
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceToward realistic computer modeling of paraffin-based composite materials: critical assessment of atomic-scale models of paraffins†
Artyom D. Glova a,
Igor V. Volgina,
Victor M. Nazarycheva,
Sergey V. Larina,
Sergey V. Lyulinab and
Andrey A. Gurtovenko*ab
a,
Igor V. Volgina,
Victor M. Nazarycheva,
Sergey V. Larina,
Sergey V. Lyulinab and
Andrey A. Gurtovenko*ab
aInstitute of Macromolecular Compounds, Russian Academy of Sciences, Bolshoi Prospect V.O. 31, St. Petersburg, 199004, Russia. E-mail: a.gurtovenko@gmail.com
bFaculty of Physics, St. Petersburg State University, Ulyanovskaya Street 3, Petrodvorets, St. Petersburg, 198504, Russia
First published on 27th November 2019
Abstract
Paraffin-based composites represent a promising class of materials with numerous practical applications such as e.g. heat storage. Computer modeling of these complex multicomponent systems requires a proper theoretical description of both the n-alkane matrix and the non-alkane filler molecules. The latter can be modeled with the use of a state-of-the-art general-purpose force field such as GAFF, CHARMM, OPLS-AA and GROMOS, while the paraffin matrix is traditionally described in the frame of relatively old, alkane-specific force fields (TraPPE, NERD, and PYS). In this paper we link these two types of models and evaluate the performance of several general-purpose force fields in computer modeling of paraffin by their systematic comparison with earlier alkane-specific models as well as with experimental data. To this end, we have performed molecular dynamics simulations of n-eicosane bulk samples with the use of 10 different force fields: TraPPE, NERD, PYS, OPLS-UA, GROMOS, GAFF, GAFF2, OPLS-AA, L-OPLS-AA, and CHARMM36. For each force field we calculated several thermal, structural and dynamic characteristics of n-eicosane over a wide temperature range. Overall, our findings show that the general-purpose force fields such as CHARMM36, L-OPLS-AA and GAFF/GAFF2 are able to provide a realistic description of n-eicosane samples. While alkane-specific models outperform most general-purpose force fields as far as the temperature dependence of mass density, the coefficient of volumetric thermal expansion in the liquid state, and the crystallization temperature are concerned, L-OPLS-AA, CHARMM36 and GAFF2 force fields provide a better match with experiment for the shear viscosity and the diffusion coefficient in melt. Furthermore, we show that most general-purpose force fields are able to reproduce qualitatively the experimental triclinic crystal structure of n-eicosane at low temperatures.
1. Introduction
Paraffins, being an important product of refinement of crude oil, represent a promising class of compounds with numerous practical applications.1 The use of paraffins is especially attractive since they are cheap and abundant. One of the most important applications of paraffins is closely related to their ability to adsorb and release a large amount of heat upon melting and crystallization.2–4 The high latent heat of fusion makes paraffins an inexpensive heat storage material with great potential. Nevertheless, the practical use of pure paraffins in heat storage devices is rather limited by their low thermal conductivity, which renders it difficult to achieve a high-rate transfer of heat during melting/crystallization cycles.3 This issue can be solved e.g. by introducing nanofillers with high thermal conductivity (such as graphene or carbon nanotubes) into the paraffin matrix.5–8 In other words, to be efficient in heat storage, pure paraffins should be replaced with paraffin-based composite materials.Computer modeling has become nowadays a universal tool for gaining a molecular-level insight into the structure and dynamics of molecular systems; such insight often is not accessible by any other experimental techniques. Paraffins, being a mixture of relatively short alkane chains (15–40 monomer units), are not an exception in this respect: alkane chains have been studied by computer simulations for decades. In fact, first simulation studies of linear polymers focused mostly on n-alkanes, owing to their simple chemical structure. Starting from pioneering studies by Williams9 and by Ryckaert and Bellemans,10,11 a dozen different force fields of n-alkanes were developed in 1980–1990s.12–29 Most of these force fields treated nonpolar hydrogen atoms implicitly, i.e. each methylene (CH2) or methyl (CH3) group was described by a single (united) atom, leading to a so-called united-atom representation.12 Since n-alkanes do not have polar hydrogens, such an approach is very effective in terms of required computational resources, as it allows one to reduce the total number of atoms in n-alkane systems by a factor of three. However, the corresponding gain in performance apparently comes at a certain price, and a precise relationship between all-atom and united-atom models of n-alkanes is still under debate.30,31 In particular, a series of seminal studies by Ryckaert, McDonald and Klein32–34 showed that hydrogen atoms should explicitly be included into the computational model to reproduce the low-temperature crystalline phase of n-tricosane and n-nonadecane.
The united-atom force fields that were developed for n-alkanes decades ago have been extensively validated against experimental data; they generally provide a realistic description as far as n-alkane systems themselves are concerned. However, when it comes to complex multicomponent systems such as paraffin-based composites, n-alkanes represent just one component of a heterogeneous system. In this case the use of alkane-specific force fields can be problematic since the theoretical description of the rest of the system (e.g. nanoparticles, filler molecules, etc) has to be done on the same footing as the one for n-alkanes. On the other hand, several state-of-the-art general-purpose force fields (such as AMBER,35 CHARMM,36 GROMOS37,38 or OPLS-AA39,40) are currently available and can routinely be used for modeling a wide range of molecular systems, including n-alkanes. And here one faces an obvious dilemma: either we develop a model for non-alkane components, which would be compatible with relatively old, alkane-specific force fields, or we employ a modern general-purpose force field for both alkane and non-alkane parts of the system. The latter is a much better (and sometimes the only possible) solution because most alkane-specific models are united-atom; this level of resolution may be insufficient for certain classes of molecules such e.g. nucleic acids and polysaccharides.41
The main goal of this work is to link alkane-specific models with the state-of-the-art general-purpose force fields in computer modeling of paraffins. Once it has been done, one can make a rational choice of an appropriate computational model for more complex systems such as paraffin-based composites. To the best of our knowledge, no systematic comparison between alkane-specific and general-purpose force fields has been done by far. Most relevant studies have focused mainly on a relative performance of alkane-specific united-atom models. For example, Muller et al.42 explored thermodynamic properties of various n-alkanes (including n-C10, n-C20, n-C60, and n-C100) for united-atom force-fields SKS, NERD, and TraPPE. Other groups reported several studies on longer n-alkanes (polyethylene). Ramos et al.43 compared systematically a wide range of properties of n-C192 in united-atom PYS and TraPPE force-fields. Hagita et al.44 examined a single polyethylene chain during quenching in various united-atom force fields and highlighted the differences in resulting structures. A good overview of performance of united-atom force fields in simulations of polyethylene can be found in a recent review paper.45
To meet a lack of a systematic comparison between alkane-specific and general-purpose force fields, we selected 10 most popular force fields and assessed carefully their performance in computer simulations of paraffins.
All force fields considered in this study can be divided in several groups. First, these are united-atom force fields developed primarily for modeling n-alkanes: NERD,19–21 TraPPE,16–18 and PYS.22–25 The NERD force field was proposed by Nath, Escobedo and de Pablo to describe the properties for relatively long n-alkanes.19 The authors demonstrated that their model was able to reproduce experimental data for the phase equilibrium and second virial coefficient for n-alkanes. The transferable potential for phase equilibria (TraPPE), which was introduced by Martin and Siepmann, was based on calculations of vapor–liquid coexistence curves.16 It was established that the TraPPE force field could reproduce correctly the densities of n-alkane melts over a wide range of temperatures. We note that an all-atom version of the TraPPE force-field for n-alkanes is also available.46 Furthermore, the TraPPE force-field was recently extended to naphthenic, aromatic, and thiophenic compounds.47 Since any classification of force fields is somewhat arbitrary and since we focus here on the united-atom TraPPE force-field that was originally developed for alkanes, we chose to consider the TraPPE force-field as alkane-specific one. Finally, the PYS model by Paul, Yoon and Smith was initially developed for polyethylene by calibrating the polyethylene model against experimental data and quantum chemical calculations.22 More recently, the PYS force field was applied to study crystal growth rates in bulk n-eicosane systems.24
The second group of models comprises the state-of-the-art general-purpose force fields: GAFF48–50 (and its most recent modification GAFF2 (ref. 51 and 52)), CHARMM,53,54 and GROMOS;37,38 first three are all-atom force fields, while GROMOS treats nonpolar hydrogens implicitly. GAFF (general Amber force field) is an all-atom general-purpose force field developed for a wide range of organic molecules.48–50 The GAFF force field is fully compatible with the AMBER family of force fields,35 extending thereby the AMBER force field to the classes of compounds beyond biomolecules (such as e.g. drug-like molecules). GAFF2 is an improved version of the GAFF force field, whose development is still ongoing.51,52 CHARMM is another family of all-atom general-purpose force fields that covers a full range of biomolecules. As far as n-alkanes are concerned, the most relevant CHARMM force field is CHARMM36.53,54 This force field was developed to model phospholipid molecules and was proved to reproduce experimental properties of lipid membranes in computer simulations.53 Since acyl tails of lipid molecules are hydrocarbon chains of fatty acid residues, one could expect that the CHARMM36 force field is also relevant for the description of n-alkane systems. A united-atom general-purpose force field GROMOS was initially developed for biomolecules (proteins and lipids); it includes a parameter set (GROMOS96 45A3) that was specifically calibrated for aliphatic carbons.37 It was shown37,38 that experimental liquid densities, heat of vaporization and free energy of hydration of n-alkane were reasonably captured by the GROMOS96 45A3 parameter set (in the following this parameter set will be referred to as the GROMOS force field).
The OPLS (Optimized Potentials for Liquid Simulations) family of force fields can be put between the above two groups as it includes both united-atom (OPLS-UA) and all-atom (OPLS-AA and L-OPLS-AA) versions. The united-atom force field OPLS-UA is one of the earliest models built up to study n-alkanes;12 its development goes back to 1984. The all-atom OPLS version (OPLS-AA) was released 12 years later39 and aimed at modeling organic liquids. The OPLS-AA force field was shown to be reasonably good in reproducing the thermodynamic properties of relatively short alkanes (up to 6 carbon atoms).55 However, it turned out that for longer alkane chains the OPLS-AA force field had to be recalibrated. This was done in ref. 56 by Böckmann et al., who introduced a new force field L-OPLS-AA optimized for relatively long n-alkanes (up to n-pentadecane).
For all considered force fields we studied a wide range of thermal, structural and dynamic properties of bulk paraffin samples. These characteristics were critically compared with each other as well as with available experimental data and the conclusions were made regarding the advantages and pitfalls of the considered force fields in computer modeling of paraffins.
2. Methods
2.1. Force fields
We have performed molecular dynamics simulations of paraffins in bulk. Since our primary interest is in the heat storage materials, here we focused on relatively short n-alkane chains of n-eicosane (C20H42). This n-alkane is considered as one of the most promising paraffins for the use in domestic heat storage devices, as its melting point (36.7 °C) is close to the physiological temperature.4To explore the effect of a force field on the thermal, structural and dynamic properties of n-eicosane, we considered 10 different force fields. All-atom models included GAFF,48–50 GAFF2,51,52 OPLS-AA,39,40,55 L-OPLS-AA,56 and CHARMM36 (ref. 53 and 54) force fields, while OPLS-UA,12,13 NERD,19–21 TraPPE,16–18 PYS,22–25 and GROMOS96 45A3 (ref. 37 and 38) were chosen as united-atom force fields. An eicosane chain consisted of either 62 atoms (all-atom models) or 20 atoms (united-atom models). A complete description of 10 force fields used for n-eicosane is presented in ESI.†
2.2. Paraffin sample preparation
The initial configurations of bulk paraffin samples were prepared as follows. We started with an all-atom description of paraffin and used the GAFF force field for n-eicosane (C20H42). With the use of the GROMACS routine gmx insert-molecules57 1000 n-eicosane chains were put randomly in a cubic simulation box with a box length of 12 nm, see Fig. 1(a). The temperature T was set to 450 K, which was well above the melting temperature of n-eicosane (309.7 K),4 so that the resulting sample would be in the liquid state. The system was then compressed with a high pressure (P = 50 bar) for 5 ns in order to reach the experimental values of density. After the compression step, the pressure was reduced to 1 bar and the eicosane sample was equilibrated for 200 ns at T = 450 K (the equilibration was monitored via the time evolution of the sample density), see Fig. 1(b). We repeated this procedure three times with different initial conditions. The three resulting configurations were used as starting structures in cooling-rate simulations for each all-atom force field. This way we assured that the simulations with different force fields have the same starting structures. The initial configurations for simulations of united-atom systems were prepared on the basis of all-atom samples by removing the coordinates of hydrogen atoms. Again, the simulations with different united-atom force fields utilized the same three starting configurations. All in all, the overall number of atoms in the systems amounted to 62![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (all-atom force fields) and 20000 (united-atom force-fields). More detail on the polymer sample preparation protocol can be found elsewhere.58
000 (all-atom force fields) and 20000 (united-atom force-fields). More detail on the polymer sample preparation protocol can be found elsewhere.58
 | ||
| Fig. 1 Snapshots of a simulation box with 1000 n-eicosane molecules (a) after the molecules were added in the box and (b) after compression and equilibration of the eicosane system. | ||
2.3. Simulation details
All molecular dynamics simulations were performed with the use of the GROMACS package (v. 5).57 The time step was set to 2 fs. Periodic boundary conditions were applied in all three dimensions. In all-atom simulations the bonds between carbon and hydrogen atoms were constrained with the P-LINCS algorithm,59 while in united-atom simulations this algorithm was applied to constrain the C–C bonds in the case of TraPPE,16–18 OPLS-UA,12,13 and GROMOS37,38 force fields following the original parameterization of these models. To handle electrostatic interactions in all-atom simulations we employed the particle-mesh Ewald method.60 In united-atom models atoms have zero partial charges and do not interact with electrostatic interactions.The van der Waals interactions were cut off at a force field-specific distance rvdw. The value of rvdw was defined by the original parameterization of each force field and was set to 0.9 nm (GAFF and GAFF2 (ref. 48,49,51 and 52)), 1.2 nm (CHARMM36 (ref. 53) and PYS22–25), 1.3 nm (OPLS-AA39,40,55 and L-OPLS-AA56), 1.38 nm (NERD19–21), 1.4 nm (GROMOS37,38 and TraPPE16–18), and 1.5 nm (OPLS-UA12,13). Furthermore, in CHARMM36, OPLS-AA and L-OPLS-AA force fields the van der Waals interactions were treated with the use of a switch function. The CHARMM36 force field uses a function that smoothly switches the forces to zero between 1.0 nm and rvdw = 1.2 nm.53 In the case of OPLS-AA and it's modification, L-OPLS-AA, the interaction energy is switched to zero between 1.1 nm and rvdw = 1.3 nm.39 In line with original parameterization of force fields considered, beyond the cutoff distance either the correction in the interaction energy (GAFF, GAFF2, NERD, and OPLS-UA force fields) or the correction in both the energy and pressure (OPLS-AA, L-OPLS-AA, and TraPPE force fields) were applied. As for the electrostatic interactions in all-atom models, the cut-off radius in the direct space sum was set to equal rvdw. The neighbor list was updated every 5 and 10 steps for united-atom and all-atom force fields, respectively. To update the neighbor lists we used the Verlet buffer cutoff scheme. The Verlet scheme implies buffering, so that the interaction energy is smoothly shifted to zero at the cut-off distance for the short-range neighbor list.57 It should be emphasized that most force fields developed in 1990s were parameterized with the use of unbuffered cutoff schemes such as a so-called group scheme. Overall, the Verlet scheme is much more efficient as compared to the group scheme; it is also crucial that the Verlet scheme has a GPU support, while the group scheme does not. Because of that, the group scheme is deprecated in some of popular simulation packages such as GROMACS. Several studies reported that the outcome of the simulations performed with the two types of cutoff schemes was rather similar.61,62 Therefore, we decided to employ the Verlet buffer cutoff scheme for every force field considered.
The cooling-rate simulations were performed in the NPT ensemble at P = 1 bar and at the temperature T that was varied for most systems in the range from 450 K to 250 K (the cooling for selected systems was extended beyond 250 K, see below). Pressure was controlled isotropically with the use of the Parrinello–Rahman barostat63 for both the equilibration phase and cooling-rate simulations. For most systems the time constant τp was set to 5 ps except OPLS-AA/L-OPLS-AA (τp = 4 ps). For OPLS-AA and L-OPLS-AA the temperature was kept constant with the use of the velocity-rescaling thermostat,64 while the Nosé–Hoover thermostat65,66 was employed for the rest of the systems. In most cases the choice of the thermostat was dictated by the original parameterization of a specific force field. Exceptions include OPLS-UA, TraPPE, and NERD force fields which were originally developed using Monte Carlo simulations. For the sake of consistency, the Nosé–Hoover thermostat was employed for these three systems.44,45 The time constant τT was set to 1 ps with the exceptions of OPLS-AA/L-OPLS-AA systems, for which τT was 0.1 ps.
In order to explore the crystallization processes in paraffin bulk samples, we cooled down the eicosane samples in a stepwise manner. Starting from a relatively high temperature (T = 450 K), for most systems the sample was cooled down to T = 250 K with steps of 10 K (all in all, 21 steps was considered). To make definitive conclusions regarding the crystallization process, the cooling of GROMOS, PYS and L-OPLS-AA samples was extended to T = 200 K, while the NERD system was studied in the temperature range 240–450 K. On every step the paraffin system was simulated for 100 ns, so that the cooling rate was 0.1 K ns−1 or 0.6 × 1010 K min−1. It has to be emphasized that the experimental cooling rates are much smaller: 0.5–1 K min−1, i.e. the cooling in experiments is ten orders of magnitude slower than in simulations.67 Such a situation is usual for atomic-scale computer simulations and occurs due to severe limitations in time and length scales when the models of high resolution are employed. The cooling rate value employed in our computational study is rather close to what can currently be achieved in atomic-scale simulations:68 a single cooling simulation in the temperature range 250–450 K with the cooling rate of 0.6 × 1010 K min−1 required 2.1 microseconds of simulated time. Furthermore, the cooling was carried out independently for three starting configurations of each system, amounting to 30 cooling-rate simulations.
To calculate the shear viscosity of n-eicosane in the liquid state at T = 450 K, additional simulations in the NVT ensemble were carried out. Following the approach of ref. 69, we performed 99 short independent simulations (5 ns each) for every force field considered. All in all, the total simulation time in our study exceeded 70 microseconds. Table S1† summarizes computational performance of all force fields studied, see ESI.†
3. Results and discussion
3.1. Thermal properties of bulk n-eicosane samples
Since paraffins represent a promising class of materials for heat storage, we start with thermal properties of bulk n-eicosane samples. To this end, here we present several structural characteristics of eicosane as a function of temperature.Eicosane is a relatively short-chain n-alkane, so that eicosane melt demonstrates an abrupt transition into the crystalline phase upon cooling.67,70 To study crystallization of eicosane bulk samples, we performed a series of cooling-rate simulations. For most systems the eicosane samples were cooled down in a stepwise manner from 450 K to 250 K. Wider temperature ranges were employed for GROMOS, PYS, L-OPLS-AA, and NERD systems, see Methods. The temperature steps were set to 10 K; at each temperature the sample was simulated for 100 ns. The corresponding temperature dependence of the mass density of eicosane samples is shown in Fig. 2 for all 10 force fields considered. Note that for each force field the eicosane density was averaged over 3 independent simulation runs with different initial configurations, so that every curve in Fig. 2 accumulates data from a simulation which is 6.3–7.8 μs long. For the sake of comparison, in Fig. 2 we also showed experimental data available for eicosane bulk samples.67,70,71 Furthermore, for clarity's sake, in Fig. S1† we present the mass density data as a ratio between computational and experimental67 values of density.
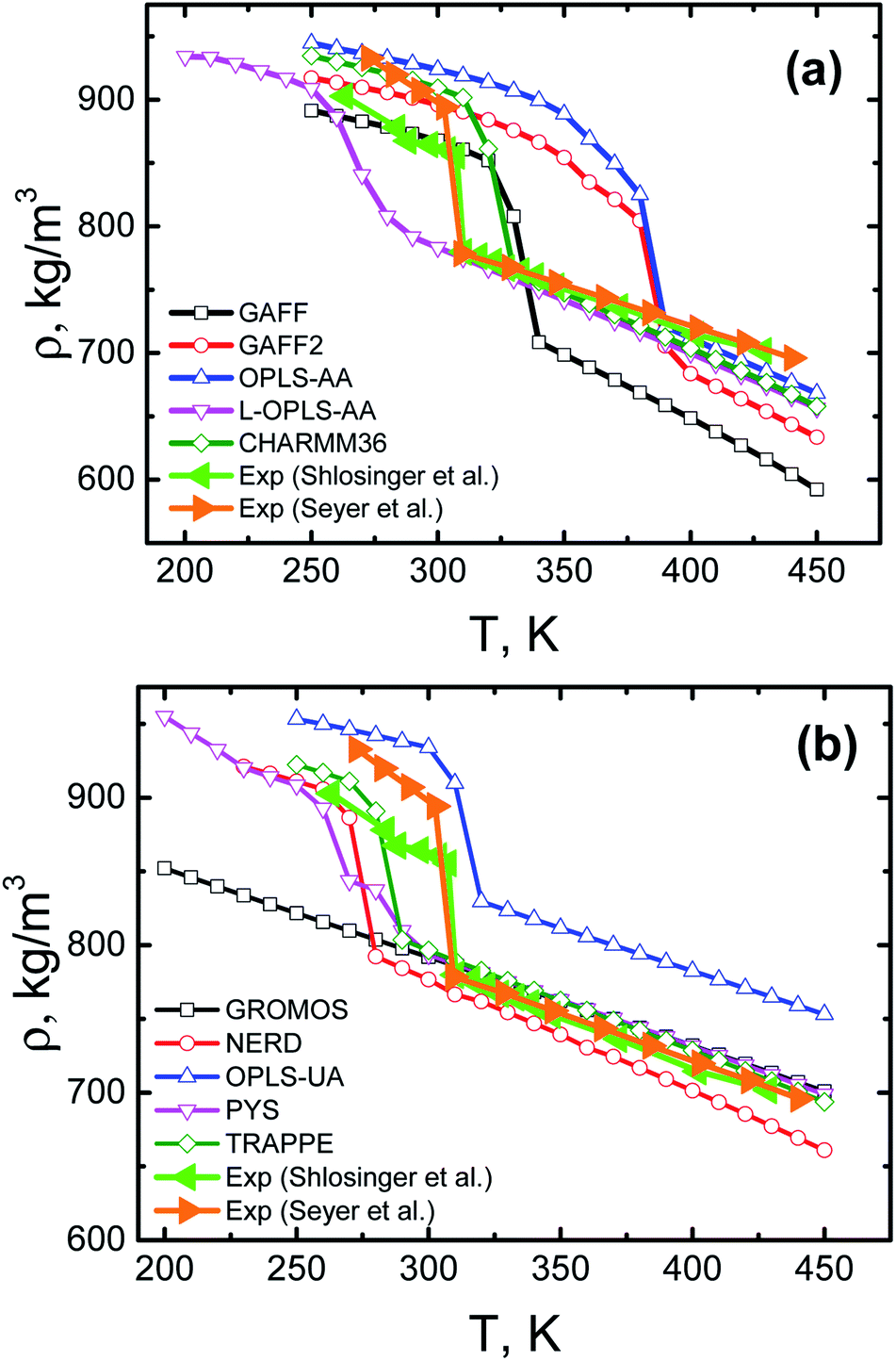 | ||
| Fig. 2 Mass density of n-eicosane samples as a function of temperature. Shown are results for all-atom (a) and united-atom (b) force fields. Error bars (standard errors) are of the size of symbols. For the sake of comparison, the experimental data67,70 is also presented. | ||
As evident from Fig. 2, the high-temperature domain of the density–temperature curves (which corresponds to the liquid state) is best described by united-atom force fields. More specifically, TraPPE, PYS, and GROMOS force fields demonstrate an excellent agreement with experimental data, see Fig. 2(b). The NERD force field is also rather close to experiment, while the OPLS-UA force field overestimates the experimentally measured density of n-eicosane melt. This conclusion is in line with the general expectations, given that TraPPE, PYS and NERD are alkane-specific force field, aimed to reproduce the experimental data as close as possible. In contrast, none of the all-atom general-purpose force field performs in the high-temperature domain as good as united-atom models. As seen in Fig. 2(a), the all-atom force fields systematically underestimate the density of eicosane melt. The best agreement with experimental values is observed for OPLS-AA, L-OPLS-AA, and CHARMM36 force fields. The GAFF force field gives the density that is around 15% smaller that the experimental value. GAFF2, the most recent modification of GAFF, improves the situation to some extent but still gives too low values of density, see Fig. 2(a). In turn, the simulation data in the low-temperature domain scatters considerably for all the considered force fields. United-atom force fields TraPPE, NERD and PYS (and – to some extent – the all-atom force-field GAFF) provide the best agreement with experimental curves, see Fig. 2.
Experimentally, the transition from liquid to crystalline state is accompanied by an abrupt change in the temperature dependence of the n-eicosane density,67,70 see Fig. 2. Remarkably, most force fields (GAFF, GAFF2, CHARMM36, OPLS-AA, TraPPE, NERD, and OPLS-UA) are able to reproduce such a transition. In turn, L-OPLS-AA and PYS force fields demonstrate a much smoother transition to the crystalline state over a wider temperature range of 30–40 K. Nevertheless, crystallization of n-eicosane does take place for L-OPLS-AA and PYS samples, despite the lack of an abrupt transition. Finally, we found that n-eicosane samples described through the GROMOS force field do not crystallize over an extended temperatures range (200–450 K), see Fig. 2(b). It should be noted that recently the GROMOS force field was successfully used to model the initial stages of crystallization in polyimide and polyetherimide nanocomposite materials.72,73 The lack of crystallization which is observed in our study for GROMOS eicosane samples could be due to excessively high cooling rates employed (see below). A difference in the chemical structure of the polymers also could play role.
An abrupt change in the temperature dependence of the eicosane density can be used to evaluate the temperature of the phase transition, i.e. the crystallization temperature Tc. Experimentally, it is known that the value of Tc for n-eicosane is approximately 310 K.4,67,70 In Table 1 we presented the values of crystallization temperature, which were estimated from cooling-rate simulations in different force fields. The crystallization temperature was defined as a temperature in the middle of the interval of the change in the density curves, see Fig. 2.
| Force field | Tc, K | CTE × 10−4, K−1 | Rg, nm (T = 250 K) | Rg, nm (T = 450 K) | H, nm (T = 250 K) | H, nm (T = 450 K) |
|---|---|---|---|---|---|---|
| GAFF | 330 ± 1 | 17.4 ± 0.1 | 0.75 ± 0.01 | 0.60 ± 0.01 | 2.43 ± 0.01 | 1.70 ± 0.01 |
| GAFF2 | 365 ± 2 | 14.3 ± 0.1 | 0.75 ± 0.01 | 0.62 ± 0.01 | 2.43 ± 0.01 | 1.82 ± 0.01 |
| OPLS-AA | 365 ± 1 | 14.7 ± 0.1 | 0.74 ± 0.01 | 0.62 ± 0.01 | 2.42 ± 0.01 | 1.80 ± 0.01 |
| L-OPLS-AA | 265 ± 3 | 12.3 ± 0.1 | 0.72 ± 0.01 | 0.57 ± 0.01 | 2.29 ± 0.01 | 1.63 ± 0.01 |
| CHARMM36 | 320 ± 1 | 12.4 ± 0.1 | 0.74 ± 0.01 | 0.59 ± 0.01 | 2.39 ± 0.01 | 1.69 ± 0.01 |
| GROMOS | — | 8.5 ± 0.1 | 0.56 ± 0.01 | 0.54 ± 0.01 | 1.59 ± 0.01 | 1.52 ± 0.01 |
| NERD | 270 ± 1 | 11.6 ± 0.1 | 0.74 ± 0.01 | 0.58 ± 0.01 | 2.41 ± 0.01 | 1.64 ± 0.01 |
| OPLS-UA | 310 ± 1 | 7.6 ± 0.1 | 0.73 ± 0.01 | 0.57 ± 0.01 | 2.38 ± 0.01 | 1.63 ± 0.01 |
| PYS | 270 ± 10 | 9.0 ± 0.1 | 0.72 ± 0.01 | 0.55 ± 0.01 | 2.34 ± 0.01 | 1.57 ± 0.01 |
| TraPPE | 280 ± 1 | 9.6 ± 0.1 | 0.74 ± 0.01 | 0.58 ± 0.01 | 2.41 ± 0.01 | 1.65 ± 0.01 |
| Experiment | 310 ref. 4 | 8.8–8.9 ref. 67 and 70 | — | — | 2.43 ref. 76 | — |
Remarkably, the crystallization of n-eicosane samples occurs at smaller temperatures for united-atom models compared to all-atom ones, see Table 1. In other words, an explicit treatment of hydrogen atoms in n-eicosane chains seems to facilitate nucleation of a crystal structure of upon cooling.
As far as a comparison with experiment is concerned, it is not straightforward due to an enormous difference in cooling rates employed in simulations and experiments. As mentioned in section “Methods”, the cooling in atomic-scale simulations is several orders of magnitude faster than in experiment. Earlier cooling-rate simulations of polyethylene samples showed that the liquid-to-crystal transition shifted to higher temperatures when the cooling rate decreased.43,74 This is most likely due to the fact that a slower cooling provides more time for the crystal structure nucleation to happen. Assuming similar behavior for n-eicosane samples considered in our study, one can expect that the slowing down of cooling in simulations also shifts the simulated crystallization temperatures to higher values. This implies that most united-atom models (except OPLS-UA) show a better agreement with experiment as compared to their all-atom counter-parts. In the case of the GROMOS force field it is possible that at a given cooling rate the crystallization occurs at temperatures lower than 200 K. For all-atom models we can expect that the deviation of the crystallization temperature from experimental values will increase, if the cooling rate becomes more and more realistic. Nevertheless, among all-atom force fields the best agreement with experiment is achieved with L-OPLS-AA, CHARMM36, and GAFF force fields.
It has to be emphasized that the crystallization temperatures presented in Table 1 were evaluated from the cooling-rate simulations. In principle, the transition temperature could also be studied through heating-rate simulations, starting from crystalline samples. Since the rates of cooling and heating in simulations exceed considerably the experimental rates, a precise relation between simulated temperatures of crystallization and melting is non-trivial. Indeed, as shown in ref. 75 for n-alkane samples described within the NERD force field, a hysteresis is observed upon heating and cooling the samples in simulations, leading to a difference in the melting and crystallization temperatures. Such a hysteresis as well as the impact of the rate of cooling and heating on the transition temperature are to be a subject of a separate simulation study.
We also estimated the coefficient of volumetric thermal expansion (CTE) of n-eicosane, which characterizes the ability of a sample to expand with temperature:68
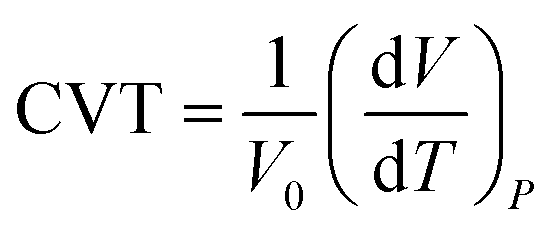 | (1) |
In Table 1 we present the CTE values calculated for n-eicosane samples in the temperature interval from 400 K to 450 K. A good agreement of computational results with experimental data is observed for most united-atom models (except NERD). On the other hand, all-atom force fields considerably overestimate the experimental value of CTE. This conclusion can also be drawn from the fact that the slopes of ρ(T)-curves for all-atom models in the high-temperature domain deviate noticeably from what was observed in experiment, see Fig. 2. Among all-atom models, the best match with experiment is seen for CHARMM36 and L-OPLS-AA systems, whose values of CTE exceed the experimental value by around 30%, see Table 1.
We conclude this subsection with the results for the size, the shape and conformations of n-eicosane chains both in liquid and crystalline states. To characterize the changes in the chain size upon cooling, we calculated the radius of gyration Rg and the end-to-end distance H of n-eicosane chains. In Fig. 3(a) we present the end-to-end distance H as a function of temperature for all force fields considered (for all-atom models only carbon atoms were taken into account for calculating H). The temperature dependence of H follows closely the pattern observed for density (see Fig. 2): one has an abrupt increase in the end-to-end distance of eicosane chains upon cooling, which is a signature of the transition from liquid to crystalline state. In terms of the end-to-end distance, this corresponds to a transition from a coiled chain conformation to a more elongated state. The only exception is the GROMOS force field which does not show the transition from liquid to crystalline phase in the temperature range from 450 K to 200 K. Interestingly, at high temperatures the values of H scatter considerably for the systems described with different force fields, while the end-to-end distances in the crystalline state turn out to be rather similar for all models, see Fig. 3(a). What is more, the end-to-end distance for eicosane chains in the crystalline state was found to be close to that of an alkane chain in the all-trans conformation, see Table 1. The latter can be calculated as nlcos(θ/2), where l (the C–C bond length) and θ (the tetrahedral angle between neighboring bonds) are defined from experiment.76
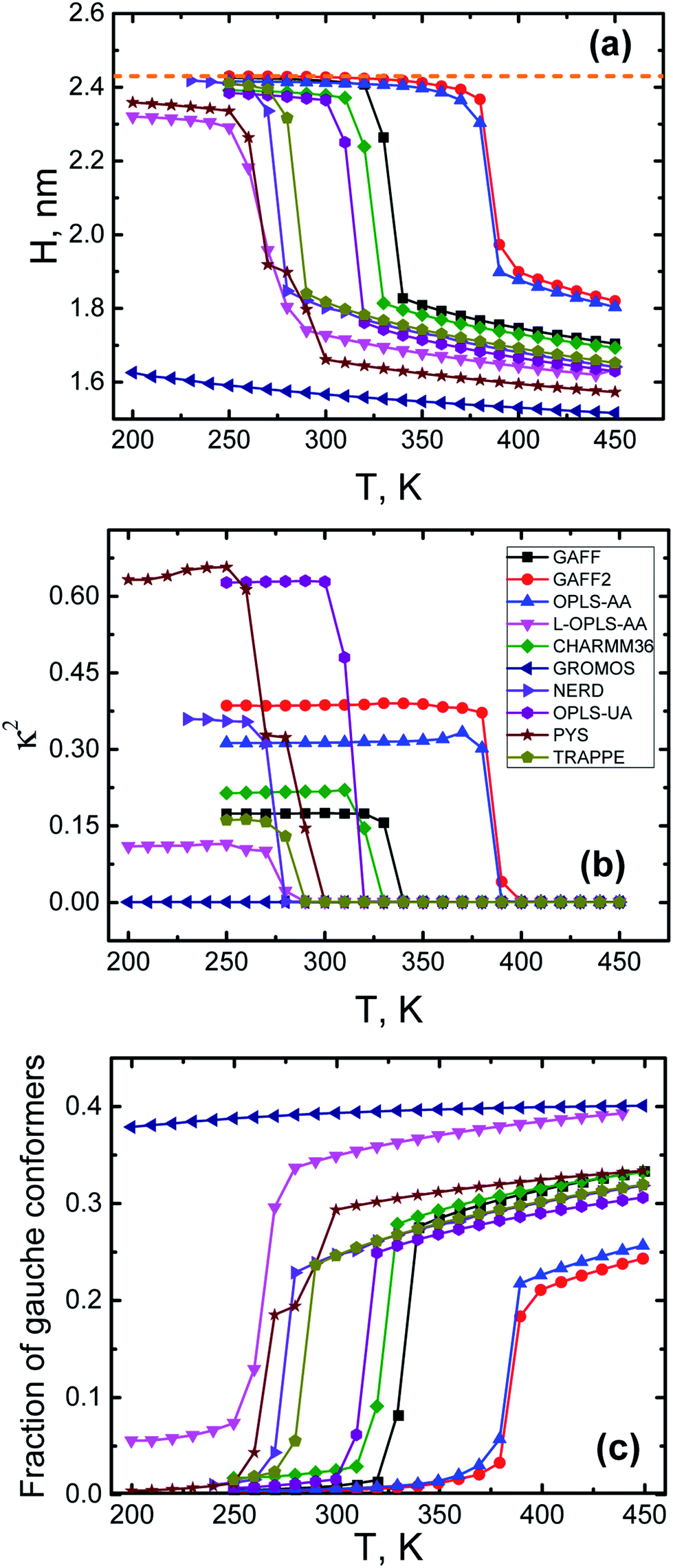 | ||
| Fig. 3 The end-to-end distance H (a), the relative anisotropy κ2 (b) of n-eicosane chains and the average fraction of gauche conformers (c) as a function of temperature in different force fields. A dashed orange line corresponds to the end-to-end distance of an eicosane chain in the all-trans conformation.76 | ||
In line with the results for the end-to-end distance, similar conclusions can also be drawn for the radius of gyration Rg of eicosane chains. In Table 1 we present the average values of Rg in liquid (T = 450 K) and crystalline (T = 250 K) states for all force fields considered. Again, one can witness a systematic increase of the chain size upon the phase transition for all models except the GROMOS force field.
To study the shape of eicosane chains in bulk samples, we considered the relative shape anisotropy κ2 that was calculated as:77
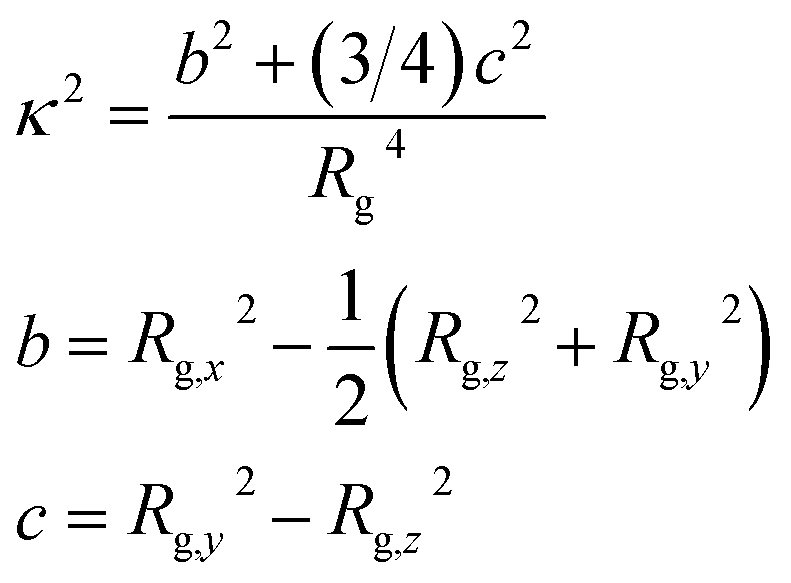 | (2) |
To characterize conformational changes of n-eicosane chains upon cooling, in Fig. 3(c) we present the average fraction of gauche conformers as a function of temperature for different force fields. In the liquid state the fraction of gauche conformers is relatively high and varies in the range from 0.25 to 0.4, depending on a force field. Cooling gradually reduces the fraction of gauche conformers up to the crystallization temperature, where n-eicosane chains undergo an abrupt transition to elongated conformations (this is not applicable to GROMOS samples which do not crystallize). As a result, the average fraction of gauche conformers at very low temperatures becomes practically zero with the exception of the L-OPLS-AA force field for which this fraction does not drop below 0.05, see Fig. 3(c). We also analyzed the fraction of gauche conformers along the hydrocarbon chain at different temperatures, see Fig. S2 and S3.† It turns out that the above conclusions hold for monomers in the middle of the backbone. However, a small fraction of end monomers is always in the gauche conformation,56 even at very low temperatures. In addition, we calculated the probability distribution of dihedral angles for different force fields at T = 450 K (see Fig. S4†) and did not find any clear correlations between the probability of the gauche-trans transition and the crystallization temperatures.
3.2. Structural and dynamic properties of n-eicosane in the liquid state
To characterize the structure of n-eicosane in the liquid state, in this subsection we focus on the static structure factor S(k). In addition to a comparison of the outcome of different force fields, this physical quantity allows one to link the results of computer simulations with experimental data.78,79 Experimentally, the static structure factor can be measured by means of X-ray diffraction.67 The structure factor S(k) is linked to the Fourier transform of the pair correlation function g(r) as:78
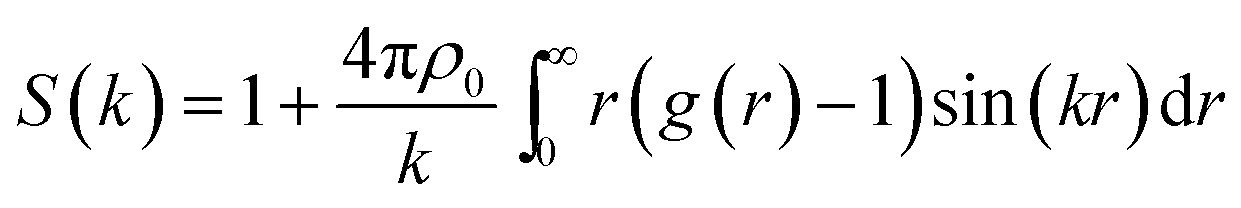 | (3) |
The most relevant X-ray diffraction study80 of n-eicosane melt was performed at T = 315 K (we recall that the transition temperature for eicosane4,67 is around 310 K). In simulations the crystallization temperature was found to scatter considerably for different models, see Table 1. The highest transition temperature (365 K) was observed for GAFF2 and OPLS-AA force fields. Therefore, for calculating S(k) we chose to set the temperature of an eicosane sample to 400 K, ensuring thereby that all our systems are in the liquid phase.
In Fig. 4 we present the static structure factor calculated for all the force fields at hand. First, it is seen that all computational models provide S(k)-curves of a very similar shape. What is more, one can distinguish three peaks in all calculated static structure factors; the positions of these peaks (k = 13, 28, and 50 nm−1) are found to be the same for all force fields. The geometrical interpretation of the peaks can be given81 in terms of the corresponding maxima of the pair correlation function g(r) at r = 2π/k. The first peak is well defined and located at k = 13 nm−1 (or r = 0.48 nm), which corresponds to either two neighboring carbon atoms of different eicosane chains or two methylene groups of the same chain, which are separated by three CH2 groups.80 Two other S(k)-peaks are considerably wider. The one that spans the interval 24–32 nm−1 (or r = 0.20–0.26 nm) can be related to two CH2 groups of a chain, which are separated by another methylene group. Finally, the widest maximum in the interval 43–57 nm−1 (or r = 0.11–0.15 nm) corresponds to two neighboring carbon atoms of an n-eicosane chain, which are covalently bonded. Remarkably, the positions of the three peaks are in good agreement with experimental measurements80 of the structure factor S(k), see Fig. 4. Therefore, one can conclude that all the considered force fields provide a very similar description of the structure of n-eicosane in the liquid state and are able to reproduce closely the experimental data.
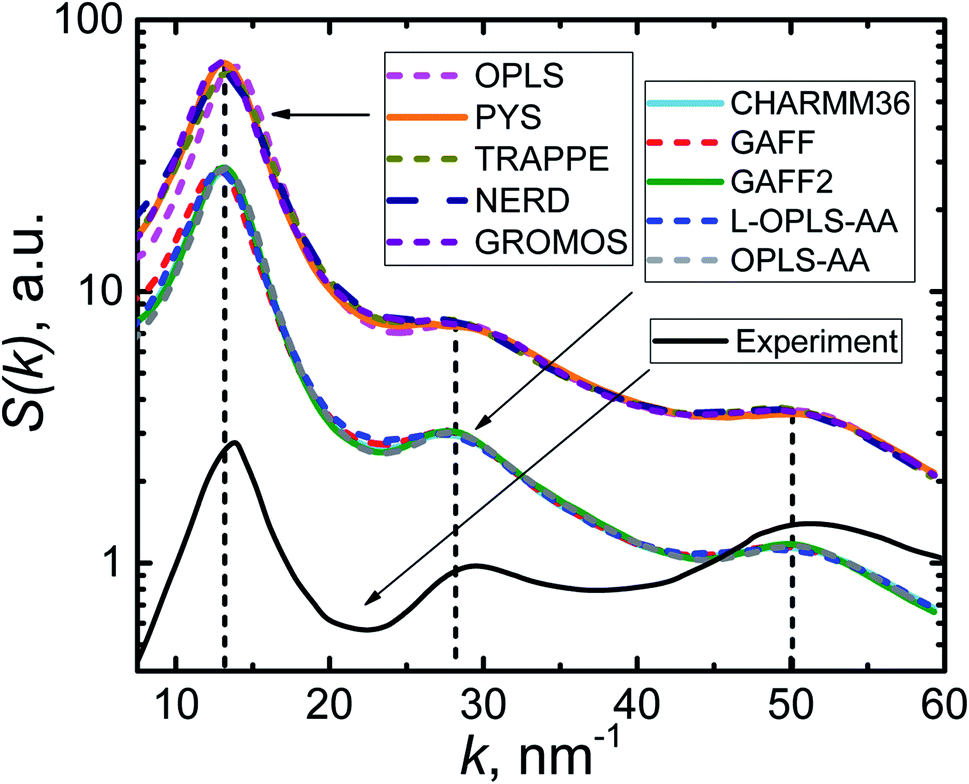 | ||
| Fig. 4 Static structure factor S(k) of n-eicosane at T = 400 K in different force fields. The experimental S(k)-curve at T = 315 K is shown by a solid black line.80 | ||
The shear viscosity η is another important characteristic of an eicosane sample in the liquid state, which can also be linked to experimental data. In equilibrium MD simulations the shear viscosity can be calculated according to the Green–Kubo approach by integrating the autocorrelation function SACF(t) of the non-diagonal components of the pressure tensor:82,83
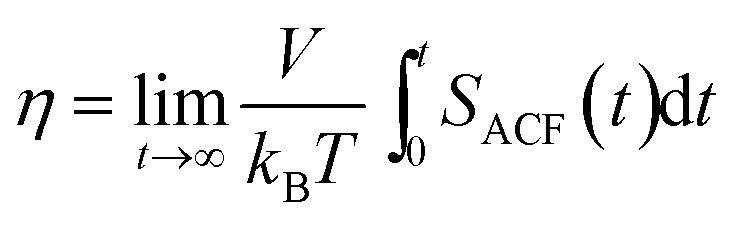 | (4) |
To calculate the shear viscosity from eqn (4), here we used a time decomposition method proposed recently by Zhang et al.69 This method implies a large number of short independent simulation runs to improve the reliability of the viscosity calculation. Correspondingly, for each force field we prepared 99 initial configurations of an n-eicosane sample at T = 450 K on the basis of three 100 ns trajectories of cooling-rate simulations (33 configurations were extracted from each trajectory). For each initial configuration we performed a 5 ns-long MD simulation that was used then for calculating the shear viscosity. A special care has been taken to ensure that 99 simulation runs provided an accurate measurement of the viscosity (data not shown).
In Table 2 we list the calculated values of the shear viscosity for each force field along with the experimental value84 measured at T = 453 K. It is seen that most united-atom models (except OPLS-UA) underestimate the shear viscosity. The closest match to the experimental data is observed for PYS and GROMOS force fields, whose viscosities are 10% and 14% smaller than what was measured in experiment.84 In turn, the largest deviation from the experimental value of viscosity is seen for OPLS-UA (33%) and NERD (46%) force fields, see Table 2.
| Force field | η, mPa s | D, 10−5 cm2 s−1 | ρ, kg m−3 |
|---|---|---|---|
| GAFF | 0.42 ± 0.04 | 2.9 ± 0.2 | 592.3 ± 0.1 |
| GAFF2 | 0.59 ± 0.01 | 2.1 ± 0.1 | 633.5 ± 0.1 |
| OPLS-AA | 0.83 ± 0.07 | 1.6 ± 0.1 | 668.2 ± 0.1 |
| L-OPLS-AA | 0.64 ± 0.08 | 2.2 ± 0.2 | 656.4 ± 0.1 |
| CHARMM36 | 0.60 ± 0.04 | 2.1 ± 0.2 | 658.1 ± 0.1 |
| GROMOS | 0.51 ± 0.01 | 2.5 ± 0.1 | 700.9 ± 0.1 |
| NERD | 0.32 ± 0.01 | 3.6 ± 0.1 | 661.0 ± 0.1 |
| OPLS-UA | 0.79 ± 0.02 | 1.8 ± 0.1 | 753.1 ± 0.1 |
| PYS | 0.53 ± 0.01 | 2.6 ± 0.1 | 698.8 ± 0.1 |
| TraPPE | 0.45 ± 0.04 | 2.9 ± 0.1 | 693.7 ± 0.1 |
| Experiment | 0.594 (T = 453 K)84 | 2.2 (T = 443 K)85 | 696.0 (T = 440 K)67 |
In turn, all-atom force fields are found to suit much better for describing the shear viscosity of eicosane in the liquid state as compared to the united-atom counter-parts: GAFF2, CHARMM36 and L-OPLS-AA force fields demonstrate an excellent agreement with experimental data, see Table 2. Two other all-atom force fields (GAFF and OPLS-AA) show considerable deviations (around 30%) from the experimental value for the shear viscosity of eicosane melt (Table 2).
As far as the dynamics of n-eicosane chains in the liquid state is concerned, it can be characterized by the diffusion coefficient of a chain. For diffusion in three-dimensional space the diffusion coefficient D can be extracted from the mean-squared displacement (MSD) via the Einstein relation:
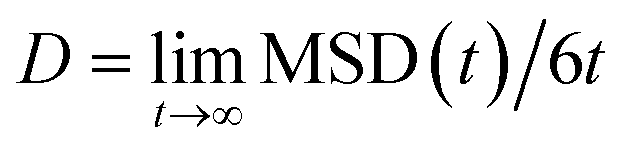 | (5) |
In Table 2 we present the diffusion coefficients calculated for all considered force fields. We also included the experimental value of D, which was measured in n-eicosane melt at T = 443 K by the pulsed-gradient spin-echo NMR method.85 Note that different experimental techniques can give the diffusion coefficients that scatter considerably. For instance, the diffusion coefficient of n-eicosane chains at T = 423 K was found to be 1.1 × 10−5 in NMR experiments and 2.0 × 10−5 cm2 s−1 when the time-of-flight quasielastic neutron scattering was used.86 Nevertheless, as one can see from Table 1, the diffusion coefficients computed for all force fields are of the same order as the experimental value.85 In general, all-atom models provide a closer match to the experimental data as compared to their united-atom counter-parts. The best agreement with experiment is achieved when L-OPLS-AA, CHARMM36 and GAFF2 force fields are used. In turn, the GAFF (OPLS-AA) force field overestimates (underestimates) the experimental value of the diffusion coefficient. Interestingly, most united-atom models (except OPLS-UA) overestimate the diffusion coefficient, see Table 2. This can be due to the implicit treatment of hydrogen atoms in these force fields, which weakens inter-atomic cohesive interactions and speeds up the dynamics.87 Among the united-atom models, the best agreement with experiment is seen for PYS and GROMOS force fields. As a concluding remark, it has to be emphasized that the results for the shear viscosity and diffusion are closely related to each other: the larger the viscosity, the slower the diffusion.
3.3. Structure of the eicosane crystalline phase
Cooling n-eicosane to the temperatures close to the phase transition temperature leads to crystallization of the n-eicosane sample. Computationally, the crystalline phase is not easy to characterize because it undergoes several transformations with decreasing temperature. In particular, it is known that at temperatures just below the melting point one can observe a so-called rotator phase.88 In this phase the system demonstrates a three-dimensional positional order but not an orientational order, i.e. the chains are still able to rotate about their long axes. Further cooling leads to the appearance of the orientational order of alkane chains. In the case of n-eicosane it is well established that its crystalline phase at sufficiently low temperatures has a triclinic crystal structure.89,90 To link our computational findings with experiment, here we also focus on the crystalline structure of n-eicosane samples at a low temperature (250 K) that is well below the transition temperature of n-eicosane.To characterize the crystal structure of eicosane, we used the approach developed in ref. 91. It is based on the analysis of the probability distribution P(θ1,θ2) of relative orientation angles θ1 and θ2 for neighboring eicosane molecules, see Fig. 5 for the definition of angles θ1 and θ2. In Fig. 6 we present the calculated probability distribution P(θ1,θ2) of orientation angles θ1 and θ2 for all-atom models of n-eicosane. The outcome of all 5 force fields demonstrates a common feature: the maxima of the P(θ1,θ2)-distribution are well localized on the main diagonal as well as on two sub-diagonals. The main diagonal of P(θ1,θ2) runs from (−2π,−2π) to (2π,2π) and corresponds to the situation when C–C vectors of monomers of neighboring eicosane chains have the same orientation with respect to the vector R between the two monomers, i.e. θ1 = θ2, see Fig. 6. This feature was observed for monoclinic crystal structures of paraffins and polyethylene.91,92 In addition, for the n-eicosane crystal one can distinguish maxima localized on two more sub-diagonals that run from (−2π,0) to (0,2π) and from (0,−2π) to (2π,0), see Fig. 6. These sub-diagonals correspond to θ1 = θ2 ± 2π and are not observed in monoclinic crystal structures. The appearance of these sub-diagonals could indicate that some n-eicosane chains are shifted by one methylene group with respect to the monoclinic crystal structure, which is typical for triclinic crystals.93 Such a shift of n-eicosane chains is equivalent to the rotation of C–C bonds by 2π in the plane normal to the long axis of the chains, leading thereby to the relation θ1 = θ2 ± 2π observed in Fig. 6. Therefore, all considered all-atom models reproduce qualitatively the triclinic crystal structure of eicosane samples, which was indeed observed experimentally at low temperatures.89,90 It is also noteworthy that there are additional maxima in the probability distribution P(θ1,θ2) outside the main diagonal and sub-diagonals, especially for OPLS-AA, L-OPLS-AA, and GAFF force fields, see Fig. 6. This could be due to the presence of eicosane chains which could be still in the rotator phase mentioned above.
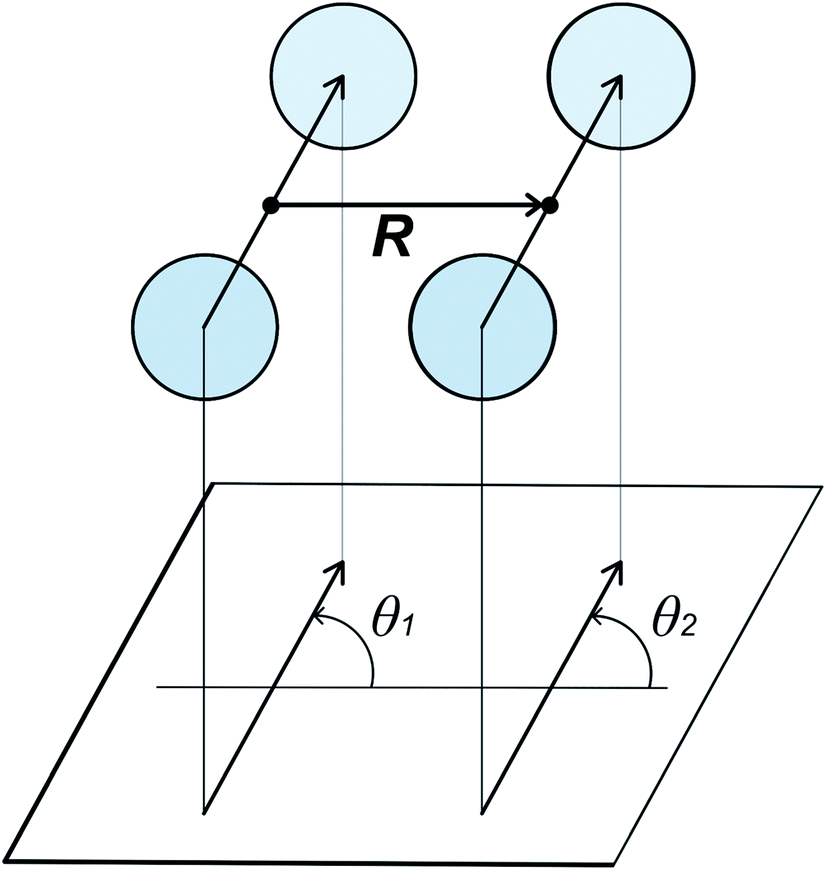 | ||
| Fig. 5 Definition of relative orientation angles θ1 and θ2 for neighboring monomers of two n-eicosane molecules in a crystal.91 | ||
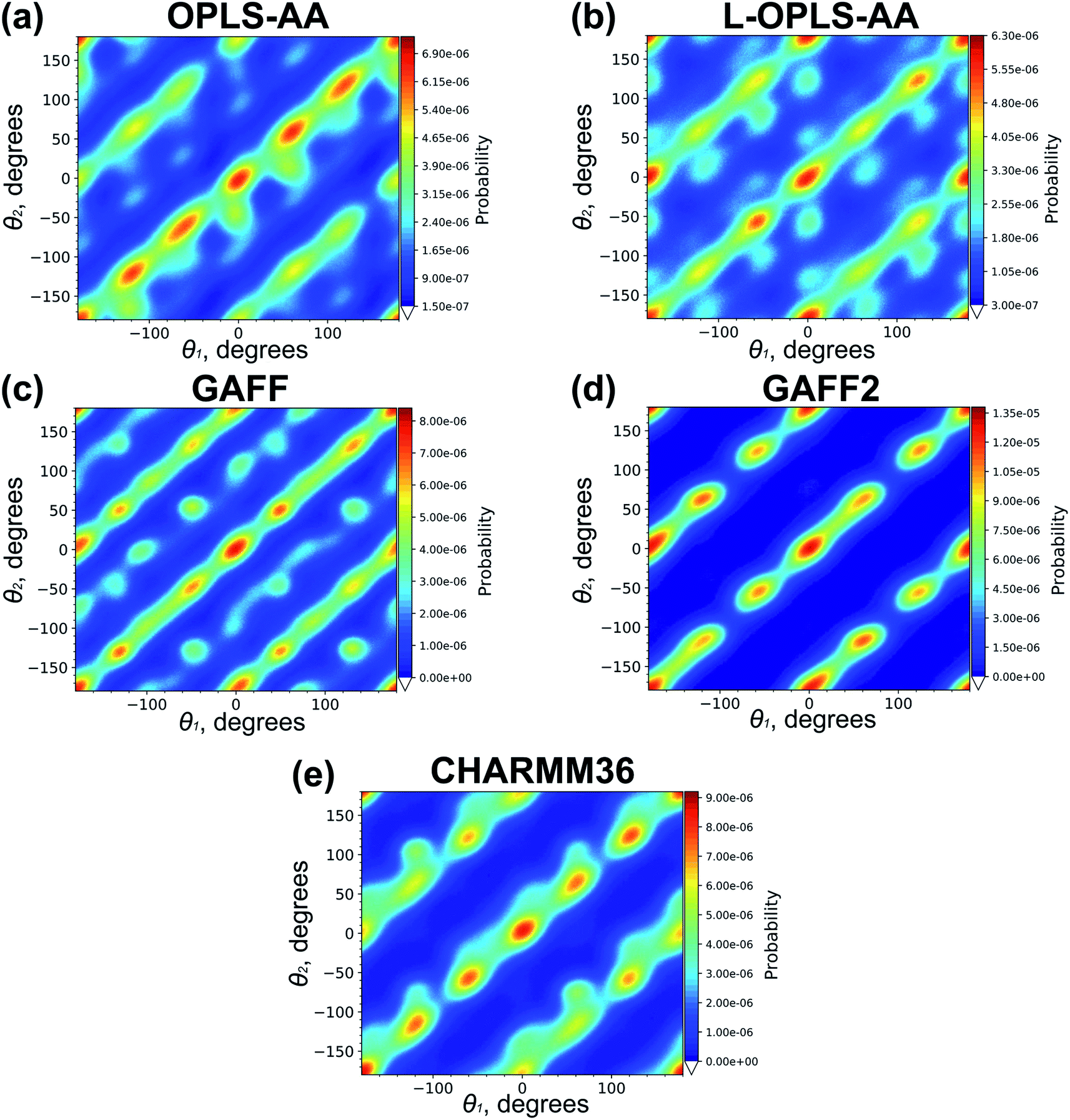 | ||
| Fig. 6 The probability distribution P(θ1,θ2) for the n-eicosane crystalline phase simulated with the use of all-atom models. For each force field the results for one initial configuration is shown; the results for two other configurations are similar and presented in Fig. S5 and S6.† | ||
We repeated the calculations of the probability distribution P(θ1,θ2) of relative orientation angles θ1 and θ2 also for united-atom models (with the exception of GROMOS samples which did not crystallize); the corresponding results are shown in Fig. 7. It is seen that the overall picture (the triclinic crystal structure) is similar to what was observed for all-atom force fields, although the distribution maxima on the main diagonal and sub-diagonals are rather indistinct. Such a difference between united-atom and all-atom models can indicate that eicosane chains in united-atom crystals are more mobile. We recall that all-atom force fields include small partial charges on carbon and hydrogen atoms. The electrostatic interactions between these partial charges, being absent in united-atom models, could additionally stabilize the crystal structure of eicosane samples.
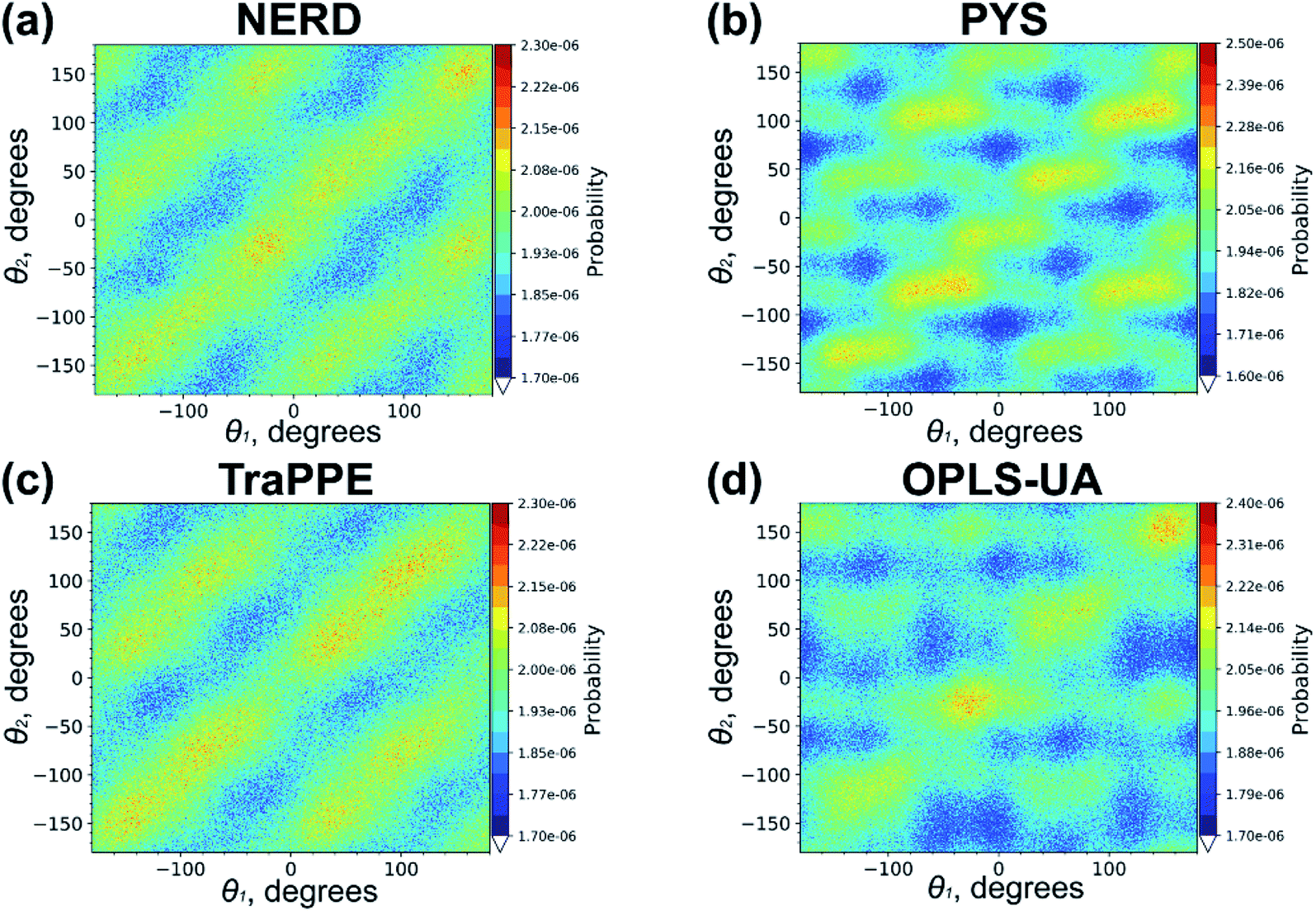 | ||
| Fig. 7 The probability distribution P(θ1,θ2) for the n-eicosane crystalline phase simulated with the use of united-atom models. For each force field the results for one initial configuration is shown; the results for two other configurations are similar and presented in Fig. S7 and S8.† | ||
To conclude, we note that the above analysis of the structure of the crystalline phases was carried out at the same absolute temperature of 250 K. However, the crystallization temperature scatters considerably for different force fields: most all-atom models are characterized by higher crystallization temperatures compared to their united-atom counter-parts, see Table 1. To eliminate the difference in the crystallization temperatures, we repeated the analysis at a model-specific temperature which was set 30–35 K lower than the crystallization temperature. More specifically, the new analysis was done at T = 230 K (L-OPLS-AA and PYS), 240 K (NERD), 250 K (TraPPE), 280 K (OPLS-UA), 290 K (CHARMM36), 300 K (GAFF), and 330 K (GAFF2 and OPLS-AA). The probability distributions P(θ1,θ2) presented in Fig. S9–S14† clearly show that the model-specific temperatures do not have significant impact on the distribution patterns, supporting thereby the main conclusions of this Subsection.
4. Conclusions
The main goal of our study was to evaluate the performance of the state-of-the-art general-purpose force fields such as GAFF, OPLS-AA, GROMOS and CHARMM in computer modeling of paraffins. To this end, we have carried out a critical assessment of these computational models through their systematic comparison with earlier alkane-specific force fields (TraPPE, NERD, and PYS) as well as with available experimental data. This allowed us to estimate the reliability of various theoretical descriptions of n-alkane components to be used in simulations of complex multicomponent systems such as e.g. paraffin-based composite materials.For our purposes we focused on n-eicosane (C20H42), one of the major components of paraffins, and selected 10 different force fields, both all-atom and united-atom ones. The all-atom force fields included GAFF, GAFF2, OPLS-AA, L-OPLS-AA, and CHARMM36. In turn, TraPPE, NERD, PYS, OPLS-UA, and GROMOS were chosen as the united-atom force fields. For each force field we calculated several thermal, structural and dynamic characteristics of eicosane samples in a wide temperature range.
Our results for the temperature dependence of the mass density show that in the liquid state the united-atom alkane-specific force fields TraPPE and PYS outperform most general-purpose force fields, except the GROMOS force field which is also a united-atom one. These three models show an excellent agreement with experimental data for the density in the high-temperature domain. Among all-atom force fields, the closest match with experiment is observed for OPLS-AA, L-OPLS-AA, and CHARMM36 force fields. We also calculated the coefficient of volumetric thermal expansion in the liquid state and found that most united-atom models (TraPPE, PYS, OPLS-UA, and GROMOS) are able to reproduce experimental data. In turn, all-atom force fields overestimate systematically the experimental value of CTE in the liquid state (minimal deviation of 30% is witnessed for CHARMM36 and L-OPLS-AA models).
Remarkably, most force fields (TraPPE, NERD, OPLS-UA, GAFF, GAFF2, CHARMM36, and OPLS-AA) are able to reproduce an abrupt change in the temperature dependence of the n-eicosane density upon cooling (the liquid–crystalline transition). Other two force fields (PYS and L-OPLS-AA) demonstrate a smooth transition to the crystalline state over a wide temperature range of tens of degrees. Finally, it turns out that the n-eicosane sample described within the GROMOS force field does not crystallize upon cooling from 450 K to 200 K at a considered cooling rate. As far as the crystallization temperature is concerned, it is noteworthy that most united-atom models (except OPLS-UA) show a better agreement with experiment as compared to all-atom force fields. The transition temperatures measured for all-atom models are systematically higher than those for their united-atom counter-parts most likely due to the fact that explicit treatment of hydrogen atoms facilitates the initial nucleation of a crystal structure upon cooling.
Calculations of the static structure factor for n-eicosane in the liquid state showed that all the considered force fields give very similar results and are able to capture the experimentally measured positions of main peaks of S(k) in the liquid state.
As far as the temperature dependence of the mass density at low temperatures is concerned, the simulation data is found to scatter considerably. Again, the best agreement with experimental data is observed for alkane-specific models such as TraPPE, NERD and PYS. On the other hand, the GAFF force field performs better in this region as compared to the rest of the general-purpose force fields.
For the shear viscosity the best performance was shown by all-atom models (GAFF2, CHARMM36, and L-OPLS-AA). In turn, almost all united-atom models underestimate the shear viscosity of n-eicosane in the liquid state. The results for the viscosity are closely related to the diffusion coefficients of n-eicosane chains in the liquid state: the larger the viscosity, the slower the diffusion. Therefore, it is not surprising that most united-atom force fields give the diffusion coefficients that exceed the experimental value. Among all-atom models, the best agreement with experiment is observed for GAFF2, CHARMM36, and L-OPLS-AA force fields. Finally, we showed that most computational models considered are able to reproduce qualitatively the experimentally observed triclinic crystal structure of n-eicosane at low temperatures. The only exception is the GROMOS force field that was not able to describe both the crystallization process and the crystal structure of eicosane samples within the considered temperature range (200–450 K) at cooling rates currently accessible in atomic-scale simulations.
All in all, the computational findings outlined above can be used as a guide for making a rational choice of an appropriate model for computer simulations of complex multicomponent systems such as paraffin-based composite materials.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Russian Science Foundation (State Agreement No. 19-13-00178). Computer simulations were performed using the computational resources of the Institute of Macromolecular Compounds, Russian Academy of Sciences, the equipment of the shared research facilities of HPC computing resources at Lomonosov Moscow State University, the resources of the Federal collective usage center “Complex for Simulation and Data Processing for Mega-science Facilities” at the NRC “Kurchatov Institute” (http://ckp.nrcki.ru/), and supercomputers at Joint Supercomputer Center of the Russian Academy of Sciences (JSCC RAS).References
- M. Freund and G. Mózes, Paraffin products: properties, technologies, applications, Elsevier Scientific Publishing Company, New York, U.S.A., 1982 Search PubMed.
- B. He, V. Martin and F. Setterwall, Energy, 2004, 29, 1785–1804 CrossRef CAS.
- M. M. Farid, A. M. Khudhair, S. A. K. Razack and S. Al-Hallaj, Energy Convers. Manage., 2004, 45, 1597–1615 CrossRef CAS.
- A. Sharma, V. V. Tyagi, C. R. Chen and D. Buddhi, Renewable Sustainable Energy Rev., 2009, 13, 318–345 CrossRef CAS.
- A. Elgafy and K. Lafdi, Carbon, 2005, 43, 3067–3074 CrossRef CAS.
- K. Tumuluri, J. L. Alvarado, H. Taherian and C. Marsh, Int. J. Heat Mass Transfer, 2011, 54, 5554–5567 CrossRef CAS.
- M. Li, Appl. Energy, 2013, 106, 25–30 CrossRef CAS.
- L. W. Fan, X. Fang, X. Wang, Y. Zeng, Y. Q. Xiao, Z. T. Yu, X. Xu, Y. C. Hu and K. F. Cen, Appl. Energy, 2013, 110, 163–172 CrossRef CAS.
- D. E. Williams, J. Chem. Phys., 1967, 47, 4680–4684 CrossRef CAS.
- J. P. Ryckaert and A. Bellemans, Chem. Phys. Lett., 1975, 30, 123–125 CrossRef CAS.
- J. P. Ryckaert and A. Bellemans, Faraday Discuss., 1978, 66, 95–106 RSC.
- W. L. Jorgensen, J. D. Madura and C. J. Swenson, J. Am. Chem. Soc., 1984, 106, 6638–6646 CrossRef CAS.
- W. L. Jorgensen and J. Tirado-Rives, J. Am. Chem. Soc., 1988, 110, 1657–1666 CrossRef CAS PubMed.
- J. I. Siepmann, S. Karaborni and B. Smit, Nature, 1993, 365, 330–332 CrossRef CAS.
- S. Chynoweth and Y. Michopoulos, Mol. Phys., 1994, 81, 133–141 CrossRef CAS.
- M. G. Martin and J. I. Siepmann, J. Phys. Chem. B, 1998, 102, 2569–2577 CrossRef CAS.
- M. G. Martin and J. I. Siepmann, J. Am. Chem. Soc., 1997, 119, 8921–8924 CrossRef CAS.
- M. G. Martin and J. I. Siepmann, J. Phys. Chem. B, 1999, 103, 4508–4517 CrossRef CAS.
- S. K. Nath, F. A. Escobedo and J. J. de Pablo, J. Chem. Phys., 1998, 108, 9905–9911 CrossRef CAS.
- S. K. Nath and R. Khare, J. Chem. Phys., 2001, 115, 10837–10844 CrossRef CAS.
- S. K. Nath, B. J. Banaszak and J. J. de Pablo, J. Chem. Phys., 2001, 114, 3612–3616 CrossRef CAS.
- W. Paul, D. Y. Yoon and G. D. Smith, J. Chem. Phys., 1995, 103, 1702–1709 CrossRef CAS.
- G. D. Smith, W. Paul, D. Y. Yoon, A. Zirkel, J. Hendricks, D. Richter and H. Schober, J. Chem. Phys., 1997, 107, 4751–4755 CrossRef CAS.
- N. Waheed, M. S. Lavine and G. C. Rutledge, J. Chem. Phys., 2002, 116, 2301–2309 CrossRef CAS.
- N. Waheed, M. J. Ko and G. C. Rutledge, Polymer, 2005, 46, 8689–8702 CrossRef CAS.
- D. Rigby and R. J. Roe, J. Chem. Phys., 1987, 87, 7285–7292 CrossRef CAS.
- V. G. Mavrantzas and D. N. Theodorou, Macromolecules, 1998, 31, 6310–6332 CrossRef CAS.
- V. A. Harmandaris, V. G. Mavrantzas and D. N. Theodorou, Macromolecules, 1998, 31, 7934–7943 CrossRef CAS.
- N. C. Karayiannis, V. G. Mavrantzas and D. N. Theodorou, Phys. Rev. Lett., 2002, 88, 105503 CrossRef PubMed.
- N. D. Kondratyuk, G. E. Norman and V. V. Stegailov, J. Chem. Phys., 2016, 145, 204504 CrossRef PubMed.
- W. Zhang and R. G. Larson, Macromolecules, 2018, 51, 4762–4769 CrossRef CAS.
- J.-P. Ryckaert, M. L. Klein and I. R. McDonald, Phys. Rev. Lett., 1987, 58, 698–701 CrossRef PubMed.
- J.-P. Ryckaert, I. R. McDonald and M. L. Klein, Mol. Phys., 1989, 67, 957–979 CrossRef CAS.
- J.-P. Ryckaert, M. L. Klein and I. R. McDonald, Mol. Phys., 1994, 83, 439–458 CrossRef CAS.
- W. D. Cornell, P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz Jr, D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell and P. A. Kollman, J. Am. Chem. Soc., 1995, 117, 5179–5197 CrossRef CAS.
- A. D. MacKerell Jr, D. Bashford, M. Bellott, R. L. Dunbrack Jr, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher III, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, D. Yin and M. Karplus, J. Phys. Chem. B, 1998, 102, 3586–3616 CrossRef PubMed.
- L. D. Schuler, X. Daura and W. F. van Gunsteren, J. Comput. Chem., 2001, 22, 1205–1218 CrossRef CAS.
- C. Oostenbrink, A. Villa, A. E. Mark and W. F. van Gunsteren, J. Comput. Chem., 2004, 25, 1656–1676 CrossRef CAS PubMed.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
- M. L. P. Price, D. Ostrovsky and W. L. Jorgensen, J. Comput. Chem., 2001, 22, 1340–1352 CrossRef CAS.
- C. G. Ricci, A. S. C. de Andrade, M. Mottin and P. A. Netz, J. Phys. Chem. B, 2010, 114, 9882–9893 CrossRef CAS PubMed.
- E. A. Muller and A. Mejía, J. Phys. Chem. B, 2011, 115, 12822–12834 CrossRef PubMed.
- J. Ramos, J. F. Vega and J. Martínez-Salazar, Macromolecules, 2015, 48, 5016–5027 CrossRef CAS.
- K. Hagita, S. Fujiwara and N. Iwaoka, AIP Adv., 2018, 8, 115108 CrossRef.
- J. Ramos, J. F. Vega and J. Martínez-Salazar, Eur. Polym. J., 2018, 99, 298–331 CrossRef CAS.
- B. Chen and J. I. Siepmann, J. Phys. Chem. B, 1999, 103, 5370–5379 CrossRef CAS.
- M. Yiannourakou, P. Ungerer, V. Lachet, B. Rousseau and J.-M. Teuler, Fluid Phase Equilib., 2019, 481, 28–43 CrossRef CAS.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- J. Wang and T. Hou, J. Chem. Theory Comput., 2011, 7, 2151–2165 CrossRef CAS PubMed.
- J. P. M. Jambeck and A. P. Lyubartsev, J. Phys. Chem. B, 2014, 118, 3793–3804 CrossRef PubMed.
- GAFF2 is part of the AmberTools19 distribution, available for download at, http://ambermd.org, accessed August 2019.
- D. Vassetti, M. Pagliai and P. Procacci, J. Chem. Theory Comput., 2019, 15, 1983–1995 CrossRef CAS PubMed.
- J. B. Klauda, R. M. Venable, J. A. Freites, J. W. O'Connor, D. J. Tobias, C. Mondragon-Ramirez, I. Vorobyov, A. D. MacKerell and R. W. Pastor, J. Phys. Chem. B, 2010, 114, 7830–7843 CrossRef CAS PubMed.
- J. B. Klauda, B. R. Brooks, A. D. MacKerell Jr, R. M. Venable and R. W. Pastor, J. Phys. Chem. B, 2005, 109, 5300–5311 CrossRef CAS PubMed.
- L. L. Thomas, T. J. Christakis and W. L. Jorgensen, J. Phys. Chem. B, 2006, 110, 21198–21204 CrossRef CAS PubMed.
- S. W. I. Siu, K. Pluhackova and R. A. Böckmann, J. Chem. Theory Comput., 2012, 8, 1459–1470 CrossRef CAS PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Pall, J. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- S. V. Lyulin, A. A. Gurtovenko, S. V. Larin, V. M. Nazarychev and A. V. Lyulin, Macromolecules, 2013, 46, 6357–6363 CrossRef CAS.
- B. Hess, J. Chem. Theory Comput., 2008, 4, 116–122 CrossRef CAS PubMed.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8592 CrossRef CAS.
- T. F. D. Silva, D. Vila-Vicosa, P. B. P. S. Reis, B. L. Victor, M. Diem, C. Oostenbrink and M. Machuqueiro, J. Chem. Theory Comput., 2018, 14, 5823–5833 CrossRef CAS PubMed.
- E. Panizon, D. Bochicchio, L. Monticelli and G. Rossi, J. Phys. Chem. B, 2015, 119, 8209–8216 CrossRef CAS PubMed.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- S. Nosé, Mol. Phys., 1984, 52, 255–268 CrossRef.
- W. G. Hoover, Phys. Rev. A, 1985, 31, 1695–1697 CrossRef PubMed.
- W. F. Seyer, R. F. Patterson and J. L. Keays, J. Am. Chem. Soc., 1944, 66, 179–182 CrossRef CAS.
- S. V. Lyulin, S. V. Larin, A. A. Gurtovenko, V. M. Nazarychev, S. G. Falkovich, V. E. Yudin, V. M. Svetlichnyi, I. V. Gofman and A. V. Lyulin, Soft Matter, 2014, 10, 1224–1232 RSC.
- Y. Zhang, A. Otani and E. J. Maginn, J. Chem. Theory Comput., 2015, 11, 3537–3546 CrossRef CAS PubMed.
- A. P. Shlosinger and E. W. Bentilla, Research and development study on thermal control by use of fusible materials, NASA Technical Report, NASA-CR-67695, 1965 Search PubMed.
- A. Jabbarzadeh and R. I. Tanner, Fluid Mech., 2009, 160, 11–21 CAS.
- V. M. Nazarychev, S. V. Larin, A. V. Lyulin, T. Dingemans, J. M. Kenny and S. V. Lyulin, Polymers, 2017, 9, 548 CrossRef PubMed.
- S. V. Larin, V. M. Nazarychev, A. Y. Dobrovskiy, A. V. Lyulin and S. V. Lyulin, Polymers, 2018, 10, 1245 CrossRef PubMed.
- T. Yamamoto, J. Chem. Phys., 2010, 133, 034904 CrossRef PubMed.
- R. Rastgarkafshgarkolaei, Y. Zeng and J. M. Khodadadi, J. Appl. Phys., 2016, 119, 205107 CrossRef.
- M. Rubinstein and R. H. Colby, Polymer physics, Oxford University Press, Oxford, 2003 Search PubMed.
- S. G. Falkovich, S. V. Larin, A. V. Lyulin, V. E. Yudin, J. M. Kenny and S. V. Lyulin, RSC Adv., 2014, 4, 48606–48612 RSC.
- T. C. Ionescu, C. Baig, B. J. Edwards, D. J. Keffer and A. Habenschuss, Phys. Rev. Lett., 2006, 96, 037802 CrossRef CAS PubMed.
- G. Venturi, F. Formisano, G. J. Cuello, M. R. Johnson, E. Pellegrini, U. Bafile and E. Guarini, J. Chem. Phys., 2009, 131, 034508 CrossRef CAS PubMed.
- A. Habenschuss and A. H. Narten, J. Chem. Phys., 1990, 92, 5692–5699 CrossRef CAS.
- K. Binder and W. Kob, Glassy materials and disordered solids, World Scientific, 2005 Search PubMed.
- M. P. Allen and D. J. Tildesley, Computer simulation of liquids, Oxford University Press, Oxford, 1987 Search PubMed.
- G. S. Fanourgakis, J. S. Medina and R. Prosmiti, J. Phys. Chem. A, 2012, 116, 2564–2570 CrossRef CAS PubMed.
- P. H. Gross and H. K. Zimmerman, Rheol. Acta, 1964, 3, 290–294 CrossRef CAS.
- E. von Meerwall, S. Beckman, J. Jang and W. L. Mattice, J. Chem. Phys., 1998, 108, 4299–4304 CrossRef CAS.
- C. Smuda, S. Busch, G. Gemmecker and T. Unruh, J. Chem. Phys., 2008, 129, 014513 CrossRef PubMed.
- I. V. Volgin, S. V. Larin, A. V. Lyulin and S. V. Lyulin, Polymer, 2018, 145, 80–87 CrossRef CAS.
- P. K. Mukherjee, Phys. Rep., 2015, 588, 1–54 CrossRef CAS.
- S. C. Nyburg and A. R. Gerson, Acta Crystallogr., Sect. B: Struct. Sci., 1992, 48, 103–106 CrossRef.
- S. R. Craig, G. P. Hastie, K. J. Roberts and J. N. Sherwood, J. Mater. Chem., 1994, 4, 977–981 RSC.
- N. Wentzel and S. T. Milner, J. Chem. Phys., 2011, 134, 224504 CrossRef PubMed.
- V. I. Sultanov, V. V. Atrazhev, D. V. Dmitriev, N. S. Erikhman, D. U. Furrer and S. F. Burlatsky, Macromolecules, 2019, 52, 5925–5936 CrossRef CAS.
- E. A. Zubova, in Encyclopedia of Polymers and Composites, Springer Berlin Heidelberg, Berlin, Heidelberg, 2013, pp. 1–17 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra07325f |
| This journal is © The Royal Society of Chemistry 2019 |