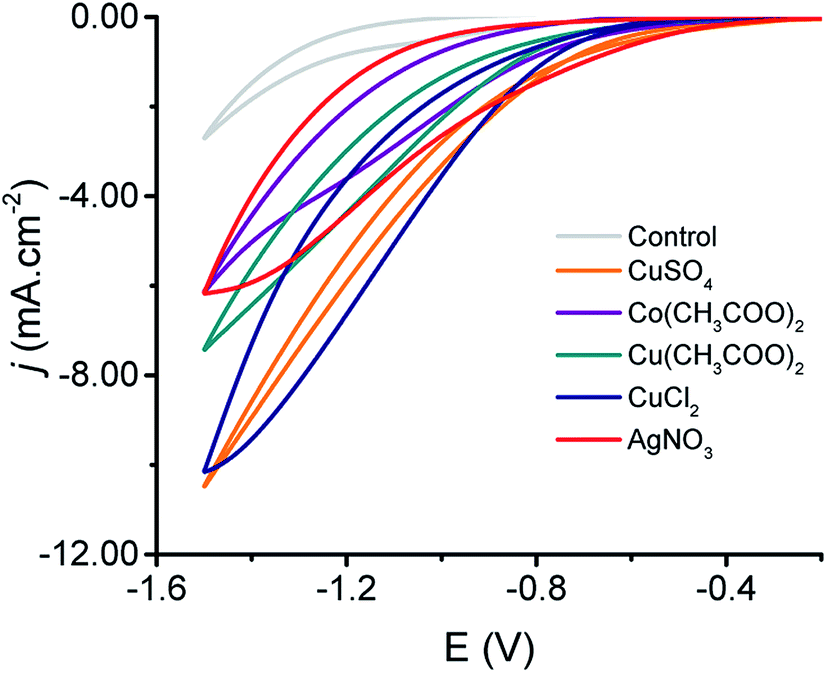 | ||
| Fig. 3 Cyclic voltammograms obtained with the produced electrodes using a solution containing 0.5 M NaHCO3 saturated with CO2. | ||
Based on the affinity for either *CO or *H, the reduction reactions observed on the produced MeNPs@CE can be attributed to the formation of either CO and H2 (on AgNPs@CE and CoNPs@CE)19,41,42 or “>2e− products” (on CuNPs@CE).28 While the reaction rate and pathway can be influenced by a number of variables (material, topography, pH, solvent, temperature, potential, counterions, etc.43), it is reasonable for the reduction reaction on the CuNPs@CE to proceed upon the initial one electron transfer to form a surface-bound CO2˙− intermediate.44,45 After that, the balance between the reactivity of both the surface46 and the radical formed (half-life ∼10 ns)47 is expected to dominate the distribution of final products.
Considering these results and the fact that copper is known for catalyzing the formation of significant amounts of hydrocarbons (at high reaction rates) over sustained periods of time, electrodes modified with CuNP were considered most advantageous and used for all further experiments.
It is also important to mention that control experiments, performed under identical conditions but without purging the solution with CO2 did not yield significant signals, confirming that CO2 was the carbon source for the obtained products. It is also worth mentioning that plain electrodes (paper-derived electrodes prepared in the same way but without metallic nanoparticles) displayed marginal electrochemical activity and thus their performance towards the formation of organic compounds was not investigated.
3.4 Resistivity and contact angle
The effect of the synthesized CuNPs on the resistivity (ρ) of CuNPs@CE was calculated as ρ = ΩA/l, where Ω, A, and l are measured resistance, the cross sectional area (thickness of the material 76.4 μm), and the distance between the two measuring electrodes, respectively. The measured resistivity was 2.5 ± 0.2 mΩ cm for the CuNPs@CE, value that is slightly lower than that of pyrolyzed electrodes fabricated in the same way but without metallic salts (control, 3.4 ± 0.2 mΩ cm). This difference suggests that while the selected concentration of CuSO4 (1 M) was able to form abundant particles on the material, it was insufficient to bridge domains of conductive carbon,31 effect that is expected to significantly increase the overall electrical conductivity of the electrodes and that can be adjusted if deemed necessary.The wettability of the electrodes is an important parameter and could have a large effect on the surface chemistry, including CO2 reduction, in aqueous solutions. Thus, the wettability was determined at room temperature by measuring the contact angle between the electrode surface and a droplet of DI water. The contact angles of CuNPs@CE (123° ± 4°) and control substrates (137° ± 4°) indicates the hydrophobic character of the carbon produced. While the NPs somewhat reduce this hydrophobicity, the hydrophobic character of the electrodes can be detrimental to the electrochemistry in aqueous solutions. In such cases, pre-wetting the electrode with a drop of ethanol or treating the substrate in an air plasma (to oxidize the carbon surface without significant oxidation of CuNPs) can decrease the contact angle. It is also important to mention that these values do not consider the inherent microstructure,48 3D arrangement and porosity of the substrates49,50 and are provided with the sole purpose of comparing the produced electrodes.
3.5 Microstructure of pyrolyzed samples
It is well known that carbon has several allotropes, which can be formed during the pyrolysis step.35 To obtain structural information related to the carbon material in the samples with and without CuNPs, Raman spectroscopy was used. Fig. 4 shows Raman spectra of CuNPs@CE and plain paper (control) samples.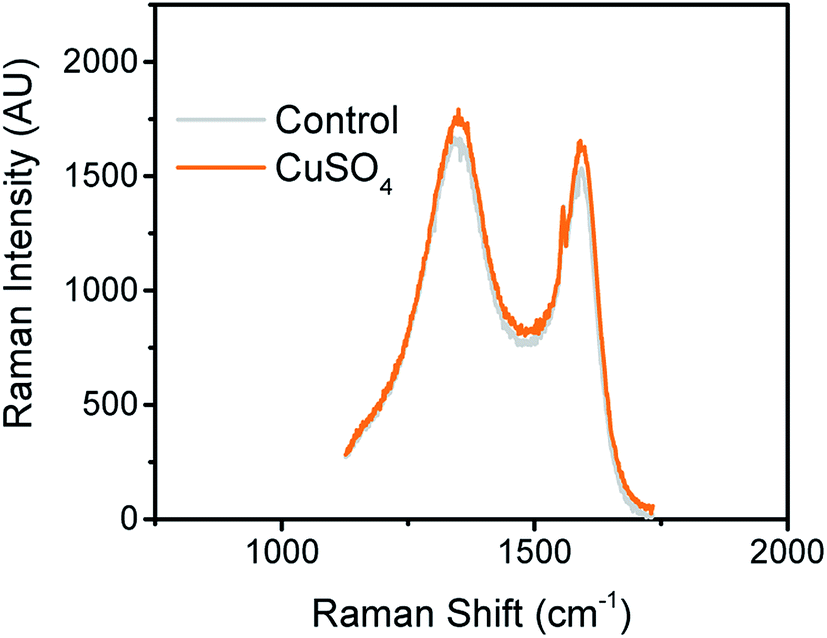 | ||
| Fig. 4 Raman spectra of pyrolyzed paper modified with CuSO4 and plain paper (control). The spectra were acquired using the conditions previously described in Experimental section. | ||
As can be seen in Fig. 4, two distinct bands were observed around 1590 cm−1 and 1362 cm−1. These bands were attributed to the corresponding breathing of C–C bonds in graphite-like carbon (sp2) and the breakdown of the symmetry (defects). The relative intensity of D and G bands was used to obtain information about the overall composition of the produced electrodes. The relative D/G ratio were 2.7 for CuNPs@CE and 2.9 for the control, suggesting the presence of a rather disordered structures in the substrates that is not significantly affected by the CuNPs. Overall, the D/G ratios reported support the resistive behavior observed by the electrochemical techniques (vide infra), also in line with other carbon-based materials commonly used as working electrodes.
3.6 Identification of the CO2 reduction products
In order to demonstrate the utility of the CuNPs@CE towards the electrochemical reduction of CO2, the proposed material was used to perform electrolysis in a CO2-saturated solution containing 0.5 M NaHCO3 (pH 7.2). These experiments were performed in a hermetically-sealed cell while applying −1.5 V (vs. Ag|AgCl|KClsat) for 30 min. While the long-term stability of the electrodes51 was not specifically assessed as part of this project, no obvious degradation of the electrodes was observed during the electrolysis (see Fig. S3†).The corresponding reaction products were analyzed by SPME-GC-MS. Table 2 summaries the compounds found in both the head space and the liquid phase. As it can be observed, the most abundant compound identified was butanoic acid (liquid phase), followed by propanoate (head space) and ethenone (liquid phase). Significantly smaller amounts of formate, butanoate and butanol were also found in the head space.
These results are not only in good agreement with the mechanisms reported in the literature for the formation of C1 and C2 compounds1,12,28,37,38 but also highlight the potential advantages of the proposed electrodes towards the formation of higher compounds with longer chains (C3 and C4) via an enzyme-like mechanism.52 As pointed by most recent reports, controlling the selectivity of this reaction is particularly tricky because it requires avoiding the evolution of hydrogen and simultaneously transferring multiple electrons and protons to CO2, processes that depend on the applied potential, the residual oxides,53,54 and the surface of the catalyst. That said, the proposed Cu-modified surfaces seem to provide a unique route for the formation of the highly energetic CO2 anion radical, which can then proceed via homogeneous chemistry and form the C2, C3 and C4 products. They also provide a convenient alternative for their integration into previously developed cells for CO2 reduction.55
4. Conclusions
This article described a simple approach for the development and fabrication of MeNPs@CE. The procedure is based on the one step in situ synthesis of MeNPs by pyrolysis of filter papers modified with different metallic salts, reduced in the presence of forming gas (5% H2) and at high temperature. The electrocatalytic activity of obtained materials were first investigated by cyclic voltammetry and the CuNPs@CE was selected. The CuNPs@CE was then characterized by a combination of techniques such as contact angle, resistivity, microscopy, Raman spectroscopy, and voltametry and successfully applied for the electrochemical reduction of CO2. The reduction reaction lead to the formation of compounds with one carbon atom (formic acid), two carbon atoms (ethenone), three carbon atoms (propanoic acid) and four carbon atoms (butanol and butanoic acid), as identified by a SPME-GC-MS technique. These reactions are critical for the applicability of the electrodes because the subsequent reactions can proceed to form either formate (that typically remains on the surface fouling the electrode) or other organic compounds of much higher chemical value.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors would like to acknowledge partial financial support to this project to Clemson University, Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET) and Facultad de Ciencias Agrarias, Universidad Nacional de Cuyo (Mendoza, Argentina). F. G. also acknowledges the support from a Fulbright Scholarship.References
- M. D. Burkart, N. Hazari, C. L. Tway and E. L. Zeitler, ACS Catal., 2019, 9, 7937–7956 CrossRef CAS.
- E. E. Benson, C. P. Kubiak, A. J. Sathrum and J. M. Smieja, Chem. Soc. Rev., 2009, 38, 89–99 RSC.
- C. Costentin, M. Robert and J.-M. Savéant, Chem. Soc. Rev., 2013, 42, 2423–2436 RSC.
- J. Rosen, G. S. Hutchings, Q. Lu, S. Rivera, Y. Zhou, D. G. Vlachos and F. Jiao, ACS Catal., 2015, 5, 4293–4299 CrossRef CAS.
- M. Rakowski Dubois and D. L. Dubois, Acc. Chem. Res., 2009, 42, 1974–1982 CrossRef CAS.
- H. Puliyalil, D. Lašič Jurković, V. D. B. C. Dasireddy and B. Likozar, RSC Adv., 2018, 8, 27481–27508 RSC.
- K. Stangeland, D. Kalai and Z. Yu, Energy Procedia, 2017, 105, 2022–2027 CrossRef CAS.
- S. Vilekar, K. Hawley, C. Junaedi, D. Walsh, S. Roychoudhury, M. Abney and J. Mansell, in 42nd International Conference on Environmental Systems, American Institute of Aeronautics and Astronautics, 2012 Search PubMed.
- W. Luc, M. Jouny, J. Rosen and F. Jiao, Energy Environ. Sci., 2018, 11, 2928–2934 RSC.
- T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS.
- R. J. Lim, M. Xie, M. A. Sk, J.-M. Lee, A. Fisher, X. Wang and K. H. Lim, Catal. Today, 2014, 233, 169–180 CrossRef CAS.
- D. T. Whipple and P. J. A. Kenis, J. Phys. Chem. Lett., 2010, 1, 3451–3458 CrossRef CAS.
- P. Tamilarasan and S. Ramaprabhu, RSC Adv., 2015, 5, 24864–24871 RSC.
- J.-H. Liu, L.-M. Yang and E. Ganz, RSC Adv., 2019, 9, 27710–27719 RSC.
- Y. Chen, Y. Huang, T. Cheng and W. A. Goddard, J. Am. Chem. Soc., 2019, 141, 11651–11657 CrossRef CAS.
- S.-Q. Liu, S.-W. Wu, M.-R. Gao, M.-S. Li, X.-Z. Fu and J.-L. Luo, ACS Sustainable Chem. Eng., 2019, 7, 14443–14450 CrossRef CAS.
- I. Reche, I. Gallardo and G. Guirado, RSC Adv., 2014, 4, 65176–65183 RSC.
- C. Zhang, S. Yang, J. Wu, M. Liu, S. Yazdi, M. Ren, J. Sha, J. Zhong, K. Nie, A. S. Jalilov, Z. Li, H. Li, B. I. Yakobson, Q. Wu, E. Ringe, H. Xu, P. M. Ajayan and J. M. Tour, Adv. Energy Mater., 2018, 8, 1703487 CrossRef.
- T. Shimoda, T. Morishima, K. Kodama, T. Hirose, D. E. Polyansky, G. F. Manbeck, J. T. Muckerman and E. Fujita, Inorg. Chem., 2018, 57, 5486–5498 CrossRef CAS.
- M. B. Solomon, A. Rawal, J. M. Hook, S. M. Cohen, C. P. Kubiak, K. A. Jolliffe and D. M. D'Alessandro, RSC Adv., 2018, 8, 24128–24142 RSC.
- S. Zhang, P. Kang and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 1734–1737 CrossRef CAS.
- X. Liu, L. Zhu, H. Wang, G. He and Z. Bian, RSC Adv., 2016, 6, 38380–38387 RSC.
- L. He, H. Yang, J. Huang, X. Lu, G.-R. Li, X. Liu, P. Fang and Y. Tong, RSC Adv., 2019, 9, 10168–10173 RSC.
- P. Su, K. Iwase, S. Nakanishi, K. Hashimoto and K. Kamiya, Small, 2016, 12, 6083–6089 CrossRef CAS PubMed.
- Q. Li, W. Zhu, J. Fu, H. Zhang, G. Wu and S. Sun, Nano Energy, 2016, 24, 1–9 CrossRef CAS.
- M. Fan, Z. Bai, Q. Zhang, C. Ma, X.-D. Zhou and J. Qiao, RSC Adv., 2014, 4, 44583–44591 RSC.
- Y. Koo, R. Malik, N. Alvarez, L. White, V. N. Shanov, M. Schulz, B. Collins, J. Sankar and Y. Yun, RSC Adv., 2014, 4, 16362–16367 RSC.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1311–1315 RSC.
- J. G. Giuliani, T. E. Benavidez, G. M. Duran, E. Vinogradova, A. Rios and C. D. Garcia, J. Electroanal. Chem., 2016, 765, 8–15 CrossRef CAS.
- G. M. Duran, T. E. Benavidez, J. G. Giuliani, A. Rios and C. D. Garcia, Sens. Actuators, B, 2016, 227, 626–633 CrossRef CAS.
- L. A. J. Silva, W. P. da Silva, J. G. Giuliani, S. C. Canobre, C. D. Garcia, R. A. A. Munoz and E. M. Richter, Talanta, 2017, 165, 33–38 CrossRef CAS.
- T. G. Cordeiro, M. S. Ferreira Santos, I. G. R. Gutz and C. D. Garcia, Food Chem., 2019, 292, 114–120 CrossRef CAS.
- M. S. Ferreira Santos, T. Gomes Cordeiro, Z. Cieslarova, I. G. R. Gutz and C. D. Garcia, Electrophoresis, 2019, 40, 2256–2262 CAS.
- T. E. Benavidez, R. Martinez-Duarte and C. D. Garcia, Anal. Methods, 2016, 8, 4163–4176 RSC.
- Q. Li, J. Fu, W. Zhu, Z. Chen, B. Shen, L. Wu, Z. Xi, T. Wang, G. Lu, J. Zhu and S. Sun, J. Am. Chem. Soc., 2017, 139, 4290–4293 CrossRef CAS.
- D. Ren, Y. Deng, A. D. Handoko, C. S. Chen, S. Malkhandi and B. S. Yeo, ACS Catal., 2015, 5, 2814–2821 CrossRef CAS.
- S. Sen, D. Liu and G. T. R. Palmore, ACS Catal., 2014, 4, 3091–3095 CrossRef CAS.
- D. Raciti, K. J. Livi and C. Wang, Nano Lett., 2015, 15, 6829–6835 CrossRef CAS.
- B. A. Rosen and I. Hod, Adv. Mater., 2018, 30, 1706238 CrossRef.
- M. Ma, B. J. Trześniewski, J. Xie and W. A. Smith, Angew. Chem., Int. Ed., 2016, 55, 9748–9752 CrossRef CAS.
- X.-M. Hu, M. H. Rønne, S. U. Pedersen, T. Skrydstrup and K. Daasbjerg, Angew. Chem., Int. Ed., 2017, 56, 6468–6472 CrossRef CAS PubMed.
- X. Nie, M. R. Esopi, M. J. Janik and A. Asthagiri, Angew. Chem., Int. Ed., 2013, 52, 2459–2462 CrossRef CAS.
- Z. Sun, T. Ma, H. Tao, Q. Fan and B. Han, Chem, 2017, 3, 560–587 CAS.
- L. Ou, RSC Adv., 2015, 5, 57361–57371 RSC.
- J. H. Montoya, A. A. Peterson and J. K. Nørskov, ChemCatChem, 2013, 5, 737–742 CrossRef CAS.
- T. Kai, M. Zhou, Z. Duan, G. A. Henkelman and A. J. Bard, J. Am. Chem. Soc., 2017, 139, 18552–18557 CrossRef CAS.
- C. Semprebon, G. McHale and H. Kusumaatmaja, Soft Matter, 2017, 13, 101–110 RSC.
- H. Lee, R. Rajagopalan, J. Robinson and C. G. Pantano, ACS Appl. Mater. Interfaces, 2009, 1, 927–933 CrossRef CAS PubMed.
- C. Tran and V. Kalra, J. Power Sources, 2013, 235, 289–296 CrossRef CAS.
- C. F. C. Lim, D. A. Harrington and A. T. Marshall, Electrochim. Acta, 2017, 238, 56–63 CrossRef CAS.
- K. D. Yang, C. W. Lee, K. Jin, S. W. Im and K. T. Nam, J. Phys. Chem. Lett., 2017, 8, 538–545 CrossRef CAS.
- Y. Lum and J. W. Ager, Angew. Chem., Int. Ed., 2018, 57, 551–554 CrossRef CAS.
- J.-P. Jones, G. K. S. Prakash and G. A. Olah, Isr. J. Chem., 2014, 54, 1451–1466 CrossRef CAS.
- K.-J. Yim, D.-K. Song, C.-S. Kim, N.-G. Kim, T. Iwaki, T. Ogi, K. Okuyama, S.-E. Lee and T.-O. Kim, RSC Adv., 2015, 5, 9278–9282 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra07430a |
| This journal is © The Royal Society of Chemistry 2019 |