
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNearly spherical CoP nanoparticle/carbon nanosheet hybrids: a high-performance trifunctional electrocatalyst for oxygen reduction and water splitting†
Wenjian Zoua,
Kunpeng Doub,
Qi Jianga,
Jiadong Xianga,
Chao-Cheng Kaun c and
Hao Tang*a
c and
Hao Tang*a
aState Key Laboratory of Advanced Technology for Materials Synthesis and Processing, International School of Materials Science and Engineering, Wuhan University of Technology, Wuhan 430070, People's Republic of China. E-mail: tanghaotc@gmail.com
bCollege of Information Science and Engineering, Ocean University of China, Qingdao 266100, People's Republic of China
cResearch Center for Applied Sciences, Academia Sinica, Taipei 11529, Taiwan
First published on 4th December 2019
Abstract
Developing active multifunctional electrocatalysts composed of earth-abundant and cheap elements is an urgent demand in energy conversion applications. This study presents a facile approach for the scalable synthesis of nanostructured cobalt phosphide embedded in carbon nanosheets (CoP NPs/CNSs). The hybrid structures show highly efficient trifunctional electrocatalytic activities toward the oxygen reduction reaction (ORR), oxygen evolution reaction (OER), and hydrogen evolution reaction (HER) under alkaline condition. The catalytic performances, which are remarkably superior to those of the previously reported CoP nanostructures enclosed by single or a few low index facets, can be attributed to the nearly spherical shape of the CoP nanoparticles with many more exposed crystal planes. Density functional theory (DFT) computations are performed to investigate the facet effects of CoP on electrocatalytic activity, and they reveal the relatively low overpotentials of (101) facets towards the OER and the free energy of water dissociation (ΔGH2O) and adsorbed H intermediates (ΔGH*) of (311) toward the HER being close to thermoneutral. This work is expected to inspire the design and fabrication of multifunctional and high-efficiency electrocatalysts by selectively exposing specific crystal planes.
Introduction
Energy conversion electrocatalysis processes, such as the oxygen reduction reaction (ORR), oxygen evolution reaction (OER), and hydrogen evolution reaction (HER), have attracted intense attention because of their prime roles in fuel cells, rechargeable metal–air batteries, and water splitting.1 HER and OER are the two key half-reactions for water splitting, while the combination of OER and ORR is required in rechargeable metal–air batteries. If trifunctional electrocatalysts toward the ORR, HER, and OER can be further realized simultaneously in the same alkaline solution, integrated metal–air battery-powered overall water splitting systems can be designed to produce H2 and O2 for the renewable energy industry. However, the sluggish kinetics and the complicated reaction pathways of the ORR/OER/HER together with the dependency on expensive noble metal catalysts (e.g., Pt or RuO2) obstruct their large-scale applications. Hence, the search for cheaper, operationally stable, and high-efficiency trifunctional electrocatalysts for ORR/OER/HER is highly desired and challenging.2Presently, transition metal–phosphides-based materials (TMPs) have attracted considerable attention due to their outstanding catalyst performance in electrochemical energy conversion and storage technologies.3–17 The morphology and electrical conductivity of electrocatalysts are important factors to affect electrocatalytic performance. The studies for improving the catalytic performance of TMPs also mainly track two ways: (a) increasing the number of active sites of the catalyst via morphology control, and (b) improving the electrical contact to these active sites. For instance, density functional theory calculations showed that the exposed Ni and P sites of Ni2P (001) surface serve as hydride acceptor and proton-acceptor center, respectively, promoting HER performance.9 The Tafel slope of the nanostructured Ni2P (001) was experimentally observed at 46 mV per decade with an exchange current density of 3.3 × 10−5 A cm−2 in 0.50 M H2SO4, which is among the highest HER activity of any non-noble metal electrocatalyst reported to date. The crystal facet-dependent electrocatalytic activities are also widely observed in cobalt phosphide (CoP). In particular, ultrathin CoP nanosheets with exposed (200) crystal facets exhibit a low overpotential (only 56 and 131 mV are required for current densities of 10 and 100 mA cm−2, respectively) and a small Tafel slope of 44 mV per decade in acidic condition.5 Single-crystalline CoP nanowires with side facets (200) have been found to be bifunctional electrocatalysts, which not only demonstrate superior catalytic activity for HER in 1.0 M KOH (an overpotential of 244 mV at 100 mA cm−2), but also exhibit exceptional electrocatalytic performance for OER in the same electrolyte (an overpotential of merely 300 mV at 100 mA cm−2 and a small Tafel slope of 78 mV dec−1), ranking among the top-performing non-precious OER catalysts.7 Urchin-like CoP nanocrystals enclosed with (011) facets have presented excellent HER catalytic performance in 0.5 M H2SO4 (a low onset overpotential of 50 mV, a small Tafel slope of 46 mV per decade) and remarkable ORR catalytic activity (a half-potential of 0.7 V and an onset potential of 0.8 V at 1600 rpm and a scan rate of 5 mV s−1) in 0.1 M KOH.8 CoP nanocrystals with abundant surface phosphide atoms on exposed (211) crystal planes are bifunctional catalysts for ORR (an onset potential of 0.92 V with a Tafel slope at 72.1 mV dec−1) and HER (a small overpotential of 62.5 mV at a current density of 10 mA cm−2) in 1 M KOH solution.9 The most common way to improve electrical conductivity of electrocatalysts is to introduce carbon nanostructures to form catalyst/carbon hybrids in order to enrich catalytic active sites and improve electrical coupling.10,11 The CoP nanoparticles/defective carbon hybrids have exhibited better ORR and OER activities in 0.1 M KOH solution comparing to the single component, which is attributed to the interfacial charge polarization with electrons gathering at the defective carbon surface and holes at the CoP surface facilitating ORR and OER.12
Despite the huge progress in TMPs-based electrocatalysts, few TMPs have been reported to demonstrate highly efficient multi-activities towards ORR, OER and HER concurrently in the same alkaline solution. The reason lays in the facts that the real adsorption sites of TMPs for HER and active sites for ORR and OER are still in debate, and that the difference of active sites and reaction pathways toward HER/ORR/OER does exist. In the present work, CoP nanoparticles embedded in carbon nanosheets (CoP NPs/CNSs) were synthesized via a one-step self-templating route.18 The as-synthesized sample exhibits as an excellent trifunctional electrocatalyst for ORR/OER/HER in alkaline media. Remarkably, the electrocatalytic activities of CoP NPs/CNSs towards ORR and OER are comparable or superior to the state-of-the-art expensive metal catalysts. By comparison to the catalytic performances and surface structures of the existing CoP counterparts5–9,11,12 and aided by density functional theory calculations, we showed that the near-sphere shape of CoP with richly exposed planes are responsible for the outstanding catalytic properties, identifying the detailed facets facilitating HER/OER/ORR. Our studies provide guidance on the development of multifunctional electrocatalysts by exposing the specific crystal planes.
Experimental section
Synthesis of materials
CoP nanoparticles embedded in carbon nanosheets (CoP NPs/CNSs) were fabricated in one-step via a self-templating and recrystallization-self-assembly route.18 In a typical synthesis, 0.249 g Co(CH3COO)2·4H2O (AR 99.5%) and 1.261 g melamine (MA, 99%) were mixed in a beaker. Subsequently, the mixture was stirred with 3 mL phytic acid solution until it was changed from a slurry to a solid sample. The precipitate was dried in an oven at 70 °C overnight and then calcined at 900 °C in flowing high-pure Ar atmosphere for 2 h to yield the final crystalline products.Characterization of CoP NPs/CNSs
Powder X-ray diffraction (XRD, Rint-2000, Rigaku, Cu Kα radiation, λ = 1.5418 Å) was used to characterize the crystalline structure of the samples. Scanning electron microscope (SEM, JEOL-7100F, 20 kV), transmission electron microscopy (TEM), high-resolution TEM (HRTEM) and HAADF-STEM SAED (JEM-2100F/Titan G260-300 transmission electron microscope) were used to obtain the morphology and structure information. EDX analysis was conducted using an FEI Talos F200X TEM at an acceleration voltage of 200 kV. X-ray photoelectron spectrometer (XPS, Kratos Model XSAM800) equipped with an Mg Kα achromatic X-ray source (1235.6 eV) was used to explore the surface elemental status. Nitrogen adsorption–desorption isotherms (a Tristar-3020 instrument) was used to determine the surface area of the hybrid structures.Electrochemical measurement
All ORR, OER and HER measurements were conducted with a three-electrode system by CHI 760D electrochemical workstation (Chenhua, China) at room temperature. Glassy carbon electrode (GCE) with a diameter of 5 mm was used as the working electrode. For ORR and OER, a platinum wire electrode and saturated calomel electrode (SCE) were used as the counter and reference electrodes, respectively. For HER, graphite electrode was used as the counter electrode.CoP NPs/CNSs electrode fabrication: 5 mg of catalyst CoP NPs/CNSs was suspended in a stock solution containing 50 μL of 5 wt% Nafion, 800 μL of isopropanol and 150 μL of deionized water by ultra-sonication for 30 min to form 1 mL homogeneous ink. Then the catalyst ink was drop-casted on GCE (loading ∼0.25 mg cm−2) for air drying.
For ORR, the electrochemical data was collected with O2 saturated 0.1 M KOH solution electrolyte. All the potentials were calibrated to a reversible hydrogen electrode (RHE) according to the equation, E(RHE) = E(SCE) + 0.0591pH + 0.24. Linear sweep voltammetry (LSV) was carried out with a rotating disk electrode (RDE) with rotation rates of 400, 625, 900, 1225 and 1600 rpm, respectively, with a sweep rate of 5 mV s−1. By analysis of the collected LSV data can conclude the ORR kinetics using the Koutecky–Levich (K–L) equation:

![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 cm−1); A is the geometric electrode area (0.196 cm2); K is the rate constant for oxygen reduction; CO is the saturated concentration of oxygen in 0.1 M KOH (1.2 × 10−6 mol cm−1); DO2 is the diffusion coefficient of oxygen (1.87 × 10−6 cm2 s−1); ν is the kinetic viscosity of the solution (0.01013 cm2 s−1); and ω is the rotation rate (rad s−1) of the electrode.
500 cm−1); A is the geometric electrode area (0.196 cm2); K is the rate constant for oxygen reduction; CO is the saturated concentration of oxygen in 0.1 M KOH (1.2 × 10−6 mol cm−1); DO2 is the diffusion coefficient of oxygen (1.87 × 10−6 cm2 s−1); ν is the kinetic viscosity of the solution (0.01013 cm2 s−1); and ω is the rotation rate (rad s−1) of the electrode.
For OER and HER, the electrochemical data were recorded in N2-saturated 1 M KOH. All the potentials were calibrated to a reversible hydrogen electrode (RHE) according to the equation, E(RHE) = E(SCE) + 0.0591pH + 0.24. LSV was carried out with a RDE with a rotation rate of 900 rpm and a sweep rate of 5 mV s−1.
Calculations
Density functional theory calculations have been performed using Perdew–Burke–Ernzerhof generalized gradient approximation (PBE-GGA) functional as implemented in VASP package.19,20 The cutoff energy for the plane wave basis was 420 eV. All atoms were relaxed until the Hellmann–Feynman forces acting on them were below 0.02 eV Å−1. The lattice constant of CoP has been determined to be a = 5.070 Å, b = 3.265 Å, and c = 5.545 Å. About 10 Å thick slabs with 15 Å of vacuum along the z-direction were used to model the adsorbate-surface systems for (101), (111), (311), (112), (210), (201), (012), (011) surfaces. The top-half atomic layers were relaxed to optimize the surface structure with all other atoms being fixed.For OER, the process is decomposed into four one-electron steps with activities evaluated by the Gibbs free energy change (ΔGi) of each reaction step, listed as below:22
| (1) H2O (l) + * → HO* + H+ + e− |
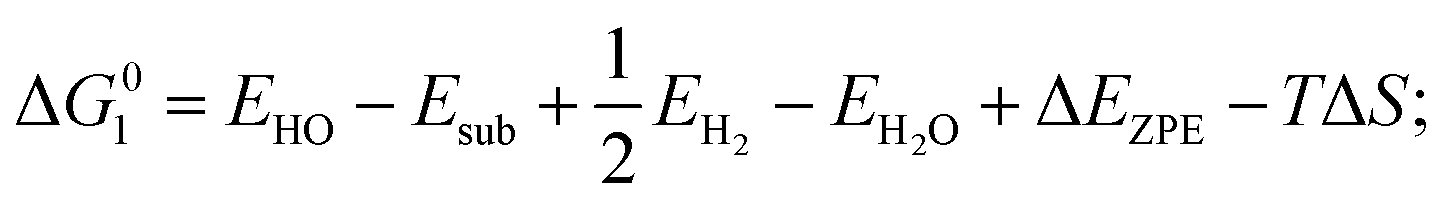
| (2) HO* → O* + H+ + e−; |
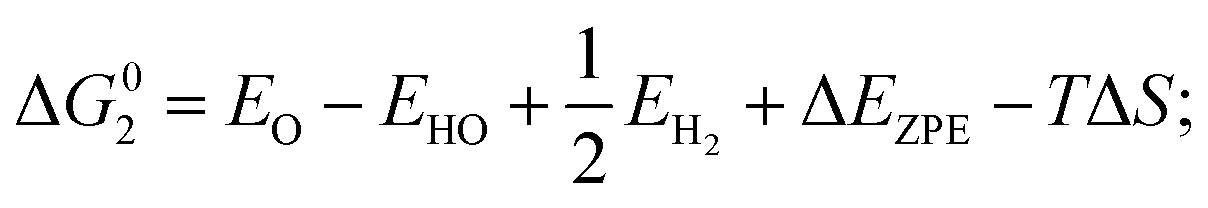
| (3) O* + H2O (l) → HOO* + H+ + e− |
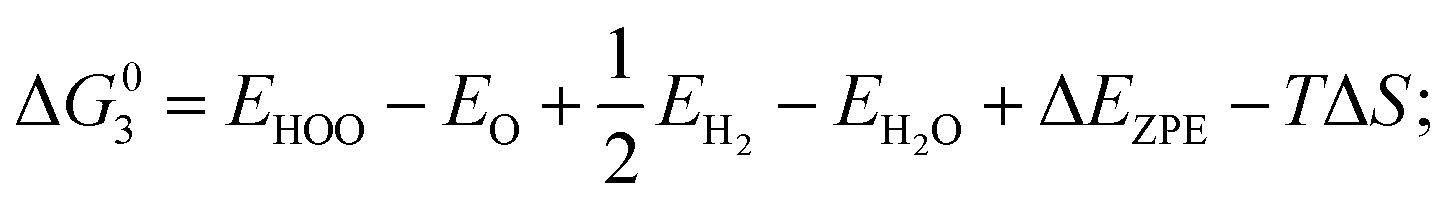
| (4) HOO* → O2 (g) + H++ e− |
where EH2 and EH2O are energies of an isolated H2 and H2O molecule, respectively, Esub, EHO, EO and EHOO are energies of clean surface and the surface with HO, O and HOO adsorption, respectively, ΔEZPE for various absorbents and TΔS are adapted from literature.21,22 As a generally-accepted OER activity descriptor, the overpotential (η) for a chemical reaction can be calculated as follow: η = max[ΔG1, ΔG2, ΔG3, ΔG4]/e − 1.23 (G1, G2, G3, G4 are the Gibbs free energy change of the OER-1, 2, 3, 4, respectively). This is independent of pH, and therefore applicable to water–alkali conditions.
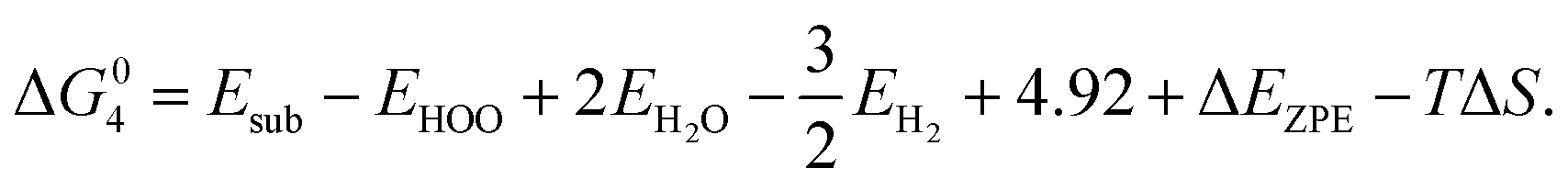
For HER in alkaline solutions, two steps are commonly involved: the first step is an initial discharge of water and the formation of hydrogen intermediates in the so-called Volmer step (H2O + e− + M = M*H + OH−, M represents active point on catalyst); the second step involves release of molecular hydrogen by either the electrochemical Heyrovsky step (H2O + e− + M*H = M + H2 + OH−) or the chemical Tafel recombination step (2M*H = 2M* + H2).23,24 The alkaline HER activity is evaluated by considering the Δ following steps:
| (1) G0 = GM + GH2O; |
| (2) G1 = GM–(H–OH)*; |
| (3) G2 = GM–H* + GOH−; |
and G3 = G0
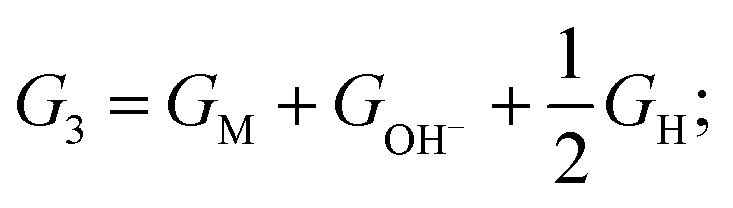
The zero point energy and entropic contributions are taken from ref. 23 and 24.
Results and discussion
Structure and morphology
The XRD pattern of the as-synthesized CoP NPs/CNSs in Fig. 1a fits well with orthorhombic (Pnma space group) CoP (JCPDS card: 29-0497) with the broad diffraction peak at 26° assigned to carbon sheet. The transmission electron microscopy (TEM) image (Fig. 1b) shows the morphology of the CoP NPs/CNSs hybrid. A large quantity of nearly spherical shape nanoparticles are embedded homogeneously in the thin nanosheets. The HAADF-STEM image (Fig. 1c) and corresponding elemental EDX mapping (Fig. 1f–g) confirm that the nearly spherical-shape nanoparticles are CoP nanoparticles and the main content of nanosheet around CoP nanoparticles is carbon with a trace amount of N and P element inherited from precursors.23 The EDS linear scan result (Fig. S2†) shows the weight ratio of phosphorus and cobalt is about 36:59 with calculating atomic ratio approaching to 1.16, indicating that the CoP nanoparticles are rich in phosphorus or carbon sheet doped with phosphorus. The HRTEM figure (Fig. 1h and S1†) with the corresponding Fourier Transform Diffraction pattern (Fig. 1i) reveals the CoP nanoparticles are of high crystal quality. Compared to the reported CoP nanostructures enclosed by single or low index facets, such as {200} or {111} or {110},11 the present CoP nanoparticles demonstrate nearly spherical shape with exposed crystal planes including (![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 1), (311), (101), (112), (201), (012), (210), (011). The nitrogen adsorption–desorption measurement demonstrates that the carbon sheets have the porous structural feature, consistent with previous report18 (Fig. S3†). The CoP NPs/CNSs have a large specific surface area of 234.25 m2 g−1 with a pore volume of 0.322 cm3 g−1 and a pore size of 1–3 nm. The porous structures would contribute to reactant diffusion during electrocatalytic processes.
1), (311), (101), (112), (201), (012), (210), (011). The nitrogen adsorption–desorption measurement demonstrates that the carbon sheets have the porous structural feature, consistent with previous report18 (Fig. S3†). The CoP NPs/CNSs have a large specific surface area of 234.25 m2 g−1 with a pore volume of 0.322 cm3 g−1 and a pore size of 1–3 nm. The porous structures would contribute to reactant diffusion during electrocatalytic processes.
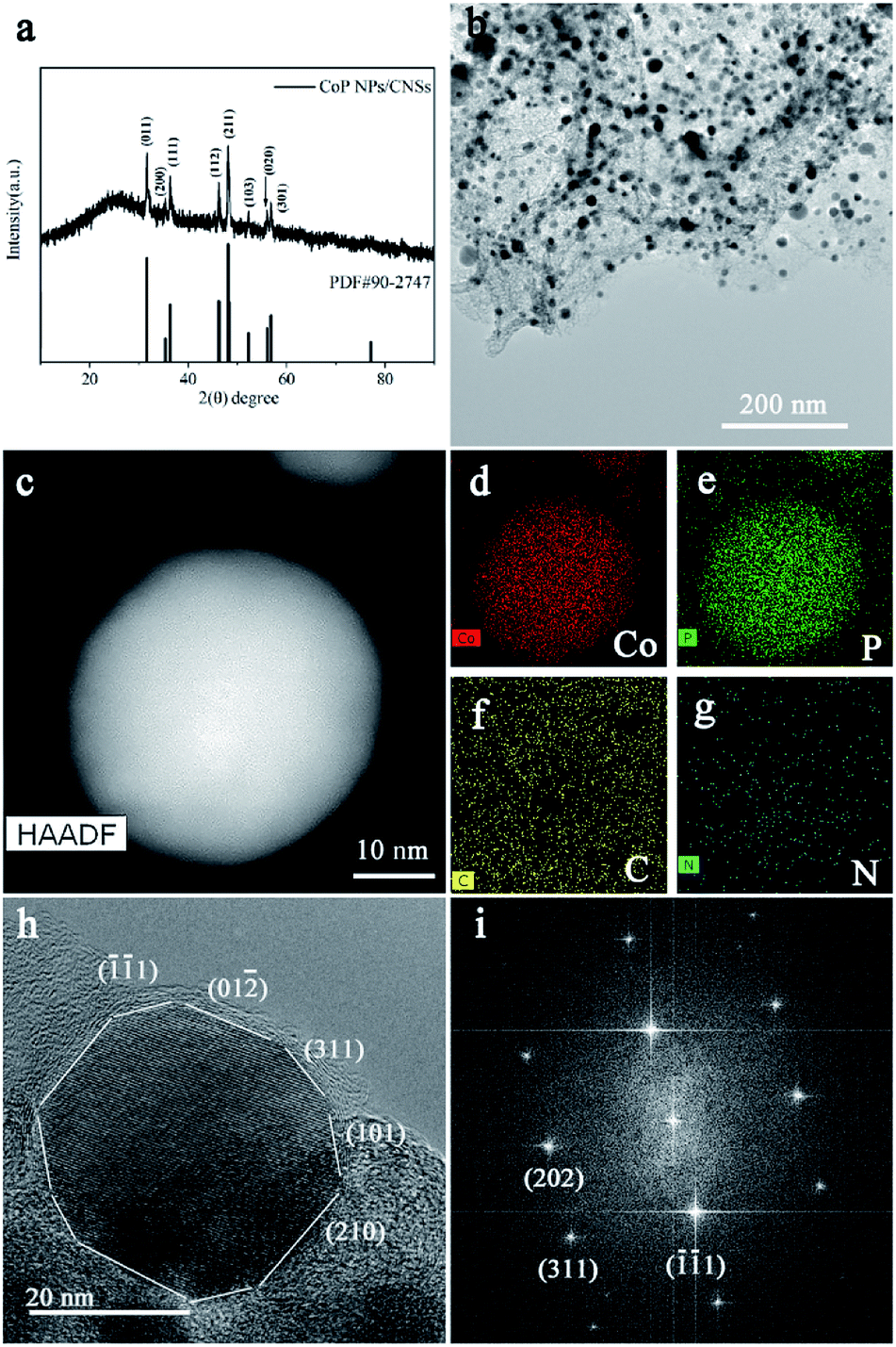 | ||
Fig. 1 Composition and structure of CoP NPs/CNSs. (a) XRD pattern of CoP NPs/CNSs. (b) TEM image of CoP NPs/CNSs. (c) STEM HAADF image of CoP NPs/CNSs and EDS mapping of (d) Co; (e) P; (f) C and (g) N. (h) HRTEM image along (1![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) ) zone axis and (i) the corresponding FFT image of (h). ) zone axis and (i) the corresponding FFT image of (h). | ||
Chemical compositions and bonding of these materials were further investigated by X-ray photoelectron spectroscopy (XPS) analysis (Fig. S4†), which confirms the coexistence of Co, O, C, N, and P. The Co 2p3/2 region displays two main peaks at 778.5 eV and 782.0 eV. And the Co 2p1/2 region contains two peaks at 794.8 eV and 801.2 eV (Fig. 2a). The prominent satellite peak located at 801.2 eV can be considered as the shakeup excitation of the high-spin Co2+ ions in the hybrid sheets.25,26 The high-resolution P 2p (Fig. 2b) has three separate peaks at 129.7 eV, 130.5 eV corresponding to P–Co bond, and 133.8 eV to P–C bond, respectively. It is worth to mention that the Co 2p3/2 peak of CoP shifted positively compared with metallic cobalt (from 778.1 eV to 779 eV) and P 2p3/2 peak shifted negatively compared with phosphorus (from 130.2 eV to 129.7 eV). The charge non-equilibrium of cobalt and phosphorus indicates that cobalt in CoP is positively charged while phosphorus is negatively charged, resulting in the electron density transfer from cobalt to phosphorus.27 The spectrum of C (Fig. 2c) can be fitted into C–C (284.7 eV), C–N (285.6 eV), C–O (285.6 eV) and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (288.5 eV).28–30 The high-resolution peaks of N 1s (Fig. 2d) can be deconvoluted into four peaks, standing for pyridinic N (398.8 eV), pyrrolic N (401.1 eV), graphitic N (402.2 eV), and oxidized N (404.8 eV).31,32 The content of each kind of nitrogen is shown in Table S1.† Aforementioned C–P bond and N peak confirm the successful doping of P and N into the carbon skeleton, such dual-doping could significantly promote electronic conductivity33,34 and contribute to catalytic efficiency.
O (288.5 eV).28–30 The high-resolution peaks of N 1s (Fig. 2d) can be deconvoluted into four peaks, standing for pyridinic N (398.8 eV), pyrrolic N (401.1 eV), graphitic N (402.2 eV), and oxidized N (404.8 eV).31,32 The content of each kind of nitrogen is shown in Table S1.† Aforementioned C–P bond and N peak confirm the successful doping of P and N into the carbon skeleton, such dual-doping could significantly promote electronic conductivity33,34 and contribute to catalytic efficiency.
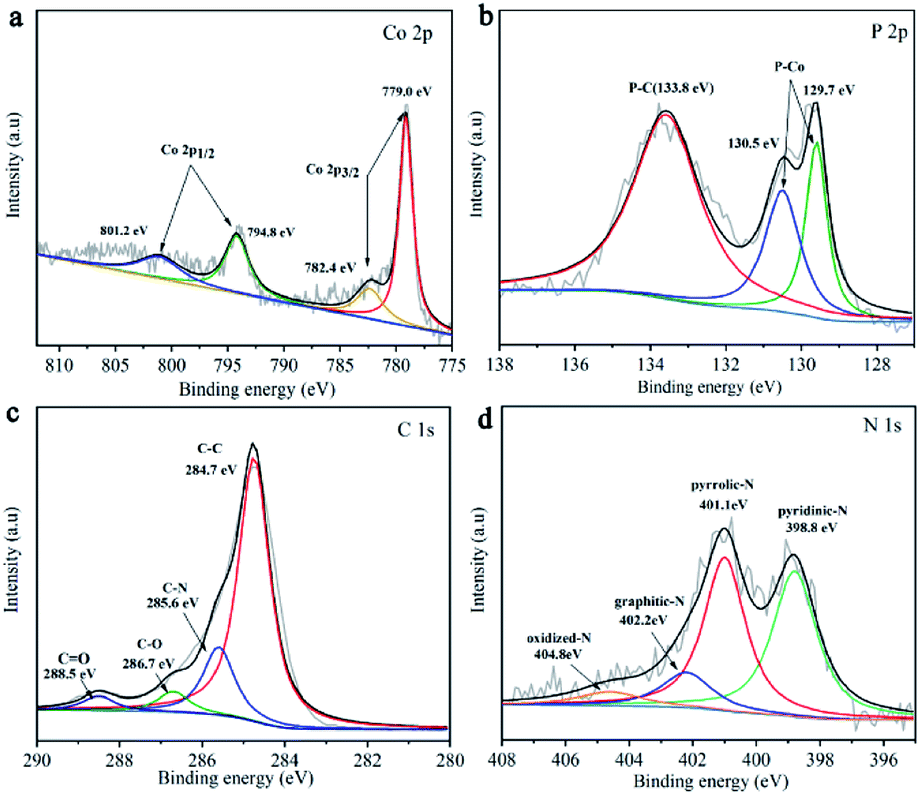 | ||
| Fig. 2 XPS of CoP NPs/CNSs. (a) High resolution Co 2p spectra. (b) High resolution P 2p spectra. (c) High resolution C 1s spectra. (d) High resolution N 1s spectra. | ||
Electrochemical activity and stability
The ORR electrocatalytic activities of as-synthesized CoP NPs/CNSs have been investigated. Previous studies35 reported that carbon-based nanomaterial, such as N, P-doped graphene, has shown moderate ORR activity in alkaline media (half-wave potential > 0.85 V). Pyrrolic-N plays an important role in ORR catalytic activity,36 which has been detected with a significant content ratio in carbon nanosheets of our samples as shown in Table S1.† Therefore, to accurately evaluate the electrochemical activity of CoP NPs/CNSs and reveal the intrinsic mechanisms, the P- and N-doped carbon nanosheets (CNSs) have been prepared by the same process for CoP NPs/CNSs, however, Co(CH3COO)2 has not been added to the reaction. The TEM image (Fig. S5†) shows the morphology of the pure CNSs with no CoP nanoparticles on it. The CV curve (Fig. S6a†) of CoP NPs/CNSs displays an apparent oxygen reduction peak, indicating a high intrinsic ORR activity. Notably, CoP NPs/CNSs exhibited a more positive onset potential (0.92 V) and peak potential (0.81 V) than those of CNSs and benchmark Pt/C in O2-saturated 0.1 M KOH. The linear sweep voltammogram (LSV) measurement (Fig. 3a) was performed on a rotating disk electrode (RDE) at 1600 rpm. CoP NPs/CNSs demonstrate a more positive half-wave potential than Pt/C (0.88 V vs. 0.91 V) with a comparable limiting current density to that of Pt/C (5.3 mA cm−2 vs. 5.6 mA cm−2). The CNSs exhibited relative poor activity, which indicated that it play minor role towards the ORR activity of CoP NPs/CNSs. Furthermore, to better reveal ORR kinetics behavior, LSV curves were measured at different rotation speeds (400–1600 rpm) as shown in Fig. 3b and S6b.† The limiting diffusion current density increases with increasing rotation speed, demonstrating that current is kinetically controlled. The Koutecky–Levich plots (j−1 vs. ω−1/2) of CoP NPs/CNSs and Pt/C (Fig. S6c and d†) obtained at 0.2–0.4 V show good linearity and near parallelism (or overlap), indicating first-order ORR kinetics with respect to O2. The slope of K–L plots were employed to measure the electron transfer number (n) per oxygen molecule during ORR. In the range of 0.3–0.4 V, the average n value of CoP NPs/CNSs and Pt/C was calculated to be 4.0, suggesting the 4e− path way. Furthermore, the Tafel slope (Fig. 4c) of CoP NPs/CNSs is slightly smaller than that of Pt/C (89.0 mV dec−1 vs. 89.3 mV dec−1), indicating better performance in this study. The durability test was carried out, showing that CoP NPs/CNSs exhibit less than 1% decay while Pt/C lost 6% activity after 10000 seconds continuous measurements (Fig. S7a†). The results suggest a promising long-term stability of CoP NPs/CNSs, which is obviously superior to the commercial Pt/C catalyst.
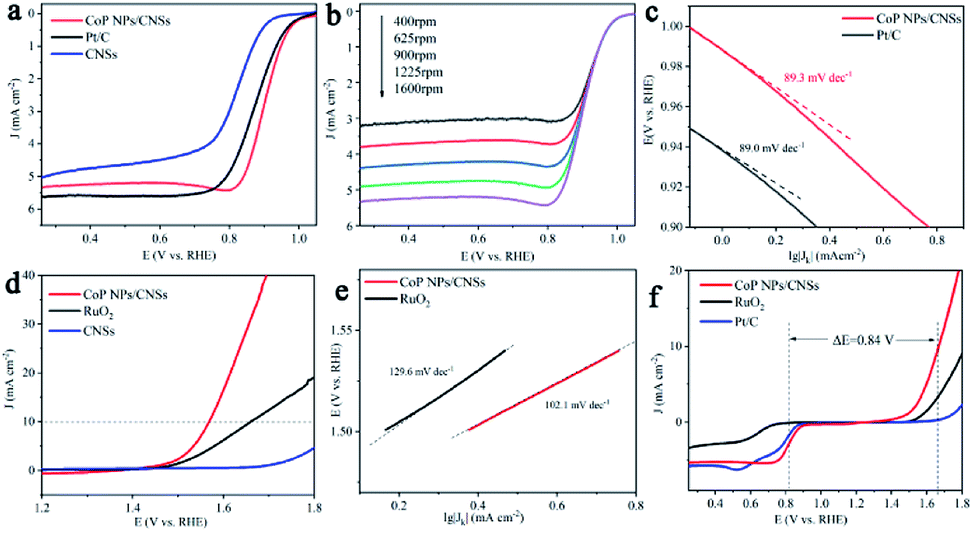 | ||
| Fig. 3 Electrocatalytic properties of the electrodes. (a–c) ORR performance: (a) LSV curves of CoP NPs/CNSs, CNSs and Pt/C catalyst in O2-saturated 0.1 M KOH solution, (b) LSV curves of CoP NPs/CNSs at different rotation rates in rpm, (c) Tafel plot of CoP NPs/CNSs and Pt/C. (d and e) OER performance: (d) LSV curves of CoP NPs/CNSs, CNSs, and RuO2 catalyst in N2-saturated 1 M KOH solution: (e) Tafel plot of CoP NPs/CNSs and RuO2, and (f) LSV curves of CoP NPs/CNSs, Pt/C and RuO2 catalyst in O2-saturated 0.1 M KOH solution. | ||
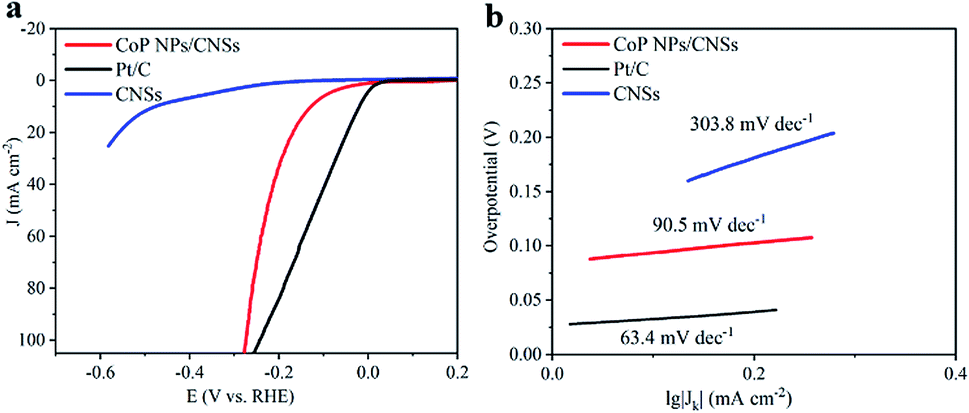 | ||
| Fig. 4 Electrocatalytic properties for HER. (a) LSV curves of CoP NPs/CNSs, CNSs and Pt/C catalyst in N2-saturated 1 M KOH solution, and (b) Tafel plot of CoP NPs/CNSs, CNSs and Pt/C. | ||
Our catalysts were also investigated for electrocatalytic OER in 1 M KOH solution. As shown in Fig. 3d, an overpotential of 0.34 V is required to obtain the current density of 10 mV cm−2 for CoP NPs/CNSs, which is 0.1 V smaller than that of RuO2. In contrast, CNSs demonstrate very poor OER activity, indicating that OER activity mainly results from CoP NPs. Meanwhile, Tafel slopes of CoP NPs/CNSs and RuO2 were calculated from LSV curves to estimate the reaction kinetics toward OER (Fig. 3e). Impressively, CoP NPs/CNSs shows a smaller value of Tafel slope (102.1 mV dec−1) than RuO2 (129.6 mV dec−1), indicating CoP NPs/CNSs had better electron transfer efficiency. The CoP NPs/CNSs hybrid demonstrated excellent stability with a slight increase (∼20 mV) in the onset potential after 1000 cycle of CV test (Fig. S7b†), which also indicate negligible structural changes during the testing time.
The overall oxygen activity of the CoP NPs/CNSs hybrid as a bifunctional catalyst could be evaluated (Fig. 3f) by the potential difference (ΔE) between ORR and OER, where ΔE = E10 − E1/2, E10 is the potential at OER current of 10 mA cm−2, and E1/2 is the half wave potential for ORR.37 However, the CoP NPs/CNSs hybrid showed the smallest ΔE of 0.84 V, and this value was markedly lower than the ΔE obtained using Pt/C and RuO2. These results signify that CoP NPs/CNSs are better reversible oxygen electrodes than Pt/C and RuO2. The results clearly indicate that the CoP NPs/CNSs hybrid catalyst is a promising low-cost and efficient catalyst for both ORR and OER.
The electrocatalytic HER activity of the CoP NPs/CNSs was examined by linear scan voltammetry in alkaline condition. For comparison, Pt/C and CNSs were also tested under the same conditions. As shown in the I–R corrected LSV polarization curves (Fig. 4a), Pt/C unquestionably exhibits the highest performance with a negligible overpotential. For the as-obtained CoP NPs/CNSs, the current densities of 10 mA cm−2 require an overpotential of 115 mV, while the bare CNSs are inactive towards HER. The Tafel slope of the CoP NPs/CNSs is calculated to be 92.4 mV dec−1 (Fig. 4b), indicating the Volmer–Heyrovsky mechanism with the Volmer step as the rate-determining step.38 Also, Fig. S8† exhibits the excellent stability of CoP NPs/CNSs through polarization curves (1st and 1000th) with negligible changes.
Based on the previous studies and our investigation as demonstrated in Table S2,† two conclusions can be reached: firstly, although CoP NPs has been widely reported to demonstrate unifunctional and bifunctional electrocatalytic activity for HER or ORR or OER, the samples in this study is a rare trifunctional electrocatalyst toward HER/ORR/OER; secondly, the ORR, OER and HER performances of CoP NPs/CNSs are much better than those of CNSs, which indicates the catalytic activities are mainly determined by CoP NPs. The catalytic reactions occur on the surface of catalysts, therefore the exposed crystal planes and atomic surface structure usually determine the catalytic activity. CoP nanoparticles in this work demonstrate a near-sphere morphology with many more exposed facets such as (1), (01), (311), (101) and (210) and so on, which is significantly different from most of the previously reported CoP nanostructures enclosed by single or few low-index facets such as {200},{011} or {111}. Therefore, to obtain a deeper insight into the mechanisms of the trifunctional electrocatalyst, density functional theory (DFT) calculations are performed to reveal the activities of different exposed planes for ORR, OER and HER, respectively.
DFT calculation
Table S3† elucidates the Gibbs free energy (ΔGi) of different planes during typical OER processes involving four elementary steps (HO*, O*, HOO* and O2). For most crystal planes, the Gibbs free energy for the formation of *OOH (ΔG3) turns out to be the highest value among all ΔGi for the four steps, and therefore regarded as the rate-determining step. However, as displayed in Fig. 5a, the potential limiting step for (101) is determined to be the O2 generation having a ΔG4 value of 1.794 eV and a corresponding calculated overpotential η of 0.564 V. The calculation results indicate that (101), (311), (012) and (210) facets contribute most to OER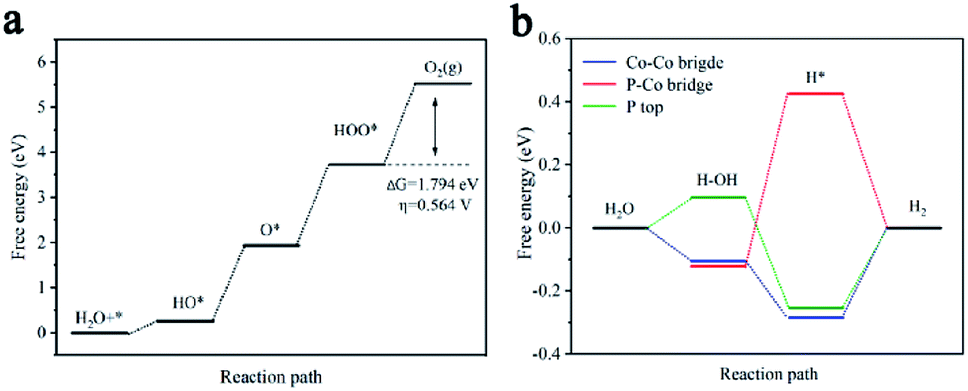 | ||
| Fig. 5 DFT calculations of CoP for OER and HER. (a) The corresponding free energy diagram for OER on (101) planes. (b) The corresponding free energy diagram for HER on Co–Co bridge, P–Co bridge, and P-top Co bridge of (311) planes. | ||
The ORR reactions are inversed from the OER reactions, but OER Gibbs free energy does not apply directly to ORR due to different pH value. Nevertheless, qualitative judgment of ORR Gibbs free energy could be obtained from ΔG1–4 of OER.
The HER pathway can be described as an initial formation of hydrogen intermediates (Volmer step) and then followed by hydrogen generation by either an electrochemical desorption step (Heyrovsky step) or a recombination step (Tafel step). The Volmer and Heyrovsky steps have different reacting species in acidic (H3O+) and alkaline (H2O/OH−) media, and the prior water dissociation to form H* intermediates for most catalysts in alkaline media are of much lower efficiency than in acid solutions.6 The acidic HER catalytic activity is determined by the optimal catalyst–hydrogen interactions with Gibbs free energy for hydrogen adsorption (ΔG0H) close to thermoneutral. While the optimal alkaline HER needs a balance between catalyst–hydrogen and catalyst–hydroxyl interactions, and correspondingly, the free energy of water dissociation (ΔGH2O = G1 − G0 = G1) for the Volmer step and the binding free energy of adsorbed H intermediates (ΔGH* = G2 − G3 = G2) for the Tafel step or Heyrovsky step, are two significant descriptors for the alkaline HER process. ΔGH2O and ΔGH* values were calculated at three different sites containing Co–Co bridge (Co br), P–Co bridge (P–Co br), and P-top as displayed in Table S4.† For example, the ΔGH* on the Co br sites of (101), P-top sites of (112), and P–Co br of (012) are 0.051, 0.029 and 0.054 eV, respectively, which are close to the optimal value (ΔGH* = 0 eV) and indicating that the adsorption of H on the these sites are more active than other sites and planes. However, the ΔGH2O onto the Co br sites of (101), P-top sites of (112), and P–Co br of (012) are 0.375, 0.498 and 0.658 eV, respectively. The relative high values of ΔGH2O indicate a significant energy barrier hindering H2O dissociation into H* intermediates and will cause HER kinetic process sluggish. In contrast, ΔGH2O onto Co br, P-top, and P–Co br sites of (311) are −0.105, 0.096, and −0.122 eV, respectively. This means that on the (311) facets, the catalyst can effectively promote cleavage of H–OH bonds to produce H* intermediates. The ΔGH* onto Co br, P-top, and P–Co br sites of (311) are −0.284, −0.253 and 0.425 eV, respectively. These appropriate ΔGH* values combining relative low ΔGH2O values confirm that all the sites of (311) are more active in alkaline condition than Co br sites of (101), P-top sites of (112), and Co–P br sites of (012). The DFT calculation results also suggest Co br on (111), (112) and Co–P br on (210) contribute to the highly reactive HER properties of CoP NPs/CNSs.
It should be emphasized that the synergistic effects originating from the different active sites (of the same facets) and different crystalline facets for the catalytic hydrogen evolution has been neglected in the DFT calculations. However, it does exist even considering qualitative factors. As shown in Table S4,† the significantly negative ΔGH2O values indicate Co br and P-top sites of CoP (201) provide active sites for hydroxyl adsorption and the binding energy are very strong, which will contribute to cleaving HO–H bonds. Therefore, these facets can act as a water dissociation promoter to produce hydrogen intermediates, which then adsorb on nearby CoP catalyst facets and sites.
In general, according to the DFT calculations, it can be reasonably deduced that the highly efficient trifunctional electrocatalytic activity of CoP NPs/CNSs is a collective effect of different crystal planes, that is, some planes dominate the OER process while some others contribute to HER process.
Conclusions
In summary, CoP NPs/CNSs hybrids are prepared via a one-step self-templating route. CoP nanoparticles own nearly spherical shape with many more exposed crystal planes than previously reported CoP nanostructures enclosed by single or few low-index facets. Electrochemical measurements demonstrate that the CoP NPs/CNSs hybrids present highly efficient trifunctional catalytic activity towards ORR, OER and HER in alkaline media. Impressively, the electrocatalytic activities of the hybrids towards ORR and OER are even superior to that of the precious metal benchmark catalysts. Comparative study and DFT calculations show the excellent trifunctional catalytic activity to be a collective effect from the different planes to ORR, OER or HER, respectively. This work is expected to inspire the design of multifunctional and high-efficiency electrocatalysts via selectively exposing specific crystal planes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Prof. S. T. Lee (FUNSOM, Soochow University) and Prof. Liqiang Mai for the critical reading. We would like to thank for the financial support from the Independent Innovation Foundation of Wuhan University of Technology, China (Grant No. 183101007, 191059001), foundation of Ocean University of China (Grant No. 3002000-861701013151), the Fundamental Research Funds for the Central Universities (WUT: 2019III172) and the Starting Research Fund from the Wuhan University of Technology.Notes and references
- W. Xia, A. Mahmood, Z. B. Liang, R. Q. Zou and S. J. Guo, Angew. Chem., Int. Ed., 2016, 55, 2650 CrossRef CAS PubMed.
- Y. J. Wang, D. P. Wilkinson and J. Zhang, Chem. Rev., 2011, 111, 7625 CrossRef CAS PubMed.
- P. Liu and J. A. Rodriguez, J. Am. Chem. Soc., 2005, 127, 14871 CrossRef CAS PubMed.
- E. J. Popczun, J. R. McKone, C. G. Read, A. J. Biacchi, A. M. Wiltrout, N. S. Lewis and R. E. Schaak, J. Am. Chem. Soc., 2013, 135, 9267 CrossRef CAS PubMed.
- C. Zhang, Y. Huang, Y. F. Yu, J. F. Zhang, S. F. i. Zhuo and B. Zhang, Chem. Sci., 2017, 8, 2769 RSC.
- H. Li, X. L. Zhao, H. L. li Liu, S. Chen, X. F. Yang, C. X. Lv, H. W. Zhang, X. L. She and D. J. Yang, Small, 2018, 28, 1668 Search PubMed.
- W. Li, X. F. Gao, D. H. Xiong, F. Xia, J. Liu, W. G. Song, J. Y. Xu, S. M. Thalluri, M. F. Cerqueira, X. l. Fue and L. F. Liu, Chem. Sci., 2017, 8, 2952 RSC.
- H. C. Yang, Y. J. Zhang, F. Hu and Q. B. Wang, Nano Lett., 2015, 1511, 7616 CrossRef PubMed.
- H. Li, Q. Li, P. Wen, T. B. Williams, S. Adhikari, C. Dun, C. Lu, D. Itanze, L. Jiang, D. L. Carroll, G. L. Donati, P. M. Lundin, Y. Qiu and S. M. Geyer, Adv. Mater., 2018, 30, 1705796 CrossRef PubMed.
- H. Tang, K. P. Dou, C. C. Kaun, Q. Kuang and S. H. Yang, J. Mater. Chem. A, 2014, 2, 360 RSC.
- Y. Pan, Y. Lin, Y. Chen, Y. Liu and C. Liu, J. Mater. Chem. A, 2016, 4, 4745 RSC.
- Y. X. Lin, L. Yang, Y. K. Zhang, H. L. Jiang, Z. J. Xiao, C. Q. Wu, G. B. Zhang, J. Jiang and L. Song, Adv. Energy Mater., 2018, 1, 1703623 CrossRef.
- H. T. Du, R. M. Kong, X. X. Guo, F. L. Qu and J. H. Li, Nanoscale, 2018, 10, 21617 RSC.
- S. P. Li, G. Zhang, X. M. Tu and J. H. Li, ChemElectroChem, 2018, 5, 701 CrossRef CAS.
- C. Wu, Y. J. Yang, D. Dong, Y. H. Zhang and J. H. Li, Small, 2017, 13, 2873 Search PubMed.
- M. Sun, H. J. Liu, J. H. Qu and J. H. Li, Adv. Energy Mater., 2016, 6, 1600087 CrossRef.
- G. Zhang, G. C. Wang, Y. Liu, H. J. Liu, J. H. Qu and J. H. Li, J. Am. Chem. Soc., 2016, 138, 14686 CrossRef CAS PubMed.
- J. Bai, B. J. Xi, H. Z. Mao, Y. Lin, X. J. Ma, J. K. Feng and S. L. Xiong, Adv. Mater., 2018, 30, 1802310 CrossRef PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- D. Voiry, H. Yamaguchi, J. Li, R. Silva, D. C. B. Alves, T. Fujita, M. Chen, T. Asefa, V. B. Shenoy, G. Eda and M. Chhowalla, Nat. Mater., 2013, 12, 850 CrossRef CAS PubMed.
- M. Li, L. Zhang, Q. Xu, J. Niu and Z. Xia, J. Catal., 2014, 314, 66 CrossRef CAS.
- C. Guan, H. J. Wu, W. Xiao, X. M. Liu, W. J. Zang, H. Zhang, J. Ding, Y. P. Feng, S. J. Pennycook and J. Wang, Nano Energy, 2018, 48, 73 CrossRef.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886 CrossRef.
- B. Zhang, J. Liu, J. S. Wang, Y. J. Ruan, X. Ji, K. Xu, C. Chen, H. Z. Wan, L. Miao and J. J. Jiang, Nano Energy, 2017, 37, 74 CrossRef CAS.
- X. Yang, A. Y. Lu, Y. Zhu, M. N. Hedhili, S. Min, K. W. Huang, Y. Han and L. J. Li, Nano Energy, 2015, 15, 634 CrossRef CAS.
- Y. P. Zhu, Y. P. Liu, T. Z. Ren and Z. Y. Yuan, Adv. Funct. Mater., 2015, 25, 7337 CrossRef CAS.
- Q. Liu, J. Q. Tian, W. Cui, P. Jiang, N. Y. Cheng, A. M. Asiri and X. P. Sun, Angew. Chem., Int. Ed., 2014, 126, 6828 CrossRef.
- Z. Wang, Y. Dong, H. Li, Z. Zhao, H. B. Wu, C. Hao, S. Liu, J. Qiu and X. W. Lou, Nat. Commun., 2014, 5, 5002 CrossRef CAS PubMed.
- M. R. Gao, X. Cao, Q. Gao, Y. Xu, Y. Zheng, J. Jiang and S. H. Yu, ACS Nano, 2014, 8, 3970 CrossRef CAS PubMed.
- H. Hu, Z. Zhao, W. Wan, Y. Gogotsi and J. Qiu, Adv. Mater., 2013, 25, 2219 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao, L. H. Li, T. Xing, Y. Chen, M. Jaroniec and S. Z. Qiao, ACS Nano, 2014, 8, 5290 CrossRef CAS PubMed.
- J. J. Duan, S. Chen, B. A. Chambers, G. G. Andersson and S. Z. Qiao, Adv. Mater., 2015, 27, 4234 CrossRef CAS PubMed.
- C. H. Choi, S. H. Park and S. I. Woo, ACS Nano, 2012, 6, 7084 CrossRef CAS PubMed.
- X. L. Ma, G. Q. Ning, C. L. Qi, C. G. Xu and J. S. Gao, ACS Appl. Mater. Interfaces, 2014, 6, 14415 CrossRef CAS PubMed.
- J. Zhang and L. Dai, Angew. Chem., Int. Ed., 2016, 55, 13296 CrossRef CAS PubMed.
- X. L. Yang, W. J. Zou, Y. H. Su, Y. H. Zhu, H. L. Jiang, J. H. Shen and C. Z. Li, J. Power Sources, 2014, 266, 36 CrossRef CAS.
- S. Chen, J. J. Duan, Z. Yao, X. M. Chen, X. W. Du, M. Jaroniec and S. Z. Qiao, Energy Storage Materials, 2015, 1, 17 CrossRef.
- Z. P. Huang, Z. Z. Chen, Z. B. Chen, C. C. Lv, M. G. Humphrey and C. Zhang, Nano Energy, 2014, 9, 373 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra07334e |
| This journal is © The Royal Society of Chemistry 2019 |