DOI:
10.1039/C9RA07335C
(Paper)
RSC Adv., 2019,
9, 33664-33673
The introduction of benzimidazole and ether moieties into poly(p-phenylene terephthalamide): effects on its microstructure, interactions and properties
Received
12th September 2019
, Accepted 25th September 2019
First published on 21st October 2019
Abstract
Co-poly(p-phenylene terephthalamide) (co-PPTA) fibers containing 4,4′-oxidianiline (4,4′-ODA) and 2-(4-aminophenyl)-5-aminobenzimidazole (BIA) in terephthaloyl chloride (TPC) and p-phenylene diamine (p-PDA) were prepared via a wet spinning method, followed by water washing and drawing at a high temperature. With the addition of a new acid-binding agent, imidazole, the solution prepared by low-temperature polycondensation had suitable viscosity for spinning. Herein, the properties of six co-PPTA fibers with different contents of BIA and 4,4′-ODA segments were studied. The mechanical properties of the co-PPTA fibers were improved with the addition of BIA and ODA; they reached the optimum tensile strength of 2.45 GPa at a p-PDA/ODA/BIA molar ratio of 2/4/4, which showed a higher degree of orientation and the highest crystallinity, and the strength further increased on increasing the thermal drawing ratio. X-ray diffraction indicated that the fibers exhibited highly ordered structures, while two-dimensional wide angle X-ray diffraction showed that molecular packing regions with highly oriented structures were formed. In addition, the co-PPTA fibers exhibited excellent thermal stability when the 5% weight loss temperature was above 492 °C under nitrogen, and glass transition occurred at about 290 °C.
1 Introduction
A poly(p-phenylene terephthalamide) (PPTA) fiber is an organic high-performance fiber obtained through polycondensation and spinning with aromatic compounds as raw materials. Kevlar® was invented and commercialized by the DuPont Company in the United States in the 1960s.1–4 The applications of para-aramid fibers have already penetrated a large number of industrial fields, from the traditional military industry to aerospace, electronics, composite materials and other new fields.5,6 The traditional preparation method of PPTA fibers is liquid crystal spinning, where a PPTA polymer with a high molecular weight and a high molecular weight PPTA polymer dissolved in concentrated sulfuric acid are used as spinning solutions.7–10 However, this process is complicated, and the solution is difficult to recover. Furthermore, pyridine is often used as an acid binding agent to adsorb the small molecule HCl generated by the reaction, and it is extremely harmful to the human body. In order to solve these problems, flexible moieties have been introduced into the rigid backbones to improve solubility. Flexible bonds can cause the molecular chains to undergo rigid twisting or flexible distortions and decrease the interactions between the benzene rings and amide groups, which eventually leads to rotation in the benzene rings and endows the molecular chain with a stable helical structure. The helical structure not only destroys the symmetry and regularity of the molecular chains but also increases the free volume, thus decreasing the crystallinity and improving the solubility.
Research on the co-poly(p-phenylene terephthalamide) (co-PPTA) fiber was first started by the Russian Research Institute of Chemical Industry.11 In 1972, Russian scientists first used benzimidazole diamine (BIA) and terephthaloyl chloride (TPC) monomer to carry out the industrial synthesis and spinning of a high-strength and high-modulus aramid fiber, SVM. Subsequently, Russian scientists introduced p-phenylene diamine (p-PDA) into the SVM molecular chain to produce Armos fiber, and they registered a patent in 1978.12–15 The tensile strength of the Armos fiber exceeded 4.9 GPa in 1987 and increased to 5.5 GPa in the 1990s with the development of spinning technology. In addition, the former Soviet Union used 3,4′-oxidianiline (3,4′-ODA) and BIA as monomers to carry out terpolymerization to obtain amide-soluble copolymers and prepare them through stock solution spinning, with strengths of 5.0–5.5 GPa and modulus of 130–140 GPa; these are comparable to high-performance carbon fibers and are the strongest among all aramid fibers today.16–22 The Japanese Ryugamine Mikado Company used 3,4′-ODA as the third monomer to obtain an industrial product with the trademark Technora®, which typically showed 3.5 GPa strength.16–19 The Institute of Chemistry Chinese Academy of Sciences and Tsinghua University used 3,4′-ODA as the third monomer, LiCl as the cosolvent and pyridine as the acid absorbent to synthesize a jelly-like copolymer by a low-temperature solution poly-condensation method. Because of their good processability and outstanding properties, copolymerized para-aramid fibers have received increasing attention from researchers.16–29 For example, Longbo Luo et al.16 comparatively studied the introduction of asymmetric heterocyclic units into PPTA and their effects on its microstructure, interactions and properties. K. Li et al.27–29 studied the mechanical properties, UV stability and the structure and property evolution during the decomplexation of hydrogen chloride from co-PPTA fibers in detail. However, current research focuses on the relationship between the structure and properties of modified PPTA fibers and the influence of the third monomer, such as BIA and 3,4′-ODA, on the structure and properties of the PPTA fibers.
Inspired by these known successes, in this study, BIA and 4,4′-ODA moieties were introduced to TPC/p-PDA rigid backbones to improve their processability. The solutions were synthesized at low temperature and spun directly into fibers via a wet-spinning process. During the synthesis process, imidazole was adopted to replace pyridine as a new acid binding agent. A series of co-PPTA fibers with different diamine molar ratios were obtained, and the effects of the 4,4′-ODA and BIA moieties on the aggregation structure, morphologies, and mechanical and thermal properties of the co-PPTA fibers were investigated in detail. In addition, the effects of the thermal drawing ratio on the mechanical properties and crystallinity of the fiber were studied. This study may serve as a rule to fabricate high-performance PPTA fibers by molecular design.
2 Experimental
2.1 Materials
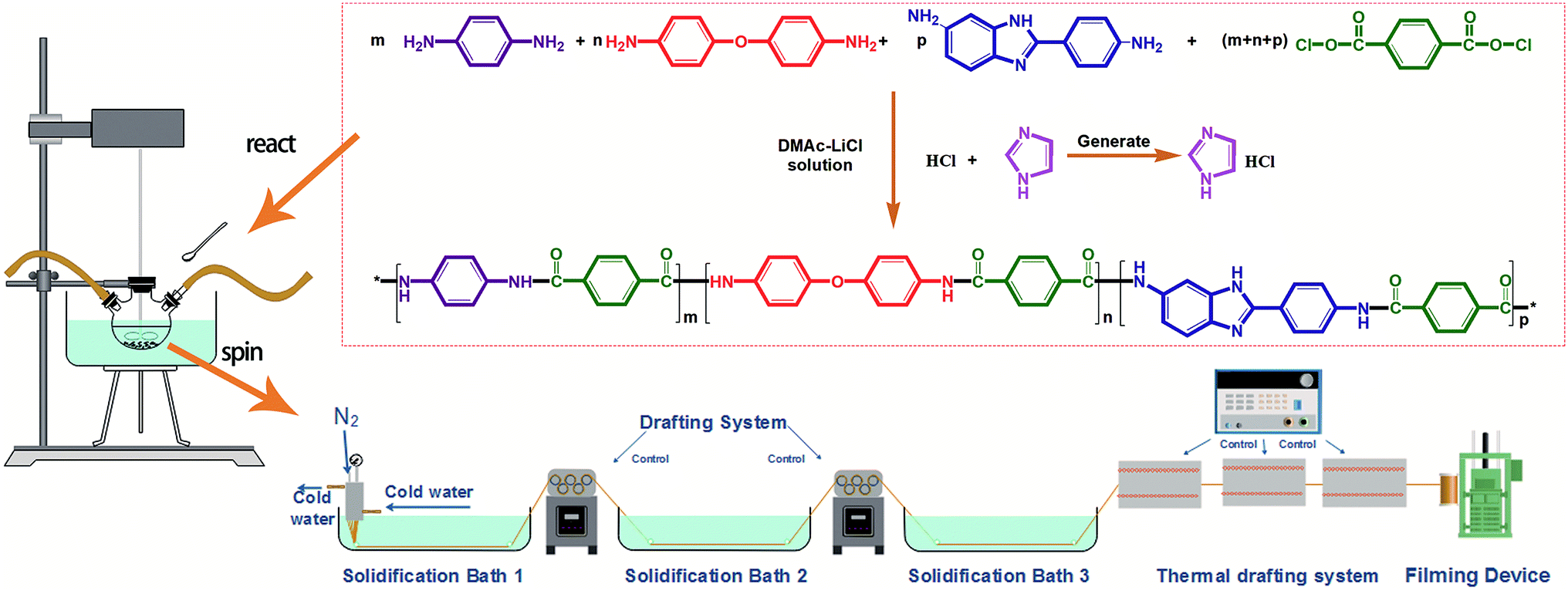
The monomer TPC was purchased from Shanghai Aladdin Biochemical Technology Co., Ltd. and used after sublimation. The monomers 4,4′-oxidianiline (4,4′-ODA), p-PDA and BIA were purchased from Changzhou Rongtuo Chemical Company and recrystallized to use. The solvent dimethylacetamide (DMAc) (analytically pure) was purchased from DuPont Co. and used after distillation. The flux lithium chloride was purchased from Tianjin Fengyue Chemical Co., Ltd. Imidazole was purchased from Saan Chemical Technology (Shanghai) Co., Ltd. The reaction principle is shown in Fig. 1.
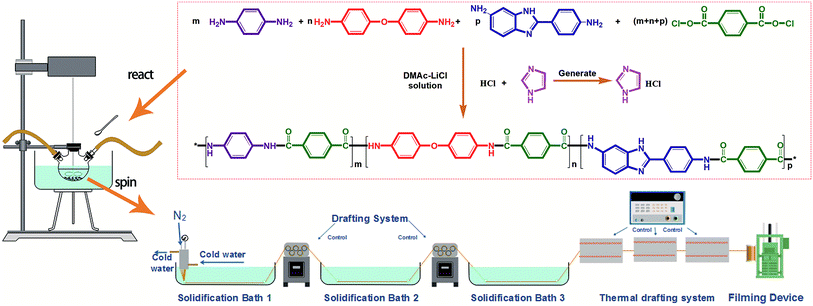 |
| | Fig. 1 Flow chart of the co-PPTA fiber preparation. | |
2.2 Preparation of the co-PPTA fibers
Co-PPTA fibers were prepared by low temperature condensation polymerization and spun by a low temperature wet spinning process. Co-PPTA fibers were synthesised by copolymerization of TPC and a mixture of three diamines, namely 4, 4′-ODA, BIA and p-PDA. Firstly, LiCl was dissolved in DMAc in a dry three-necked flask, which was evacuated and filled with nitrogen to ensure the absence of water or oxygen. Afterwards, diamine monomer BIA, p-PDA, and ODA were added to the system in sequence and were gradually dissolved as the system temperature increased from 0 °C to 20 °C. Meanwhile, imidazole was added as an acid binding agent. Then, high-purity TPC was continuously added and reacted at low temperature for 2 to 4 hours to obtain a viscous spinning solution with fluidity. Based on this method, the co-PPTA solutions with different p-PDA/ODA/BIA molar ratios (6/2/2, 4/2/4, 2/2/6, 2/4/4, 2/6/2, 4/4/2) were synthesized respectively.
The Co-PPTA solution for the wet-spinning was degassed under vacuum to remove air bubbles. Co-PPTA fibers were produced by extruding the viscous co-PPTA solution through the spinneret (100 holes, 9 μm in diameter). Co-PPTA solutions formed in DMAc solvent were primarily extruded into a coagulation bath consisting of a DMAc/H2O mixture to obtain co-PPTA fibers. After removing the residual solvents in the washing bath, the co-PPTA fibers were dried at 80 °C to 200 °C and delivered into furnaces with temperatures ranging from 200 °C to 500 °C to be converted into co-PPTA fibers. The experimental flowchart is illustrated in Fig. 1.
2.3 Characterization
The intrinsic viscosity of the co-PPTA solution was determined by an Ubbelohde viscometer (German JULABO 77960). The co-PPTA solution was diluted to 0.5 mg ml−1, 1.0 mg ml−1, 1.5 mg ml−1 and 2.0 mg ml−1 with H2SO4. The intrinsic viscosity of the co-PPTA solution was determined in a constant temperature tank at 30 °C. The intrinsic viscosity of the co-PPTA solution was obtained by extrapolation. The capillary diameter of the Ubbelohde viscometer was 0.58 mm.
Fourier transform infrared (FTIR) spectra of the fibers treated at different p-PDA/ODA/BIA molar ratios were obtained on a Nicolet Nexus 670 FTIR spectrometer in the wave number range of 4000 to 400 cm−1.
The mechanical properties of co-PPTA fibers were determined on an Instron 3344 instrument with a gauge length and extension speed of 250 mm and 125 mm min−1, respectively. For each group of fibers, at least 40 filaments were tested as representative.
The equatorial structure of the co-PPTA fibers was measured by two-dimensional wide angle X-ray diffraction (2D WAXD, Bruker AXS D8 AVANCE). The fibers were tightly wound and laid flat on the test bench. The X-ray was excited by a Kα ray from a Cu anode target with a wavelength λ of 0.154056 nm, a tube voltage of 40 kV, a tube current of 40 mA, a scanning range of 5 to 40°, and a scanning speed of 10° min−1. 2D WAXD was used to measure the meridional structures of the co-PPTA fibers. The crystallinities of the co-PPTA fibers were measured by X-ray diffraction (Ultima IV) in a scanning range of 5 to 90°.
The degree of fiber orientation can be calculated by Herman's equation:30–32
| |
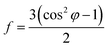 | (1) |
where
f is the orientation factor along the fiber axis and the average value of the included angle between the fiber axis and the lattice
c axis. cos
2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) φ
φ can be calculated by the below integral. Once all the values of (cos
2φ) have been obtained, the molecular orientation in the direction of (
hkl) can be evaluated.
| |
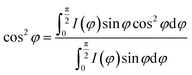 | (2) |
Scanning electron microscopy (SEM, Hitachi S-4700) was used to characterize the surface and cross-section morphologies of the co-PPTA fibers. A bunch of the co-PPTA fibers was smoothly laid and fixed on the measuring table with conductive adhesives. Epoxy resin and a curing agent were used to embed the fibers. The fibers were immersed in liquid nitrogen for 5 minutes and broken, then bonded to the side of the sample table by conductive adhesives. In vacuum, the surfaces and cross-sections of the co-PPTA fibers were coated with platinum metal particles with a diameter of 5 nm by electroplating and were sprayed with gold to render them conductive.
The thermal stabilities of the co-PPTA fibers were determined by thermogravimetric analysis (TGA, Q50, manufactured by TA Instruments). The co-PPTA fibers were cut into powder, and about 5 mg was placed in the sample table. The heating rate was 10 °C min−1, and the temperature was 800 °C in nitrogen atmosphere.
The thermos–mechanical properties of co-PPTA fibers were measured by a dynamic mechanical analyzer (DMA, Q800, manufactured by TA). A bunch of the co-PPTA fibers were fixed on the film test fixture with a heating rate of 5 °C min−1 and a frequency of 1 Hz in the temperature range from 30 °C to 450 °C.
3 Results and discussion
3.1 Chemical structure
The chemical structures of the co-PPTA fibers with different p-PDA/ODA/BIA molar ratios (6/2/2, 4/2/4, 2/2/6, 2/4/4, 2/6/2, 4/4/2) characterized by FT-IR are shown in Fig. 2. In the functional group region of 4000 to 400 cm−1 in the infrared spectra, the characteristic absorption bands of co-PPTA fibers at 1640 cm−1 (aramid-I band), 1495 cm−1 (aramid-II band) and 1248 cm−1 (aramid-III band) are revealed. There is a relatively weak absorption band at 1593 cm−1, which is the characteristic absorption band of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N in benzimidazole; thus, BIA can successfully participate in condensation polymerization. The absorption peak at 1110 cm−1 is assigned to the C–O–C of ODA. Additionally, the peak becomes more intense with increasing ODA content.
N in benzimidazole; thus, BIA can successfully participate in condensation polymerization. The absorption peak at 1110 cm−1 is assigned to the C–O–C of ODA. Additionally, the peak becomes more intense with increasing ODA content.
 |
| | Fig. 2 FT-IR curves of the co-PPTA fibers with different molar ratios of p-PDA/ODA/BIA. | |
The ATR-FTIR curves of the co-PPTA fibers with different molar ratios of p-PDA/ODA/BIA in the range of 3000 to 3600 cm−1 were carefully investigated to study the interactions of the hydrogen bonds. In Fig. 3, the bonds of free N–H (∼3405 cm−1) are reflected in the wavenumbers of band 1 (center: ∼3415 cm−1).16,29,30 Band 2 (center: ∼3350 cm−1) mainly corresponds to N–H groups participating in hydrogen bonding interactions in amide groups.16,29,30 The wavenumber of bond 3 (center: ∼3200 cm−1) represents hydrogen bonded N–H (∼3145 cm−1) stretching in polybenzimidazole, respectively.16,29,30 The center of band 3 is lower than that of band 2; therefore, band 3 can be attributed to stronger hydrogen bond stretching of the N–H groups.16 Based on the above analysis, the percentages of the free N–H, hydrogen-bonded N–H and stronger hydrogen-bonded N–H can be calculated by the relative areas of band 1, band 2 and band 3.16,30–32
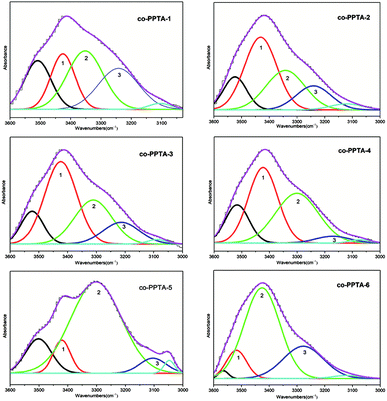 |
| | Fig. 3 The FTIR curves of the co-PPTA fibers with different molar ratios of p-PDA/ODA/BIA in the range of 3600 to 3000 cm−1; band 1, band 2 and band 3 correspond to free N–H, hydrogen-bonded N–H and strong hydrogen-bonded N–H stretching, respectively. | |
Hydrogen bonding plays an important role in the preparation process of PPTA fibers; it restricts drawing processing of polyamides. Hydrogen bonding can limit further alignment of molecular chains. The greater the amount of free N–H, the more readily the molecular chain is drawn in the process of movement, which is more conducive to the formation of fiber orientation structures. With the increase in BIA content and the decrease in PDA content, hydrogen-bonded N–H and strong hydrogen-bonded N–H decreased and free N–H increased, as shown in Fig. 4(a). Fig. 4(b) shows that with the decrease in BIA content and the increase in ODA content, free N–H and strong hydrogen-bonded N–H decreased and hydrogen-bonded N–H increased. Fig. 4(c) shows that free N–H and strong hydrogen-bonded N–H increased while hydrogen-bonded N–H decreased with decreasing ODA content and increasing PDA content. The introduction of BIA and ODA destroyed the regularity of the molecular chains. The softer the molecular chains, the more readily they can be drafted, and the more regular the arrangement between the molecular chains; this results in an increase in the formation of strong hydrogen bonds. Furthermore, the introduction of BIA leads to a certain degree of twist in the molecular chain, which in turn leads to a decrease in the formation of strong hydrogen bonds.
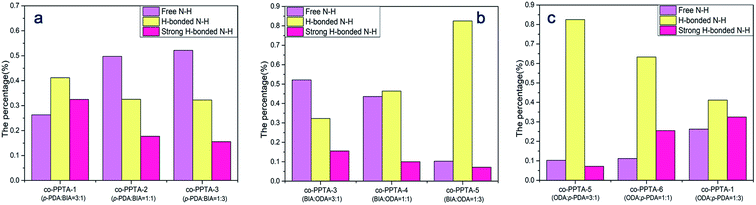 |
| | Fig. 4 Influence on the amounts of free N–H, hydrogen-bonded N–H and strong hydrogen-bonded N–H stretching in co-PPTA fibers with different molar ratios of p-PDA/ODA/BIA; (a) ODA is constant, p-PDA and BIA change; (b) p-PDA is constant, BIA and ODA change; (c) BIA is constant, BIA and p-PDA change. | |
3.2 Mechanical properties
The intrinsic viscosities, tensile strengths and moduli of the co-PPTA fibers with different molar ratios are listed in Table 1. According to the intrinsic viscosity of the fibers in the table, a spinning solution with suitable viscosity can be prepared directly by introducing ODA and BIA. The co-PPTA-4 fibers, which possess a p-PDA/ODA/BIA molar ratio of 2/4/4, possess the optimum mechanical properties with the tensile strength of 2.45 GPa. The mechanical properties of the fibers are more complicated and influence by many factors, such as intrinsic viscosities, intermolecular interactions, the rigidity of the polymer backbone, molecular packing and structural defects.30–32 Due to the presence of hydrogen bonding interactions and high orientation in the fibers, the mechanical properties of the PI fibers were improved when BIA was incorporated into the polymer chains.33 The introduction of BIA destroys the regularity of the PPTA fibers, and the tensile strength and modulus of the fibers enhance with increasing amount of BIA segments. As mentioned above, the free N–H and the rigidity of the benzimidazole rings are positive factors in improving the tensile strength and modulus of the co-PPTA fibers. Moreover, the introduction of ODA increases the flexibility of the chains and the entanglement between the molecular chains of the fibers. Therefore, as shown in Fig. 5(a), the flexibility of the molecular chains appropriately enhances the stretchability of the molecular chains, thereby improving the orientation of the fibers and the mechanical properties of the fibers. However, when the proportion of ODA increases to a large extent, the rigid structure of the molecular chains decreases, and the mechanical properties of the fibers decrease instead.
Table 1 The intrinsic viscosities and the mechanical properties of the co-PPTA fibers with different molar ratios of the diamines
| |
co-PPTA fibers |
p-PDA/ODA/BIA |
[η] (dL g−1) |
Tensile Strength (GPa) |
Fixed-length Modulus (GPa) |
Elongation (%) |
| p-PDA:BIA |
3:1 |
co-PPTA-1 |
6:2:2 |
2.21 |
1.39 ± 0.12 |
76.63 ± 3.97 |
2.18 ± 0.22 |
| 1:1 |
co-PPTA-2 |
4:2:4 |
2.76 |
1.81 ± 0.14 |
73.98 ± 2.01 |
4.04 ± 0.19 |
| 1:3 |
co-PPTA-3 |
2:2:6 |
2.81 |
2.33 ± 0.04 |
75.03 ± 2.21 |
3.33 ± 0.14 |
| BIA:ODA |
3:1 |
co-PPTA-3 |
2:2:6 |
2.81 |
2.33 ± 0.04 |
75.03 ± 2.21 |
3.33 ± 0.14 |
| 1:1 |
co-PPTA-4 |
2:4:4 |
2.68 |
2.45 ± 0.04 |
63.10 ± 0.87 |
4.83 ± 0.01 |
| 1:3 |
co-PPTA-5 |
2:6:2 |
2.05 |
1.67 ± 0.08 |
35.21 ± 1.31 |
5.38 ± 0.13 |
| ODA:p-PDA |
3:1 |
co-PPTA-5 |
2:6:2 |
2.05 |
1.67 ± 0.08 |
35.21 ± 1.31 |
5.38 ± 0.13 |
| 1:1 |
co-PPTA-6 |
4:4:2 |
2.13 |
2.00 ± 0.05 |
50.13 ± 1.09 |
4.61 ± 0.11 |
| 1:3 |
co-PPTA-1 |
6:2:2 |
2.21 |
1.39 ± 0.12 |
76.63 ± 3.97 |
2.18 ± 0.22 |
 |
| | Fig. 5 Sketch of the chain segment motion at high temperature (a) and hypothetical structure model of the co-PPTA fiber (b). | |
The co-PPTA-6 fiber was selected to carry out experimental research on different thermal drawing ratios. The mechanical properties, modulus and elongation are shown in Table 2. A hypothetical structure model of the co-PPTA fiber is shown in Fig. 5(b). As the draw ratio increases, the corresponding tensile strengths and moduli of the co-PPTA fibers increase, reaching the optimum tensile strength of 2.72 GPa and modulus of 70.13 GPa, respectively. Through thermal drawing, the molecular chains of the fibers are rearranged, and crystalline and oriented structures gradually appear above the glass transition temperature. Coupled with the existence of a large number of hydrogen bonds between molecular chains, the mechanical strength of the fibers is enhanced.
Table 2 The mechanical properties of co-PPTA-6 fibers with different drawing ratios
| Drawing ratio |
Tensile strength (GPa) |
Fixed-length modulus (GPa) |
Elongation (%) |
| 1.00 |
0.86 ± 0.07 |
16.63 ± 0.73 |
9.40 ± 0.45 |
| 1.25 |
2.00 ± 0.05 |
50.13 ± 1.09 |
4.61 ± 0.11 |
| 1.50 |
2.35 ± 0.05 |
59.55 ± 2.95 |
4.73 ± 0.07 |
| 1.75 |
2.44 ± 0.08 |
63.72 ± 2.22 |
4.56 ± 0.18 |
| 2.00 |
2.59 ± 0.03 |
69.40 ± 3.29 |
4.65 ± 0.14 |
| 2.25 |
2.72 ± 0.05 |
70.13 ± 0.77 |
4.21 ± 0.15 |
3.3 Aggregation structure
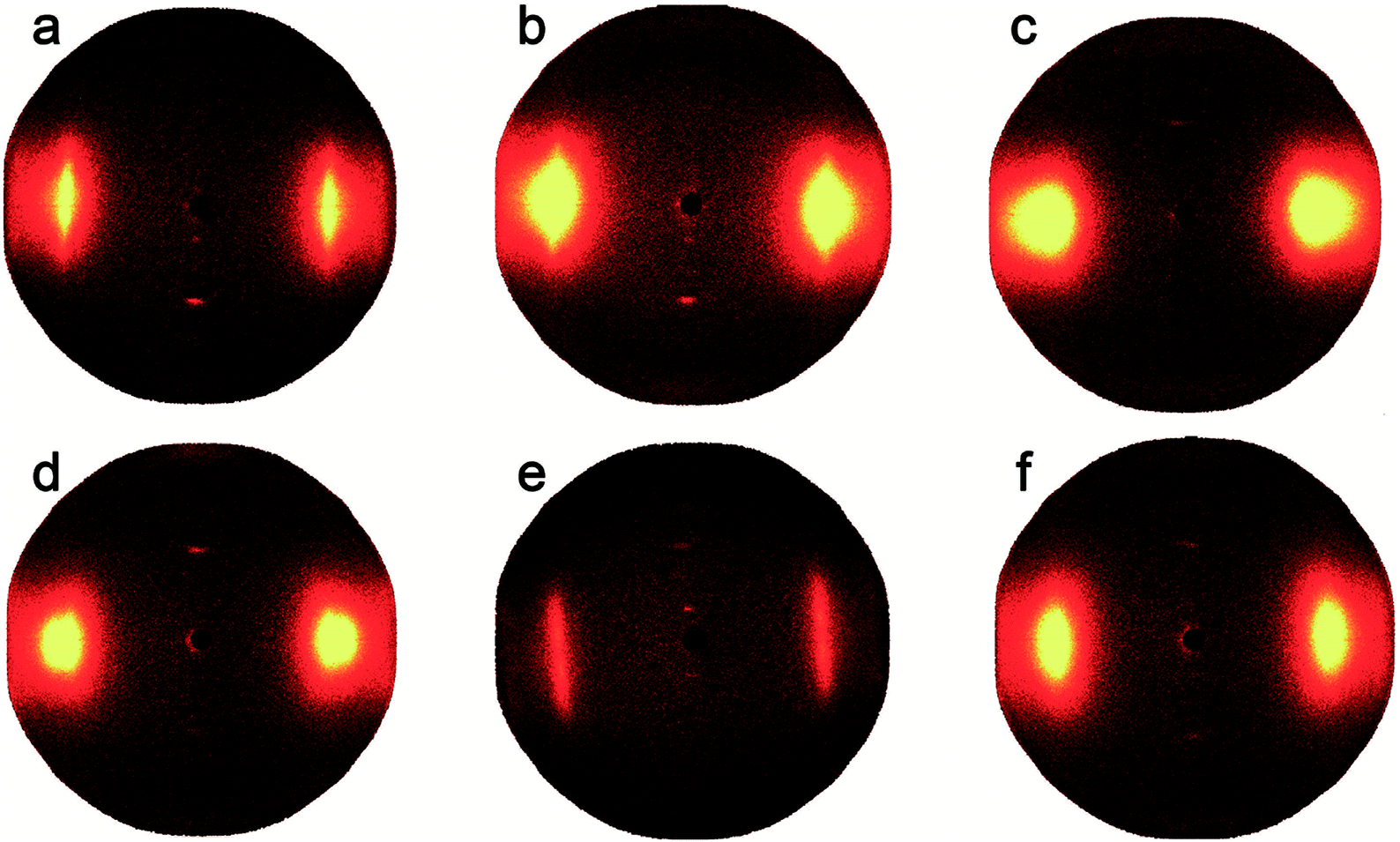
Fig. 6 shows the 2D-WAXD patterns of co-PPTA fibers with different molar ratios of the diamines. The co-PPTA fibers exhibit two clear diffraction spots in the meridional direction, indicating that a highly oriented structure is formed along the fiber axis direction. Diffuse diffraction spots appear at the fiber equator, indicating ordered intermolecular packing in the transverse fiber direction. In order to analyse the orientation degrees of the fibers in the equatorial and meridian directions, diffraction curves in the equatorial and meridian directions are shown in Fig. 7.
 |
| | Fig. 6 Set of 2D-WAXD fiber patterns with different molar ratios of the diamines; (a) co-PPTA-1, (b) co-PPTA-2, (c) co-PPTA-3, (d) co-PPTA-4, (e) co-PPTA-5, (f) co-PPTA-6. | |
 |
| | Fig. 7 Sets of WAXD fiber patterns scanned along the equatorial (a) and meridian (b) directions. | |
Two-dimensional wide angle X-ray diffraction (2D WAXD) was performed on a Bruker D8 Discover diffractometer equipped with GADDS as a 2D detector. The degrees of orientation of the fibers are shown in Table 3. The axial orientation degrees of the co-PPTA fibers can reach above 85.93%, among which the orientation degree of the co-PPTA-5 fibers is as high as 98.79%. The addition of ODA and BIA can increase the flexibility of the molecular chains. Under the same drawing ratio, the greater the flexibility of the molecular chains, the more readily they orient along the axial direction in the process of polymer transformation from random coil to ordered structures along the fiber axis. Therefore, the addition of ODA and BIA can enhance the degree of orientation. Theoretically, the degree of fiber orientation determines the changes in the mechanical properties. However, as shown in Fig. 5(a), when there are too many flexible groups, the rigid structure of the molecular chain decreases. Although the degree of orientation is very high, the mechanical properties decrease instead, as the co-PPTA-4 fiber shows.
Table 3 The degrees of orientation and crystallinities of the co-PPTA fibers with different molar ratios of the diamines
| |
co-PPTA fibers |
Degree of orientation (%) |
Amorphous peak position (°) |
Crystallinity (%) |
| p-PDA:BIA |
3:1 |
co-PPTA-1 |
85.93 |
20.244 |
33.65 |
| 1:1 |
co-PPTA-2 |
85.99 |
20.756 |
39.44 |
| 1:3 |
co-PPTA-3 |
86.04 |
20.147 |
62.11 |
| BIA:ODA |
3:1 |
co-PPTA-3 |
86.04 |
20.147 |
62.11 |
| 1:1 |
co-PPTA-4 |
90.45 |
19.267 |
66.62 |
| 1:3 |
co-PPTA-5 |
98.79 |
19.677 |
31.18 |
| ODA:p-PDA |
3:1 |
co-PPTA-5 |
98.79 |
19.677 |
31.18 |
| 1:1 |
co-PPTA-6 |
86.27 |
20.872 |
33.05 |
| 1:3 |
co-PPTA-1 |
85.93 |
20.244 |
33.65 |
The XRD spectra are shown in Fig. 8. The crystallinities of the X-ray diffraction spectra of the co-PPTA fibers with different molar ratios of the diamines were calculated by MID JADE5 software, and the average values were calculated several times. The amorphous peak positions and crystallinities are shown in Table 3. There are two diffraction peaks around 20° and 27°, corresponding to the (110) and (200) crystal planes, respectively.34–37 The intensity of the (110) crystal plane peak is higher than that of the (200) crystal plane, which indicates the high modulus aramid fiber. The changes in the peak intensity and apparent crystal plane size reflect the evolution of the crystallinity during processing. The crystallinity of the co-PPTA fibers decreases due to the introduction of the third monomers BIA and ODA. The X-ray diffraction spectra of the co-PPTA fibers show a sharp diffraction peak when 2-theta is about 20°; it is arched by an amorphous peak, indicating that the crystal structure of the co-PPTA fibers is obviously different from the amorphous structure on this crystal plane. The other diffraction peaks are not sharp, which indicates that there is a very incomplete crystalline phase in the co-PPTA fibers. There are crystalline macromolecular chains in this crystal phase; however, its aggregation arrangement is not perfect. In addition, during spinning and post-processing, the influence of the spinning draw ratio and precursor heat treatment on its crystalline structure are also quite important.
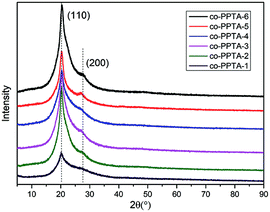 |
| | Fig. 8 X-ray diffraction spectra of co-PPTA fibers with different molar ratios of the diamines. | |
As reported previously, the drawing ratios played an important role in the molecular orientation of co-PPTA fibers. Fig. 9 shows the set of 2D-WAXD fiber patterns of the co-PPTA-6 samples with different drawing. Diffraction curves in the equatorial and meridian directions are shown in Fig. 10. As shown in Table 4, the mechanical properties and the degree of orientation of the co-PPTA fibers increase with the increase in drawing ratio. It is evident that the alignment of molecules becomes more ordered due to the stretching of the molecule chain as the draw ratio increases. When there is no drawing ratio, there is no diffraction spot in the radial direction; thus, the mechanical properties of the fiber are poor. As the thermal drawing ratio increases, the fibers gradually become orderly, which indicates that the molecular chains will rearrange after drawing. In addition, we found that the fiber with the best mechanical properties has the largest degree of orientation.
 |
| | Fig. 9 Set of 2D-WAXD fiber patterns of the co-PPTA-6 samples with different drawing ratios. (a) Ratio = 1; (b) ratio = 1.25; (c) ratio = 1.5; (d) ratio = 1.75; (e) ratio = 2; (f) ratio = 2.25. | |
 |
| | Fig. 10 Set of WAXD fiber patterns of co-PPTA-6 with different drawing ratios scanning along the equatorial (a) and meridian (b) directions. | |
Table 4 The degrees of orientation and crystallinities of the co-PPTA-6 fibers with different drawing ratios
| Drawing ratio |
Degree of orientation (%) |
Amorphous peak position (°) |
Crystallinity (%) |
| 1.00 |
— |
20.867 |
29.28 |
| 1.25 |
86.27 |
20.872 |
33.05 |
| 1.50 |
87.04 |
20.783 |
35.79 |
| 1.75 |
87.69 |
21.011 |
55.65 |
| 2.00 |
88.18 |
21.203 |
66.36 |
| 2.25 |
88.85 |
20.595 |
72.48 |
In order to clearly understand the structural evolution of the fibers during the drawing process, WAXD measurements were also performed to characterize the macromolecular packing of the co-PPTA fibers, as illustrated in Fig. 11. The crystallinities and degrees of orientation of the co-PPTA-6 fibers with different drawing ratios are shown in Table 4. The table shows that the crystallinities and orientation degrees of the fiber increase with increasing thermal drawing ratio, and the arrangement of molecular chains also tends to be more regular; therefore, the mechanical properties will also be strengthened. The fibers also had two crystal planes, (110) and (200), respectively. At low drawing ratios, the (200) crystal plane is located at about 23°. With increasing drawing rate, the fibers have three crystal planes; however, when the drawing rate increases to 1.75, the angle of the crystal plane changes to around 27°.
 |
| | Fig. 11 X-ray diffraction spectra of the co-PPTA-6 fibers with different drawing ratios. | |
3.4 Morphologies of the co-PPTA fibers
During the wet-spinning process, the coagulation of fibers is a dual diffusion process; namely, the solvent diffuses into the bath and the coagulant diffuses into the fibers.30–32 Different diffusion rates lead to the formation of a skin-core structure. Fig. 12 shows SEM characterizations of the co-PPTA fibers with different molar ratios enlarged 6000 times, respectively. The mean diameters of the co-PPTA fibers are about 8 to 12 μm; it can also be clearly seen that the co-PPTA fibers have cylindrical shapes, smooth fiber surfaces and uniform diameters. From the cross-section images, all the co-PPTA fibers possess dense morphologies without obvious microvoid structures. The SEM results showed that the fibers possess homogenous and uniform morphologies and that the incorporation of ODA and BIA had little effect on the morphologies of the final PI fibers.34 It is well known that the skin-core structure is a typical structure of fibers prepared by wet spinning processes. The skin-core structure of fibers is directly related to the coagulation conditions and spinning speed.30–32 The cross-sectional morphological photographs of the co-PPTA-4 fibers are different from those of the other fibers. This is because the tensile strength of the co-PPTA-4 fibers is optimum, and they are more ordered across along the fiber axis. Overall, the co-PPTA fibers exhibit homogeneous surfaces as well as dense cross-section morphologies, endowing the prepared PPTA fibers with excellent mechanical performance.
 |
| | Fig. 12 SEM images of the co-PPTA fibers: (a1 and a2) co-PPTA-1, (b1 and b2) co-PPTA-2, (c1 and c2) co-PPTA-3, (d1 and d2) co-PPTA-4, (e1 and e2) co-PPTA-5, (f1 and f2) co-PPTA-6. | |
3.5 Thermal gravimetric analysis
Fig. 13 and its inset table show that all the fibers exhibit 5% weight loss temperatures (Td5) over 492 °C under nitrogen and 10% weight loss temperatures (Td10) over 505 °C, demonstrating the outstanding thermal and thermo-oxidative stabilities of the co-PPTA fibers. The co-PPTA-2 fibers have the highest thermal weight loss temperature, with a Td5 value of 503 °C and a Td10 value of 522 °C. It can be clearly observed that the Td5 values of the high ODA content fibers are relatively low, indicating that the introduction of benzimidazole moiety results in improvement in the thermal and thermo-oxidative stabilities of the co–PPTA fibers; this is probably related to the increased intermolecular interactions.32 In conclusion, the introduction of ODA leads to a decrease in the thermal stability of the PI fibers because of the decreased rigidity of the polymer backbone.
 |
| | Fig. 13 TGA and DTG of the co-PPTA fibers at a heating rate of 10 °C min−1 under nitrogen atmosphere. | |
3.6 Dynamic mechanical analysis
DMA was carried out under a nitrogen environment to investigate the effects of the ODA and BIA contents on the thermal mechanical properties of the co–PPTA fibers; as illustrated in Fig. 14, two relaxation peaks appeared. The secondary glass transition occurs at about 100 °C, which is the β relaxation peak, and the glass transition occurs at about 290 °C, which is the α relaxation peak. The temperature of the glass transition (Tg) decreases with the increasing BIA content. Meanwhile, it can be observed from Fig. 4 that the strong hydrogen bond effect slightly decreased, and the Tg also decreased. In addition, due to the introduction of BIA and ODA, the loss factor tanδ increased. As shown in Fig. 14, the change of tanδ is determined by the joint action of ODA and BIA. ODA improved the mobility of the molecular chains; meanwhile, the greater the elasticity of the molecular chains, the smaller the loss factor. The loss factor tanδ of the co-PPTA-5 fibers is the largest. The co-PPTA-1 fibers have the highest Tg value of 300 °C. Therefore, the introduction of flexible groups into the main chain can decrease the glass transition temperature, and the strong hydrogen bonds between molecules can increase the glass transition temperature. The flexibility of the fiber increases the internal friction in the molecular chain, causing an increase in the internal friction resistance of the chain segment in the movement process and further causing the loss factor to increase.
 |
| | Fig. 14 DMA curves of the co-PPTA fibers with different molar ratios of the diamines at a heating rate of 5 °C min−1 under nitrogen atmosphere. (a) ODA is constant, p-PDA and BIA change; (b) p-PDA is constant, BIA and ODA change; (c) BIA is constant, BIA and p-PDA change. | |
4 Conclusions
High-performance co-PPTA fibers containing different molar ratios have been prepared by wet spinning followed by hot drawing. The mechanical properties of the co-PPTA fibers are enhanced with increasing BIA and ODA content, reaching the optimum tensile strength of 2.45 GPa at the p-PDA/ODA/BIA molar ratio of 4/2/4. Additionally, the mechanical properties of the fibers increased with increasing thermal draw ratio. BIA and ODA destroy the regularity of the molecular chains and decrease the hydrogen bonds generated by PDA. XRD indicated highly ordered structures, and two-dimensional wide-angle XRD showed that highly oriented structures in the molecular packing regions were formed in the fibers. Additionally, the co-PPTA fibers exhibited excellent thermal and thermo-oxidative stabilities, with a 5%-weight-loss temperature of over 500 °C under a nitrogen environment. The introduction of flexible groups into the main chain can decrease the glass transition temperature. The two thermal relaxation processes, β relaxation and α relaxation, were observed above room temperature. The α relaxation was verified to be hydrogen-bonding and molecular orientation-dependent. A glass transition occurred at about 290 °C, which indicated β relaxation. The introduction of flexible groups into the main chain can decrease the glass transition temperature and increase tanδ. It is believed that these co-PPTA fibers have good prospects in future research.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors gratefully acknowledge the financial support from the National Natural Science Foundation of China (51790501) and the National Key Research and Development Program of China (No. 2017YFB0307600).
Notes and references
- A. Abu-Obaid, J. M. Deitzel, J. W. Gillespie and J. Q. Zheng, J. Compos. Mater., 2011, 45, 1217–1231 CrossRef CAS.
- B. Ahmad and A. Fariborz, Eur. Polym. J., 2002, 38, 2119–2124 CrossRef.
- J. D. Menczel and J. Therm, Anal. Calorim., 2000, 59, 1023–1027 CrossRef CAS.
- K. Li, L. Luo, J. Huang and H. Wang, J. Appl. Polym. Sci., 2015, 132, 42482–42491 Search PubMed.
- K. E. Perepelkin, N. N. Machalaba and V. A. Kvartskheliya, Fibre Chem., 2001, 33, 105–114 CrossRef CAS.
- N. N. Machalaba and K. E. Perepelkin, J. Ind. Text., 2002, 31, 189–204 CrossRef CAS.
- H. Davis, Text. Res. J., 2000, 70, 945 CrossRef CAS.
- X. Hu, S. E. Jenkin and B. G. Min, Macromol. Mater. Eng., 2003, 288, 823–843 CrossRef CAS.
- Nakamura and T. C. Toyokazu, Liquid crystal compounds and compositions, 1990 Search PubMed.
- M. W. Bowen, C. King and H. H. Yang, Process for making fibers with improved hydrolytic stability, 1991 Search PubMed.
- L. Luo, J. Huang, Y. Feng, X. Wang, J. Qin and X. Liu, High Perform. Polym., 2017, 29, 58–67 CrossRef CAS.
- I. V. Tikhonov, A. V. Tokarev and S. V. Shorin, Fibre Chem., 2013, 45, 1–8 CrossRef CAS.
- S. L. Bazhenov, A. K. Rogozinskii and S. S. Mal'kov, Polym. Phys., 2011, 53, 1182–1186 CAS.
- G. B. Sklyarova, I. V. Slugin and A. I. Kashirin, Fibre Chem., 2006, 38, 25–26 CrossRef.
- A. V. Tokarev, I. V. Tikhonov and V. M. Shchetinin, Fibre Chem., 2006, 38, 450–452 CrossRef.
- L. Luo, Y. Wang, Y. Dai, Y. Yuan, C. Meng, Z. Cheng, X. Wang and X. Liu, J. Mater. Sci., 2018, 53, 13291–13303 CrossRef CAS.
- B. Herbert, High modulus, high tenacity poly(p-phenylene terephthalamide) fiber, 1975 Search PubMed.
- S. Ozawa, Polym. J., 1987, 19, 119–125 CrossRef CAS.
- Microvast power systems CO., LTD., Method for preparing aromatic polyamide porous film and aromatic polyamide porous film, World Patent Organization, 2017 Search PubMed.
- I. Han, J. Lee, J.-Y. Kim, T.-H. Park and S.-Y. Kwon, Process for making aromatic polyamide filament, 2013 Search PubMed.
- D. Ying, T. Bin and Y. Li, Mater. Sci. Forum, 2017, 4502, 2158–2165 Search PubMed.
- E. I. DuPont De Nemours and company, Carbon-containing fiber blends including aramid and modacrylic fiber, 2018 Search PubMed.
- L. Luo, Y. Wang, J. Huang, D. Hong, X. Wang and X. Liu, RSC Adv., 2016, 6, 62695–62704 RSC.
- A. A. Leal, J. M. Deitzel and J. W. Gillespie, Compos. Sci. Technol., 2007, 67, 2786–2794 CrossRef CAS.
- A. A. Leal, J. M. Deitzel, S. H. McKnight and J. W. Gillespie, Polymer, 2009, 50, 1228–1235 CrossRef.
- K. G. Lee, R. Barton and J. Schultz, J. Polym. Sci., Part B: Polym. Phys., 1995, 33, 1–14 CrossRef CAS.
- K. Li, L. Li, J. Qin and X. Liu, Polym. Degrad. Stab., 2016, 128, 278–285 CrossRef CAS.
- L. Luo, Y. Wang, J. Huang, D. Hong, X. Wang and X. Liu, High Perform. Polym., 2016, 28, 381–389 CrossRef.
- I. Abronin, A. Bandurkin, L. Volkova and M. Shablygin, Fibre Chem., 2016, 47, 377–379 CrossRef CAS.
- J. Chang, H. Niu, M. Zhang, Q. Ge, Y. Li and D. Wu, J. Mater. Sci., 2015, 50, 4104–4114 CrossRef CAS.
- H. Niu, M. Huang, S. Qi, E. Han, G. Tian, X. Wang and D. Wu, Polymer, 2013, 54, 1700–1708 CrossRef CAS.
- M. Zhang, H. Niu, J. Chang, Q. Ge, C. Li and Dezhen Wu., Polym. Eng. Sci., 2015, 55, 2615–2625 CrossRef CAS.
- Q. Ma, C. Li and M. Tian, Para-aramid fiber, 2018 Search PubMed.
- M. Sun, J. Chang, G. Tian, H. Niu and D. Wu, J. Mater. Sci., 2016, 51, 2830–2840 CrossRef CAS.
- A. M. Hindeleh, N. A. Halim and K. A. Zig, J. Macromol. Sci., Part B: Phys., 1984, 23, 289–309 CrossRef.
- C. Jackson, R. Shadt and K. Gardner, Polymer, 1994, 35, 1123–1131 CrossRef CAS.
- Y. Rao, A. J. Waddon and R. J. Farris, Polymer, 2001, 42, 5925–5935 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Hongqing Niu
a,
Hongqing Niu