
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAssembly of reduced graphene oxides into a three-dimensional porous structure via confinement within robust cellulose oligomer networks†
Yuuki Hata a,
Yoshitaka Saitob,
Toshiki Sawadaac,
Hidetoshi Matsumotob and
Takeshi Serizawa*a
a,
Yoshitaka Saitob,
Toshiki Sawadaac,
Hidetoshi Matsumotob and
Takeshi Serizawa*a
aDepartment of Chemical Science and Engineering, School of Materials and Chemical Technology, Tokyo Institute of Technology, 2-12-1 Ookayama, Meguro-ku, Tokyo 152-8550, Japan. E-mail: serizawa@polymer.titech.ac.jp
bDepartment of Materials Science and Engineering, School of Materials and Chemical Technology, Tokyo Institute of Technology, 2-12-1 Ookayama, Meguro-ku, Tokyo 152-8550, Japan
cPrecursory Research for Embryonic Science and Technology (PRESTO), Japan Science and Technology Agency (JST), 4-1-8 Honcho, Kawaguchi-shi, Saitama 332-0012, Japan
First published on 27th November 2019
Abstract
The assembly of nanomaterials into a networked superstructure is a strategy used to construct macroscopic porous materials having the functional properties of nanomaterials. However, because nanomaterials generally prefer densely packed assembled states owing to their high surface energies, the construction of a fine porous structure is still a challenge. In this study, we demonstrate the assembly of reduced graphene oxides (rGOs) into a fine porous structure via confinement within robust cellulose oligomer networks. The confinement of rGOs within cellulose oligomer networks was achieved in one step via the enzymatic synthesis of cellulose oligomers. When the resultant cellulose oligomer gels confining rGOs were reduced by hydrogen iodide, the robust cellulose oligomer networks served as a confinement space for rGOs, preventing excessive aggregation of the rGOs and thus encouraging their assembly into a fine porous structure. Electrochemical measurements revealed that the porous rGO materials could act as electrode materials for supercapacitors. Our strategy based on simple physical confinement will allow for the creation of functional porous materials with excellent nanomorphologies from various nanomaterials.
Introduction
The assembly of nanomaterials, such as metal nanoparticles, carbon nanomaterials, and polymer nanoparticles, into superstructures is the strategy used to construct mesoscopic or macroscopic materials having the functional properties of nanomaterials.1–10 Precise control of nanomaterial assembly has been achieved by employing DNA origami technology,11–13 templates with well-defined morphology,9,14–16 electric17,18 or magnetic19,20 fields, and other techniques. Meanwhile, fuzzy control based on the self-assembly of neat nanomaterials under designed conditions has widely been investigated owing to its simplicity, which offers versatility, scalability, and cost effectiveness. It has been demonstrated that the destabilization of nanomaterial dispersions by methods, such as the removal of surface stabilizing agents or functional groups from the nanomaterial surface, can induce the formation of three-dimensional assembled structures, thereby producing porous macroscopic materials.4,6,8,10,21,22 Although their high surface energies could direct nanomaterials to assemble into densely packed structures to reduce the surface area, under appropriate conditions (e.g., high nanomaterial concentrations), such voluminous porous structures are formed as kinetically trapped states.23 In general, it is desirable for resultant macroscopic materials to inherit the nanoscale dimensions of their raw nanomaterials because a nanomorphology and high surface area are essential for their functional properties. However, as nanomaterials prefer to form densely packed structures, the construction of such a fine porous structure is still a challenge.We recently demonstrated the confinement of nanomaterials, including naturally derived cellulose nanocrystals (CNCs), within robust crystalline cellulose oligomer networks.24,25 When cellulose oligomers were synthesized via cellodextrin phosphorylase (CDP)-catalyzed oligomerization of α-D-glucose 1-phosphate (αG1P) monomers from D-glucose primers in colloidal dispersions of CNCs, the synthesized oligomers self-assembled in situ into nanoribbon network structures for the formation of hydrogels.24 Within the robust crystalline nanoribbon networks, the CNCs were confined in a well-dispersed state. Furthermore, we found that the confined CNCs were prevented from aggregation into densely packed structures. In fact, the water solvent of the hydrogels confining CNCs was successfully exchanged with a low polar solvent, namely toluene, while the gels maintained structural integrity without apparent changes in their shape because of the robustness of cellulose oligomer networks. Then, the organogels were immersed in solutions of hydrophobic polymers with toluene solvent, followed by heat dry to obtain polymer nanocomposites. The CNCs in the nanocomposites were found to be in a well-dispersed state despite incompatibility between hydrophilic CNCs and hydrophobic polymers. The finding indicates that confinement within robust crystalline nanoribbon networks is a promising strategy for maintaining a dispersed state of nanomaterials under various conditions.
Herein, the assembly of reduced graphene oxides (rGOs) into a fine porous structure via confinement within cellulose oligomer nanoribbon networks is reported (Fig. 1). The enzymatic synthesis of cellulose oligomers in dispersions of graphene oxides (GOs) resulted in confinement of GOs within cellulose oligomer gels. Simultaneously, the GOs were reduced to be rGOs as a side reaction. The water solvent of the nanoribbon network hydrogels confining the rGOs was successfully exchanged with acetic acid owing to the robustness of crystalline cellulose oligomer assemblies, allowing for further chemical reduction of the confined rGOs by hydrogen iodide in acetic acid. As a result, the rGOs assembled into a porous structure while the cellulose oligomer nanoribbons were decomposed owing to the harsh reaction conditions. The resultant rGO gels had a pore size smaller than that of previously reported rGO gels produced by destabilization of GO dispersions by reduction reactions.26–28 The smaller pore size was attributed to the confinement effect that prevented excessive aggregation of rGOs in the initial stage of the reduction reaction. The rGO gels exhibited electrical conductivity owing to the formation of a percolation network of rGOs. Electrochemical measurements revealed that the rGO gels could act as electrode materials for supercapacitors.
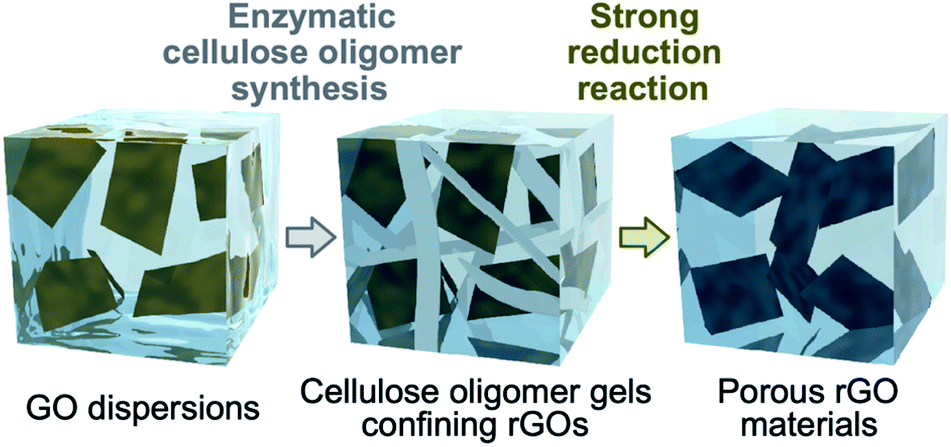 | ||
| Fig. 1 Schematic illustration of this study. | ||
Experimental
Materials
αG1P disodium salt n-hydrate and 40% sodium deuteroxide (NaOD)/deuterium oxide (D2O) solution were purchased from Wako Pure Chemical Industries. Aqueous GO dispersion (monolayer content >95%), ProteoMass MALDI-MS standard, 1% trifluoroacetic acid, acetonitrile, 2,5-dihydroxybenzoic acid, and D2O were purchased from Sigma-Aldrich. All other reagents were purchased from Nacalai Tesque. Ultrapure water with a resistivity greater than 18.2 MΩ cm was supplied by a Milli-Q Advantage A-10 apparatus (Merck Millipore) and used throughout all experiments.Enzymatic synthesis of cellulose oligomers in GO dispersions
Following a previous report, CDP from Clostridium thermocellum YM4 was prepared with a genetically engineered Escherichia coli containing a plasmid with the cdp gene.29 For the synthesis of cellulose oligomers, αG1P monomer (0.2 M) and D-glucose primer (0.05 M) were incubated with CDP (0.2 U mL−1) in a 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer solution (0.5 M, pH 7.5) containing GOs (4 mg mL−1) at 60 °C for 3 d.To readily assess gelation, vials containing the mixtures (0.3 mL) after the reaction were inverted. For Raman spectroscopy and scanning electron microscopy (SEM), the gelled products (i.e., the hybrid gels composed of cellulose oligomers and rGOs) were purified by immersion in ultrapure water (exchanged each day) at 4 °C for at least 1 week. For the other characterization techniques, particulate product dispersions were obtained by pipetting the mixtures after the reaction. The particulate products were purified with ultrapure water via at least five centrifugation (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400g)/redispersion cycles to remove more than 99.999% of the soluble fraction of the reaction mixtures. Immediately after the purification or after subsequent freeze-drying, the purified products were stored at 4 °C until use. For quantification of the insoluble products, a volume of the product dispersions was dried at 105 °C for 24 h, followed by weighing.
400g)/redispersion cycles to remove more than 99.999% of the soluble fraction of the reaction mixtures. Immediately after the purification or after subsequent freeze-drying, the purified products were stored at 4 °C until use. For quantification of the insoluble products, a volume of the product dispersions was dried at 105 °C for 24 h, followed by weighing.
Characterization of the hybrid gels
For Raman spectroscopy, the hybrid gels (≤25 μL) were immersed three times in 1 N NaOH aqueous solutions for more than 1 h to remove cellulose oligomers through dissolution. Note that the gel state was maintained after the treatment. The resultant gels were purified by immersion in ultrapure water for 1 h five times and then freeze-dried. An NRS-4100 instrument (JASCO) equipped with a diode laser (532 nm) was operated at a laser intensity of 1% to obtain Raman spectra. Cubic function curves were fitted to the spectra in the 680–860 and 2000–2250 cm−1 ranges, where no peak was observed, to obtain baselines. The baselines were used to calculate the peak intensities of the G band (∼1596 cm−1) and D band (∼1346 cm−1) (IG and ID, respectively).For 1H NMR spectroscopy, the freeze-dried products were added to 600 μL of 4% NaOD/D2O. After centrifugation (20400g), 500 μL of the supernatants containing dissolved cellulose oligomers (≥20 mg mL−1) was collected. An AVANCE III HD500 spectrometer (500 MHz, Bruker) was operated at ambient temperature to obtain the 1H NMR spectra. The spectra were calibrated using the signal of residual water (δ = 4.79) as an internal standard. The average degree of polymerization (DP) was calculated using the following equation: average DP = H1′,1′′/(H1α + H1β) + 1, where H1′,1′′, H1α, and H1β are the integrals of the corresponding protons (see the chemical structure of cellulose oligomer in Fig. S3†).
For X-ray diffraction (XRD) analysis, the freeze-dried products were pressed into pellets using a hand press. A D8 DISCOVER instrument (Bruker) with Cu Kα radiation (λ = 1.542 Å) was operated under ambient conditions to obtain the transmission XRD patterns of the products using a two-dimensional (2D) detector. The 2D diffraction patterns were converted into 1D profiles in the 2θ (θ is the Bragg angle) range of 10–30°.
For attenuated total reflection-Fourier transform infrared (ATR-FTIR) absorption spectroscopy, the freeze-dried products were mounted on the sample stage. An FT/IR-4100 instrument (JASCO) was operated at a cumulative measurement number of 100 and a resolution of 2.0 cm−1 under ambient conditions to obtain IR absorption spectra.
For SEM, the water solvent of the hybrid hydrogels was exchanged stepwise with 10, 20, 30, 40, 50, 60, 70, 80, and 90 vol% ethanol, ethanol, ethanol/tert-butyl alcohol (1:1, v/v), and then tert-butyl alcohol by immersion. The resultant organogels with tert-butyl alcohol solvent were freeze-fractured using liquid nitrogen and a razor blade. After freeze-drying, the resultant freeze-dried gels were mounted on substrates using Dotite (Nisshin EM Corporation) and then coated with osmium. A field-emission scanning electron microscope (JSM-7500F, JEOL) was operated at an accelerating voltage of 5 kV to observe the fracture surface corresponding to the inside of the original hybrid hydrogels.
For atomic force microscopy (AFM), the product dispersions (cellulose oligomer concentrations of ∼0.01 mg mL−1) were spin-cast on mica at 600 rpm for 30 min and then dried for more than 12 h in a dessicator. An SPM-9600 instrument (Shimadzu) was operated in the tapping mode under ambient conditions to observe nanostructures.
Chemical reduction of the hybrid gels
The hybrid gels with a thickness of 1 mm were synthesized in a gap between two glass slides and then cut into circles with a diameter of 7.0 mm. The water solvent of the obtained disk-shaped hybrid gels was exchanged with 50 vol% acetic acid and then acetic acid by immersion at room temperature. The resultant organogels were immersed in 7 mL of a mixture containing 95% acetic acid and 2.8% hydrogen iodide at room temperature, followed by incubation at 95 °C using an oil bath for 3 h for the reduction reaction. Then, the reduction reaction was performed again in the same way (i.e., two times in total). The resultant disk-shaped rGO gels were purified by immersion in 50% acetic acid and then ultrapure water. The porosity of the rGO gels was calculated from a previously reported density of rGO (2.2 g cm−3)30 and the rGO concentration in the gels.To investigate effects of the reduction reaction on cellulose oligomers, cellulose oligomer gels without rGOs were prepared according to a previous report31 and applied to the reduction reaction as described above. Note that the gels disappeared after the reaction. For matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry, aliquots of the resultant solutions were dried at 60 °C to remove hydrogen iodide and acetic acid, followed by the addition of 2,5-dihydroxybenzoic acid, trifluoroacetic acid, and acetonitrile at concentrations of 1.7 mg mL−1, 0.02% (v/v), and 50% (v/v), respectively. The mixtures were deposited onto an AXIMA 384-well plate and then dried under ambient conditions. An AXIMA-performance instrument (Shimadzu) equipped with a nitrogen laser (λ = 337 nm) and pulsed ion extraction was operated at an accelerating potential of 20 kV in linear positive ion mode. Peptide standards (ProteoMass MALDI-MS Standard) were used to calibrate the spectra at 757.3997 Da (bradykinin fragment 1–7), 1533.8582 Da (P14R), and 2465.1989 Da (ACTH fragment 18–39).
Characterization of the rGO gels
For SEM, freeze-dried gels were prepared from the rGO hydrogels as described above with the exception of omission of the fracture step. Regions corresponding to the surface of the original rGO hydrogels were observed by the scanning electron microscope under the same conditions mentioned above. The obtained images were analyzed by the ImageJ software (US National Institutes of Health) to estimate pore sizes: images were binarized at appropriate thresholds, pores with diameters less than 5 μm were picked, and the average diameter was calculated from more than 200 pores.Raman spectroscopy measurements were performed as mentioned above.
For current–voltage analysis, the disk-shaped rGO gel was put between two platinum plates as electrodes and pressed into a thickness of 0.1 mm. The measurements were conducted under ambient conditions using a 4200A-SCS instrument (Keithley) in voltage linear sweep mode (0–1 V) at a compliance of 0.1 A. The current–voltage curves were subjected to linear fitting to obtain slopes, which corresponded to conductance (G). Electrical conductivity (σ) was calculated using the following equation: σ = G (l/A), where l and A are the thickness and area of the gels, respectively.
For cyclic voltammetry (CV), the water solvent of the disk-shaped rGO gels was exchanged by immersion with a 1 M sulfuric acid aqueous solution that had been bubbled with dry nitrogen gas. To perform the measurements in the two-electrode system, two pieces of the disk-shaped rGO gels impregnated with 1 M sulfuric acid were separated by a glass microfiber filter (Whatman GF/A) as a separator, followed by putting them between two platinum plates as current collectors. The gels were pressed into a thickness of 0.1 mm. The measurements were conducted under ambient conditions using a potentiostat/galvanostat (1287A, Solartron) and a frequency response analyzer (1255B, Solartron) in a voltage range of 0–1 V at scan rates of 5, 20, 60, and 100 mV s−1.
Charge/discharge measurements were conducted in the same setup for three specimens under ambient conditions using an HJ1001SD8 instrument (Hokuto Denko) in a voltage range of 0–1 V at current densities of 5, 10, 20, 50, and then 100 A g−1 for each specimen. Ten charge/discharge cycles were performed at each current density. Capacitance (C) was calculated by linear approximation for the discharge steps using the following equation: C = IΔt/ΔV, where I, Δt, and ΔV are discharging current, time, and voltage, respectively. The specific capacitance values were shown as those for the rGO mass and one electrode.
Results and discussion
Confinement of rGOs within cellulose oligomer networks
We focused on GOs as a nanomaterial in this study because of ease of their assembly into porous structures via chemical reduction and attractive potential applications of resultant rGO porous materials.6 Commercially available monolayer GOs of several micrometers in diameter were used in this study (Fig. S1†). Cellulose oligomers were enzymatically synthesized in 4 mg mL−1 GO dispersions at pH 7.5 at 60 °C for 3 d, leading to gel formation (Fig. 2a and b). The relatively high GO concentration of 4 mg mL−1 was chosen as higher GO concentrations have been shown to be preferable in the construction of rGO porous structures via the reduction reaction of GO dispersions.6 Note that GO dispersions with concentrations above 4 mg mL−1 were too viscous (i.e., pasty) to be used to prepare reaction mixtures. The gel formation was clearly different from the production of nanosheet-shaped crystals during the cellulose oligomer synthesis without GOs.25,29 In other words, the presence of GOs caused the gel formation, leading to confinement of GOs in the gels.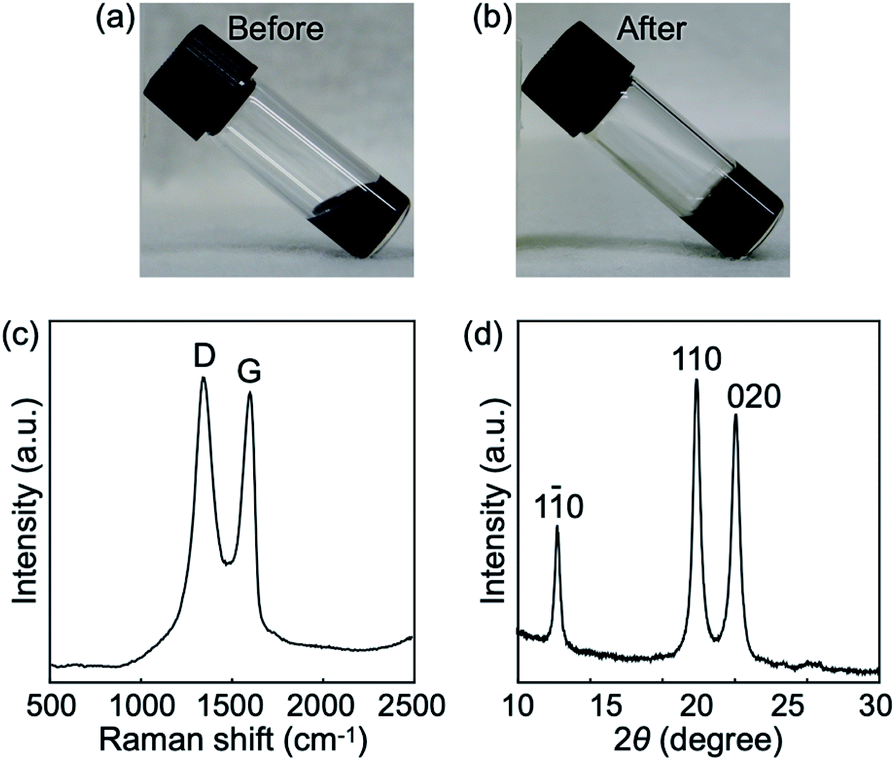 | ||
| Fig. 2 Photographs of the reaction mixtures (a) before and (b) after the reaction, which are sol and gel states, respectively. (c) Raman spectrum of the confined rGOs. (d) XRD profile of the products. Miller indices for cellulose II32 are shown above the peaks. | ||
The oxidation degree of GOs after the confinement was investigated by Raman spectroscopy. The spectrum showed two characteristic peaks of the G and D bands (Fig. 2c), which were derived from the in-plane sp2 carbon–carbon bond stretching motion and the breathing modes of six-atom rings with activation by defects, respectively.33 The intensity ratio of the two peaks (ID/IG) was calculated to be 1.08, which was higher than that of the raw GOs (1.00). Because ID/IG is known to vary inversely with the sp2 cluster size in carbon materials,33 the result suggests the emergence of new small sp2 clusters in GOs due to reduction as a side reaction of the cellulose oligomer synthesis. Incubation of GOs at various pHs adjusted by buffer species that do not act as reducing agents revealed a tendency toward more blackish color of GOs at higher pHs (Fig. S2†), indicating more extended sp2 clusters. Hydroxide ion appeared to catalyze reduction of GOs.34,35 In addition, aldehyde group in glucose36 and piperazine nitrogen atoms in HEPES37,38 (the buffer species used for the cellulose oligomer synthesis reaction) might contribute to the reduction of GOs as well. Taken together, these results reveal that the GOs confined within the gels had been reduced to be hydrophobic rGOs.
The synthesized cellulose oligomers were structurally characterized. The 1H NMR spectrum showed the average DP to be 10 (Fig. S3†). The XRD profile (Fig. 2d) and IR absorption spectrum (Fig. S4†) revealed that the allomorph was cellulose II. The results were the same with those of the cellulose oligomers produced without GOs,25,29 indicating that GOs hardly influenced the chemical and crystal structure of cellulose oligomers. The concentration of the synthesized cellulose oligomers was 4.8 mg mL−1. The αG1P monomer conversion was calculated as ∼15% based on the average DP of 10. The monomer conversion was slightly lower than that for the reaction without GOs (∼35%).29 The lower monomer conversion with GOs was attributed to the capability of GOs to denature39 and inhibit40 enzymes. In addition, the fact that the XRD profile did not show any peak derived from stacking of rGOs41,42 indicates a well-dispersed state of the confined hydrophobic rGOs even in water (Fig. 2d).
SEM observations for the freeze-dried gels revealed a well-grown network structure composed of nanoribbon-shaped fibers (Fig. 3). AFM observations for the mechanically crushed products showed the nanoribbon thickness to be 5.6 ± 0.3 nm (Fig. S5†), which was consistent with the chain length of a cellulose oligomer with DP of 10 in the cellulose II allomorph (5.2 nm).24 The consistency indicates that the nanoribbons were lamellar crystals of cellulose oligomers. Therefore, GOs appeared to prevent cellulose oligomers from aggregating and subsequently precipitating by acting as obstacles, leading to further development of cellulose oligomer assemblies into well-grown nanoribbon networks. This plausible mechanism for the nanoribbon network formation is shared by the cellulose oligomer synthesis in water-soluble polymer solutions31,43,44 and in CNC dispersions.24 Within the nanoribbon networks, the confined rGOs were observed in a well-dispersed state (Fig. 3). This means that the dispersed state of the hydrophobic rGOs was maintained even in water owing to the confinement effect by the robust cellulose oligomer networks.
 | ||
| Fig. 3 SEM images of the freeze-dried gels prepared from the hybrid hydrogels at relatively (a) low and (b) high magnifications. | ||
Assembly of the confined rGOs into three-dimensional porous structures
The hybrid gels composed of the cellulose oligomer nanoribbons and rGOs did not exhibit electrical conductivity owing to the absence of a percolation network of well-dispersed rGOs and/or an insufficient reduction degree of the rGOs. Therefore, we intended to subject the hybrid gels to a strong chemical reduction for constructing conductive rGO gels. The hybrid gels maintained their gel state even after solvent exchange from water to acetic acid under ambient conditions because of the robustness of the crystalline cellulose oligomer assemblies, allowing for a strong reduction reaction by hydrogen iodide in acetic acid at a higher temperature.27,45 The reduction reaction was performed twice to achieve sufficient reduction of rGOs (see below). Even after the reaction, gel state was maintained while shrinkage occurred to some extent (Fig. 4a and b). The resultant gel volume, rGO concentration, and porosity were 22% of that of the raw hybrid gels, 18 mg mL−1, and 99.2%, respectively. The observations suggest that the reduction reaction by hydrogen iodide induced the formation of macroscopic rGO assemblies driven by an increase in the hydrophobicity of the rGOs. In addition, the reduction reaction for hybrid gels with 1 mg mL−1 rGO produced rGO gels with a larger porosity (99.6%) (Fig. S6†). The larger porosity suggests, assuming a similar extent of rGO stacking, the formation of larger pores. The result indicates controllability of porous structure by tuning rGO concentration in the precursor rGO/cellulose oligomer hybrid gels. | ||
| Fig. 4 Photographs of the hybrid gels (a) before and (b) after the reduction reaction by hydrogen iodide. (c) Raman spectrum of the rGOs after the reduction reaction. (d and e) SEM images of the freeze-dried gels prepared from the rGO hydrogels. (f) A photograph showing the rGO gels to be electrically conductive. (g) Current–voltage curve of the rGO gels. | ||
Raman spectroscopy measurements revealed that the ID/IG value of the rGOs after the reduction reaction was 1.26 (Fig. 4c), which was higher than that before the reduction reaction, showing successful further reduction of the rGOs by hydrogen iodide. SEM observations revealed a fine porous structure probably composed of rGOs (Fig. 4d and e). On the other hand, no cellulose oligomer nanoribbon was observed probably due to decomposition of cellulose oligomers under the harsh reaction conditions. In fact, subjecting cellulose oligomer gels without rGOs to the reduction reaction resulted in disappearance of not only the gels in visual observation but also signals from cellulose oligomers in mass spectra obtained by MALDI-TOF mass spectrometry (Fig. S7†). Furthermore, the mass spectra showed new peaks in relatively low m/z range, which were considered to be signals of the degradation products. We could not, however, identify the products and thus the degradation mechanism because of multiple kinds of products and limited information from mass spectrometry. The resultant rGO gels were found to be electrically conductive (Fig. 4f), indicating a percolation network structure composed of conductive rGOs. In fact, the current–voltage curve of the rGO gels exhibited linearity and showed electrical conductivity to be 0.94 ± 0.09 S m−1 (Fig. 4g). The value is comparable to that of previously reported rGO gels.26 Therefore, it was found that a strong reduction reaction for the hybrid gels produced highly conductive porous materials composed of rGOs.
The SEM images of the rGO gels revealed an average pore size of 1.26 ± 0.62 μm in diameter (Fig. 4d and e), which was even smaller than the diameters of the raw GOs (several micrometers, Fig. S1†). Thus far, various rGO gels have been constructed from GO dispersions by employing different reduction reactions (e.g., the hydrothermal reaction26 and reactions using various reducing agents27,28,46). Despite differences in reaction conditions, most of the resultant rGO gels typically had pore sizes of several micrometers. The formation of relatively small pores in this study was attributed to the confinement effect by the robust cellulose oligomer networks; although the nanoribbon networks had been decomposed in the final stage of the reduction reaction, they appeared to serve as a robust confinement space in the initial stage, preventing excessive aggregation of rGOs for the formation of the fine porous structure.
Application of the porous rGO materials to supercapacitor
The rGO gels were composed of porous structures with electrical conductivity. Therefore, we explored their potential as electrode materials for supercapacitors. The electrochemical measurements were performed in the two-electrode system (Fig. S8†) using a 1 M sulfuric acid aqueous solution as the electrolyte. Cyclic voltammograms were nearly rectangular in shape at scan rates of 5–100 mV s−1 (Fig. 5a). The behavior is typical of supercapacitors.47 The fact that no notable peak was observed in the cyclic voltammograms indicates almost no redox reaction in the voltage range of 0–1 V. In addition, the maintenance of rectangular shape in the scan rate range suggests good ion diffusion within the porous rGO materials. Meanwhile, rGO gels produced via only once reduction reaction showed notable peaks in the cyclic voltammograms (Fig. S9†). The result indicates that the twice reduction reactions allowed for the removal of most of redox active groups on rGOs.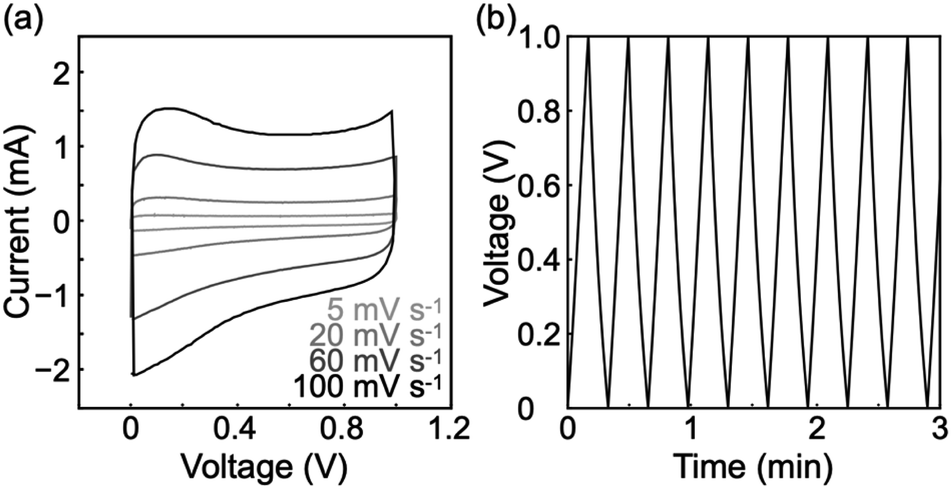 | ||
| Fig. 5 (a) Cyclic voltammograms of the rGO gels at scan rates of 5–100 mV s−1. (b) Charge/discharge curves of the rGO gels at a current density of 5 A g−1. | ||
Galvanostatic charge/discharge measurements showed successful charge and discharge cycles (Fig. 5b and S10†), demonstrating that the porous rGO materials could act as electrode materials for supercapacitors. Symmetry in each charge/discharge cycle supports the absence of a redox reaction and good ion diffusion within the porous rGO materials. The specific capacitance was calculated from the discharge curves in the first 10 cycles at a current density of 5 A g−1 to be 111 ± 10 F g−1, while increases in the current density caused lower specific capacitance values (Table S1†). The specific capacitance and coulombic efficiency after 1000 cycles were 78% of the initial and 98%, respectively (Fig. S11†), showing a good cycle stability of the rGO gels. The specific capacitance of 111 ± 10 F g−1 was comparable to that of commercial supercapacitors made from activated carbons6 and 30% lower than that previously reported for rGO gels produced via a hydrothermal process.26 Nevertheless, the fine porous structure revealed by SEM observations implies a high specific surface area, which had the potential to yield a higher performance as a supercapacitor. In fact, the conditions for constructing the rGO gels and for the electrochemical measurements can still be optimized.
The rGO gels were compared with previously reported rGO/cellulose composites.48 The specific capacitance per rGO mass was higher for the rGO/cellulose composites (172.8 F g−1). However, the specific capacitance per total mass of the materials, which is important in practical applications, was higher for our rGO gels than the rGO/cellulose composites (71.2 F g−1) because cellulose components had been removed in our rGO gels. The advantage of the absence of cellulose components appeared to be derived from the low DP of cellulose oligomers; cellulose oligomers with DP of 10 are easy to be degraded into oligomers with DPs of less than 7, which are water-soluble and thus removable.49 In summary, the porous rGO materials constructed via confinement within the robust cellulose oligomer networks showed promise as electrochemical energy storage devices.
Conclusions
We demonstrated that the robust cellulose oligomer networks served as a confinement space preventing excessive aggregation of rGOs during the assembling process, which led to the formation of a fine porous structure. Considering the plausible mechanism based on simple physical confinement and also the amphiphilicity and physicochemical inertness of cellulose oligomers, the confinement strategy can be applied to other nanomaterials for constructing various functional macroscopic materials with a three-dimensional porous structure. Candidates are one-dimensional nanomaterials represented by carbon nanotubes50 and two-dimensional nanomaterials represented by metal oxide nanosheets51–53 because they typically have micrometer scale lengths in at least one dimension, which should be a requirement for materials that are to be confined within the nanoribbon networks, whose apparent pore sizes are a few micrometers. Even non-water-dispersible nanomaterials can be confined via enzymatic cellulose oligomer synthesis by using surfactants to stabilize the nanomaterial dispersions. Therefore, our findings open a new avenue for the creation of nanostructured functional materials in a bottom-up manner.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors wish to thank the Ookayama Materials Analysis Division (Tokyo Tech) for Raman spectroscopy measurements, XRD analysis, and SEM observations. This study was partially supported by a Grant-in-Aid for Scientific Research (18H02029) from the Japan Society for the Promotion of Science (JSPS) for T. Serizawa and a Grant-in-Aid for JSPS Research Fellow (18J15025) for Y. H. Y. H. is grateful to JSPS for a Research Fellowship for Young Scientists.References
- O. D. Velev and S. Gupta, Adv. Mater., 2009, 21, 1897–1905 CrossRef CAS.
- M. Grzelczak, J. Vermant, E. M. Furst and L. M. Liz-Marzán, ACS Nano, 2010, 4, 3591–3605 CrossRef CAS PubMed.
- Z. Nie, A. Petukhova and E. Kumacheva, Nat. Nanotechnol., 2010, 5, 15–25 CrossRef CAS PubMed.
- N. Gaponik, A.-K. Herrmann and A. Eychmüller, J. Phys. Chem. Lett., 2012, 3, 8–17 CrossRef CAS.
- R. K. Joshi and J. J. Schneider, Chem. Soc. Rev., 2012, 41, 5285–5312 RSC.
- Y. Xu, G. Shi and X. Duan, Acc. Chem. Res., 2015, 48, 1666–1675 CrossRef CAS PubMed.
- K. Thorkelsson, P. Bai and T. Xu, Nano Today, 2015, 10, 48–66 CrossRef CAS.
- C. Ziegler, A. Wolf, W. Liu, A.-K. Herrmann, N. Gaponik and A. Eychmüller, Angew. Chem., Int. Ed., 2017, 56, 13200–13221 CrossRef CAS PubMed.
- T. Bollhorst, K. Rezwan and M. Maas, Chem. Soc. Rev., 2017, 46, 2091–2126 RSC.
- D. Wen and A. Eychmüller, Chem. Commun., 2017, 53, 12608–12621 RSC.
- L. Y. T. Chou, K. Zagorovsky and W. C. W. Chan, Nat. Nanotechnol., 2014, 9, 148–155 CrossRef CAS PubMed.
- W. Liu, J. Halverson, Y. Tian, A. V. Tkachenko and O. Gang, Nat. Chem., 2016, 8, 867–873 CrossRef CAS PubMed.
- Y. Tian, Y. Zhang, T. Wang, H. L. Xin, H. Li and O. Gang, Nat. Mater., 2016, 15, 654–661 CrossRef CAS PubMed.
- F. Caruso, R. A. Caruso and H. Möhwald, Science, 1998, 282, 1111–1114 CrossRef CAS PubMed.
- M. A. Correa-Duarte, J. Pérez-Juste, A. Sánchez-Iglesias, M. Giersig and L. M. Liz-Marzán, Angew. Chem., Int. Ed., 2005, 44, 4375–4378 CrossRef CAS PubMed.
- J. Guo, B. L. Tardy, A. J. Christofferson, Y. Dai, J. J. Richardson, W. Zhu, M. Hu, Y. Ju, J. Cui, R. R. Dagastine, I. Yarovsky and F. Caruso, Nat. Nanotechnol., 2016, 11, 1105–1111 CrossRef CAS PubMed.
- K. M. Ryan, A. Mastroianni, K. A. Stancil, H. Liu and A. P. Alivisatos, Nano Lett., 2006, 6, 1479–1482 CrossRef CAS PubMed.
- K. Wang, S.-M. Jin, J. Xu, R. Liang, K. Shezad, Z. Xue, X. Xie, E. Lee and J. Zhu, ACS Nano, 2016, 10, 4954–4960 CrossRef CAS PubMed.
- A. Ahniyaz, Y. Sakamoto and L. Bergström, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 17570–17574 CrossRef CAS PubMed.
- L. Hu, R. Zhang and Q. Chen, Nanoscale, 2014, 6, 14064–14105 RSC.
- J. R. Capadona, O. Van Den Berg, L. A. Capadona, M. Schroeter, S. J. Rowan, D. J. Tyler and C. Weder, Nat. Nanotechnol., 2007, 2, 765–769 CrossRef CAS PubMed.
- N. C. Bigall, A.-K. Herrmann, M. Vogel, M. Rose, P. Simon, W. Carrillo-Cabrera, D. Dorfs, S. Kaskel, N. Gaponik and A. Eychmüller, Angew. Chem., Int. Ed., 2009, 48, 9731–9734 CrossRef CAS PubMed.
- H. Tsurusawa, M. Leocmach, J. Russo and H. Tanaka, Sci. Adv., 2019, 5, eaav6090 CrossRef PubMed.
- Y. Hata, T. Sawada, T. Sakai and T. Serizawa, Biomacromolecules, 2018, 19, 1269–1275 CrossRef CAS PubMed.
- Y. Hata, T. Sawada, H. Marubayashi, S. Nojima and T. Serizawa, Langmuir, 2019, 35, 7026–7034 CrossRef CAS PubMed.
- Y. Xu, K. Sheng, C. Li and G. Shi, ACS Nano, 2010, 4, 4324–4330 CrossRef CAS PubMed.
- W. Chen and L. Yan, Nanoscale, 2011, 3, 3132–3137 RSC.
- K. Sheng, Y. Xu, C. Li and G. Shi, New Res. Carbon Mater., 2011, 26, 9–15 CrossRef CAS.
- T. Serizawa, M. Kato, H. Okura, T. Sawada and M. Wada, Polym. J., 2016, 48, 539–544 CrossRef CAS.
- C. Gao, S. Zhang, F. Wang, B. Wen, C. Han, Y. Ding and M. Yang, ACS Appl. Mater. Interfaces, 2014, 6, 12252–12260 CrossRef CAS PubMed.
- Y. Hata, T. Kojima, T. Koizumi, H. Okura, T. Sakai, T. Sawada and T. Serizawa, ACS Macro Lett., 2017, 6, 165–170 CrossRef CAS.
- M. Hiraishi, K. Igarashi, S. Kimura, M. Wada, M. Kitaoka and M. Samejima, Carbohydr. Res., 2009, 344, 2468–2473 CrossRef CAS PubMed.
- A. C. Ferrari and D. M. Basko, Nat. Nanotechnol., 2013, 8, 235–246 CrossRef CAS PubMed.
- X. Fan, W. Peng, Y. Li, X. Li, S. Wang, G. Zhang and F. Zhang, Adv. Mater., 2008, 20, 4490–4493 CrossRef CAS.
- S. Pei and H.-M. Cheng, Carbon, 2012, 50, 3210–3228 CrossRef CAS.
- O. Akhavan, E. Ghaderi, S. Aghayee, Y. Fereydooni and A. Talebi, J. Mater. Chem., 2012, 22, 13773–13781 RSC.
- J. K. Grady, N. D. Chasteen and D. C. Harris, Anal. Biochem., 1988, 173, 111–115 CrossRef CAS PubMed.
- A. Habib, M. Tabata and Y. G. Wu, Bull. Chem. Soc. Jpn., 2005, 78, 262–269 CrossRef CAS.
- L. Jin, K. Yang, K. Yao, S. Zhang, H. Tao, S.-T. Lee, Z. Liu and R. Peng, ACS Nano, 2012, 6, 4864–4875 CrossRef CAS PubMed.
- M. De, S. S. Chou and V. P. Dravid, J. Am. Chem. Soc., 2011, 133, 17524–17527 CrossRef CAS PubMed.
- X. Zhang, Z. Sui, B. Xu, S. Yue, Y. Luo, W. Zhan and B. Liu, J. Mater. Chem., 2011, 21, 6494–6497 RSC.
- J.-H. Cheng, Y.-H. Chen, Y.-S. Yeh, S. Hy, L.-Y. Kuo and B.-J. Hwang, Electrochim. Acta, 2016, 197, 146–151 CrossRef CAS.
- Y. Hata, T. Sawada and T. Serizawa, Polym. J., 2017, 49, 575–581 CrossRef CAS.
- Y. Hata, T. Sawada and T. Serizawa, J. Mater. Chem. B, 2018, 6, 6344–6359 RSC.
- I. K. Moon, J. Lee, R. S. Ruoff and H. Lee, Nat. Commun., 2010, 1, 73 CrossRef PubMed.
- Z. Sui, X. Zhang, Y. Lei and Y. Luo, Carbon, 2011, 49, 4314–4321 CrossRef CAS.
- Y. Wang, Y. Song and Y. Xia, Chem. Soc. Rev., 2016, 45, 5925–5950 RSC.
- W. Ouyang, J. Sun, J. Memon, C. Wang, J. Geng and Y. Huang, Carbon, 2013, 62, 501–509 CrossRef CAS.
- Y.-H. P. Zhang and L. R. Lynd, Biotechnol. Bioeng., 2004, 88, 797–824 CrossRef CAS PubMed.
- M. F. L. De Volder, S. H. Tawfick, R. H. Baughman and A. J. Hart, Science, 2013, 339, 535–539 CrossRef CAS PubMed.
- M. Osada and T. Sasaki, J. Mater. Chem., 2009, 19, 2503–2511 RSC.
- A. H. Khan, S. Ghosh, B. Pradhan, A. Dalui, L. K. Shrestha, S. Acharya and K. Ariga, Bull. Chem. Soc. Jpn., 2017, 90, 627–648 CrossRef.
- K. Maeda and T. E. Mallouk, Bull. Chem. Soc. Jpn., 2019, 92, 38–54 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra08318a |
| This journal is © The Royal Society of Chemistry 2019 |