
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCd(II) removal by Fe(II) surface chemically modified layered double hydroxide–graphene oxide: performance and mechanism†
Wei Liao,
He Wang,
Hui-qiang Li * and
Ping Yang
* and
Ping Yang
College of Architecture and Environment, Sichuan University, Chengdu 610065, China. E-mail: Lwei0314@163.com; wh_820@163.com; lhq_scu@163.com; yping63@163.com; Tel: +86 18200288011 Tel: +86 18011446490 Tel: +86 18980668020 Tel: +86 18602804508
First published on 28th November 2019
Abstract
Cd(II) adsorption onto Fe(II) modified Layered double hydroxide–graphene oxide (LDH–GO@Fe(II)) was investigated using batch experiments. With the modification of Fe(II), LDH–GO maintained its structure, while Fe(II) species formed non-crystalline iron oxide clusters on the surface of the LDH/GO. A kinetics study indicated that adsorption obeyed a pseudo-second-order rate law. The equilibrium data were fitted well with the Langmuir isotherm model. The maximum adsorption capacity of LDH–GO@Fe(II)10 was 28.98 mg g−1, higher those that of pure LDH–GO and LDH–GO@Fe(II)50. The increased sorption capacities could be explained by the increased specific surface area. Modification with Fe(II) would lead to the generation of amorphous Fe oxides and Fe could occupy the binding sites for Cd(II), thus excess Fe in the structure will restrain the adsorption of Cd(II). The XRD and XPS patterns revealed the formation of Cd(OH)2 after adsorption. Batch experiments indicated that precipitation and surface complexation were the main pathways for Cd(II) removal.
1. Introduction
With the rapid development and extensive applications of heavy metals and their composites, release of heavy metals into the environment is inevitable and harmful. Cadmium is an extremely toxic heavy metal to humans and the environment even at low levels.1 The development of methods to remove trace potentially toxic metal contaminants from aqueous wastewater is one of the most important environmental issues being investigated. Among the techniques, the precipitation of metals as insoluble metal hydroxides via increment of the pH is the most applied approach.2 However, precipitation produces a large amount of sludge, and generates secondary pollution, and is incapable of removing trace metal contaminants.3,4 In comparison, adsorption has shown superiority in the removal of heavy metals and is extensively researched due to its low cost, high efficiency and easy operation.5 Different materials have been studied intensively to eliminate heavy metals from aqueous solutions. Most materials suffer from low efficiency or low adsorption capacity, which restrict their applications.6 Fiol et al.7 prepared AC from olive stone waste to remove heavy metals from aqueous solutions, and the highest value of the Langmuir maximum adsorption capacity for Cd(II) was found to be 7.73 mg g−1. Bertagnolli et al.8 employed a calcined bentonite clay to remove Cu(II), which presented the Cu(II) maximum adsorption capacity of 11.89 mg g−1.In recent years, exploring hierarchical nanocomposites with multifunctional properties through combining various building blocks together into a well-designed structure has been a hot topic for material science.9 The composite materials composed of blocks with different physical and chemical properties can usually inherit full advantages of the component materials and even form multifunctional materials with unexpected properties for unique applications.10 Recent studies have documented that hierarchical composites have superb removal capacities for pollutants on the basis of the boosting of abundant active sites and favorable mass transfer, overcoming the demerits associated with agglomeration and low efficiency in the common nanoparticles.11 Over past few years, there are great interests in the synthesis of porous material supported layered double hydroxides (LDH) because of their high surface area, gap structure and unique property.12 The composites can provide stable sites for pristine LDH particles loading to restrain the aggregation of the particles to some extent, which thereby increases the surface area and stable sites for the removal of pollutants from environment. Among these composites, graphene materials supported LDH attracts the most research interests due to the superior physicochemical properties. With the combination, the disadvantage of aggregation can be effectively prevented, and their distinguishing properties can be integrated together. Graphene can provide the good electrical conductivity and high mechanical strength, while LDHs can provide good chemical reactivity.9 Thus, an electron pathway can be easily formed and a network of stress transfer can be also presented between the graphene and LDHs, which is an important aspect as advanced functional materials. In another aspect, such structured composites are always with a porous structure, which make the heat and mass transfers during a reaction greatly improved and make active sites easily exposed.10 Some studies on LDH–graphene composites and their use for elimination of metal ions have been reported: Pb(II),13 U(VI)14 and Cr(VI).15 Our study also showed the LDH–GO composites had an outstanding removal capacity for Fe(II).16 At the Fe(II) concentration of 100 mg L−1, Fe2O3 and FeOOH were formed in the inner space of the LDH–G and the structure of LDH–G had an obvious alteration. Introducing Fe has been reported to be one of the efficient methods for adsorbent modification.17–19 Iron-modified materials showed high affinity for metals due to their large surface area, microporous structure and internal surface.20 Doula21 synthesized clin–Fe by adding natural clinoptilolite in an iron nitrate solution under strongly basic condition, and the clin–Fe was proven for its ability to adsorb high concentration Cu(II). Yang et al.22 reported corn stalk-derived biochar impregnate with iron nanoparticles to serve as an adsorbent, which showed rapid removal and high performance in single and mixed metal solutions. To the best of our knowledge, no attempt has been made to remove Cd(II) by Fe-decorated LDH–graphene composites. Fe-decorated LDH–graphene composites probably provide us a simple method for the efficient elimination of environmental pollutants from aqueous solution.
Therefore, the objectives of this study were (i) to characterize the Fe(II)-modified LDH/GO (LDH–GO@Fe(II)); (ii) to evaluate its adsorption kinetics and adsorption capacity for Cd(II); (iii) to elucidate the mechanisms involved in adsorption of Cd(II) by LDH–GO@Fe(II). Scanning electron microscopy, X-ray photoelectron spectroscopy, and X-ray powder diffraction patterns were used to better understand the mechanisms of Cd(II) removal. It is expected that the outcomes from the study would provide understanding for the application of LDH–GO@Fe(II) as an efficient adsorbent for heavy metal removal.
2. Materials and methods
2.1. Materials
Purified natural graphite powder was purchased from Sinopharm Chemical Reagent Co. Ltd. Other reagents were analytical grade and purchased from Sigma Aldrich (Shanghai) Co. Ltd. without further purification. Glassware was soaked in 10–20% HCl solution for 24 h and rinsed sequentially three times with distilled water.2.2. Fabrication of the adsorbents
GO was synthesized by Hummers methods as described elsewhere.23 The detailed processes were illustrated in the ESI.† LDH–GO composites were synthesized via the co-precipitation methods. Briefly, 0.1 g of GO powder in 100 mL water was ultra-sonicated for 1 h and then stirred vigorously with a magnetic stirrer. A 100 mL mixed metal solution of MgCl2·6H2O and AlCl3·9H2O was prepared in deionized water with a total metal ion concentration of 0.4 mol L−1 and a Mg2+/Al3+ molar ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Another 100 mL alkaline solution of 0.6 mol L−1 NaOH and 0.2 mol L−1 Na2CO3 was also prepared. Both the mixed metal solution and alkaline solution were simultaneously dropwise added to the GO suspension under vigorous stirring at room temperature. The pH was maintained at 10 ± 0.5 during the process. The results suspension was stirred for another 4 h at room temperature, and then was aged in a water bath at 65 °C for 4 h. The precipitate was centrifuged, washed thoroughly with distill water until the washings were neutral. The precipitate was then dried at 65 °C overnight. The resulting powder was designated as LDH–GO. Hierarchical Fe-decorated LDH–GO composites were prepared by adding a certain amount of LDH–GO to Fe(II)-containing (5, 10, 50, 100 mg L−1) solution and reacting for 1 h, and noted as LDH–GO@Fe(II)x (x = 5, 10, 50, 100).
:1. Another 100 mL alkaline solution of 0.6 mol L−1 NaOH and 0.2 mol L−1 Na2CO3 was also prepared. Both the mixed metal solution and alkaline solution were simultaneously dropwise added to the GO suspension under vigorous stirring at room temperature. The pH was maintained at 10 ± 0.5 during the process. The results suspension was stirred for another 4 h at room temperature, and then was aged in a water bath at 65 °C for 4 h. The precipitate was centrifuged, washed thoroughly with distill water until the washings were neutral. The precipitate was then dried at 65 °C overnight. The resulting powder was designated as LDH–GO. Hierarchical Fe-decorated LDH–GO composites were prepared by adding a certain amount of LDH–GO to Fe(II)-containing (5, 10, 50, 100 mg L−1) solution and reacting for 1 h, and noted as LDH–GO@Fe(II)x (x = 5, 10, 50, 100).
2.3. Batch experiment
The work Cd(II) solutions were prepared at various concentrations by diluting the stock Cd(II) solution with fresh distilled water. The experiments were carried out in a 250 mL glass conical flask by adding 0.2 g of adsorbents in 200 mL of the Cd(II)-containing solution with continuous stirring over a water bath magnetic stirrer at 500 rpm. If not otherwise specified, the solution pH, temperature and initial concentration of cadmium in the cadmium solutions were 5, 25 °C and 20 mg L−1, respectively. For kinetics study, the suspensions were withdrawn at a given specific time intervals and then immediately filtrated through a 0.45 μm filter membrane, and the concentration of the residual cadmium in the filtrates was analyzed. Cadmium concentration was measured by the spectrophotometric method using 1-(2-pyrdylazo)-2-naphthol (PAN),24 the detailed operation is illustrated in the ESI.† Adsorbed cadmium was calculated from the difference between the initial cadmium concentration and the concentration that remained in the supernatant solution. Each experiment was conducted in duplicate. The dissolution of iron ions was monitored. No iron was detected during the process of Cd(II) sorption by LDH–GO@Fe(II)x. The blank checks were performed to verify the absence of adsorbate precipitation and/or adsorption to the walls of the vessels.2.4. Kinetic studies
The pseudo-first-order and pseudo-second-order kinetic were used to analyze adsorption kinetic data. The linear form of the pseudo-first-order (1) and the pseudo-second-order (2) equations can be expressed as follows:25|
ln(qe − qt) = lnqe − k1t
| (1) |
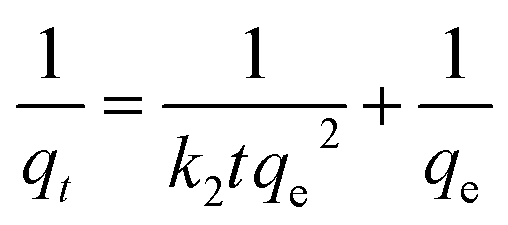 | (2) |
2.5. Adsorption isotherm
Isothermal adsorption model is an effective method to evaluate the interaction mechanism and maximum adsorption capacity.26 Generally, Langmuir and Freundlich adsorption isothermal models are applied to deal with the situation of water treatment. The Langmuir model is based on the hypothesis that the adsorption process occurs in the form of monolayer adsorption on the homogeneous surfaces, which can be depicted by the following equation:
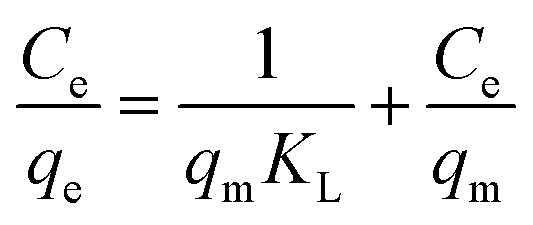 | (3) |
Freundlich model normally represents the multilayer adsorption on the heterogeneous solid surfaces, which can be described as:
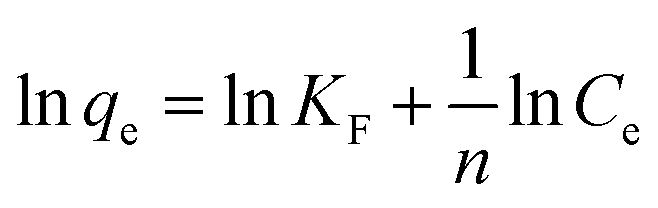 | (4) |
2.6. Characterization
The images of synthesized samples before and after adsorbing Cd(II) were captured by a scanning electron microscope (SEM) (JSM-7500F, Japan). The specific surface areas and pore structures of the samples were detected by nitrogen adsorption based on Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) methods using N2 adsorption–desorption at 77 K on a surface area analyzer (ASAP2020, USA). The X-ray powder diffraction (XRD) patterns were carried out by a powder diffractometer using Cu Kα radiation at a scanning speed of 2° min−1 (PANalytical B.V., Holland). X-ray photoelectron spectroscopy (XPS) tests were measured on an AXIS Ultra DLD (Shimadzu, Japan) using monochromatic Al Kα X-ray source.3. Results and discussion
3.1. Structure and morphologies of as-prepared particles
The structures of the obtained particles were characterized by XRD to determine the crystal structure and its integrity in Fig. S1.† The as-prepared LDH–GO particles were principally composed of a hexagonal LDHs phase and exhibited their characteristic diffractions, such as the peaks [003], [006], [012], [110] and [113], with a d-spacing of 0.780 nm.27 It has to be noted that no diffraction peaks of impurities were discerned, which suggested high purity of the sample. The hydrolyzed LDH–GO (H-LDH–GO) exhibited an XRD pattern similar to the pristine LDH–GO, indicating the good stability of the LDH–GO in the water. The LDH–GO@Fe(II)x particles (LDH–GO@Fe(II)10 and LDH–GO@Fe(II)50) showed similar diffraction peaks as LDH–GO particles but slight change in d-spacings and relative intensities of peaks. This observation matched well with the structure of LDH–GO, which suggested that the layered structures well retained after Fe(II)-decoration. Compared with the pristine LDH–GO, the diffraction peaks intensity of the H-LDH–GO and LDH–GO@Fe(II)x were lower, which was due to slightly decomposition of the LDH–GO. The change in crystal lattice parameters in XRD patterns after Fe-modification was due to isomorphic substitution of metal ions in the structure. Isomorphic substitution would lead to the change in the d-spacings and relative intensities of peaks that reflected lattice parameters of layer framework.28 This might due to partial isomorphic substitution of Mg(II) by Fe(II) during the modification. And then the Fe(II) were oxidized to Fe(III), which was confirmed by XPS.Due to the highly variable chemistry of iron, there are various possible structures of Fe species in composites. In most cases, single di- and tri-valent Fe, oxo- and hydroxo-complexes, polymeric oxidic species and iron oxide species are present simultaneously.21 The XRD patterns revealed no notable changes in the parent material diffraction peaks, which indicated no detectable damage to the LDH/GO, nor the presence of additional Fe oxidic or oxo-hydroxidic crystalline phases.21 This absence of additional peaks was due to the amorphous nature of the Fe-phases formed at LDH/GO surface sites, which was in line with the previous literatures.20,29
The wide scan XPS spectrum of H-LDH–GO and LDH–GO@Fe(II)10 composites are shown in Fig. 1(a). The most significant features in these spectrums were the Mg 1s, Al 2p, Fe 2p, O 1s, and C 1s signals. The peaks due to Fe revealed the presence of Fe species in the LDH–GO@Fe(II)10 composite. The detailed spectra of the Fe 2p is illustrated in Fig. 1(b). The peaks observed at ∼725 and ∼711 eV were assigned to Fe 2p1/2 and Fe 2p3/2, respectively.30 For stoichiometry evaluation of Fe, only Fe 2p3/2 was analyzed, and the Fe 2p1/2 was used as reference for the fitting procedure based on previous literatures.31,32 The signal of Fe(II) peak contribution was found at about 711.0 eV. Another peak observed at about the binding energy of 713.0 eV was assigned to Fe(III).33 It revealed the oxidation of Fe(II) in the structure. The broad O 1s peaks suggested the existence of various oxygen-containing compounds, referred to organic oxygen (O contained in carboxyl, carbonyl and alkoxy groups) and inorganic oxygen.34 In Fig. 1(c), three peaks at 529, 530, and 531 eV were attributed to M–O, C–O/C–OH, and H2O bonds, respectively.35
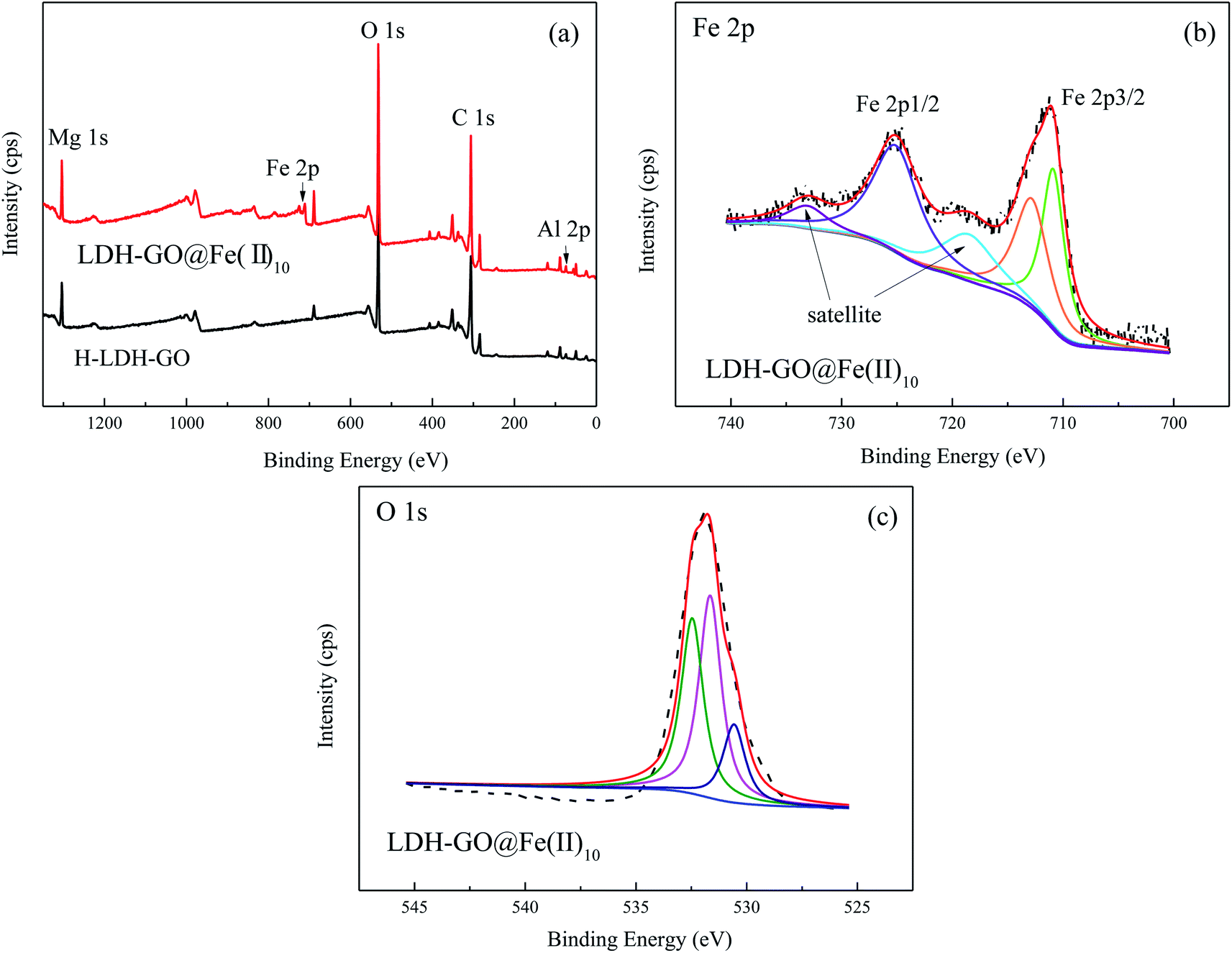 | ||
| Fig. 1 (a) Wide scan, (b) Fe 2p and (c) O 1s XPS spectra. | ||
The SEM micrograph and EDS spectra of the H-LDH–GO, LDH–GO@Fe(II)10 and LDH–GO@Fe(II)50 particles are shown in Fig. S2.† The EDS spectra results revealed that the composition of the H-LDH–GO adsorbent was predominated by Mg, Al, C and O. After modification with Fe(II), the Fe peak was clearly detected in the LDH–GO@Fe(II)10 and LDH–GO@Fe(II)50 samples. The results indicated that Fe was successfully grafted onto the LDH–GO surface during the modification process. The surface content of Fe for LDH–GO@Fe(II)50 was 34.95% (atomic%), which was about 5 times than that of LDH–GO@Fe(II)10. The formation of Fe-decorated microsphere was mainly through the following reaction: (i) the Fe(II) isomorphic substituted of Mg(II); (ii) partial divalent iron was converted to trivalent iron upon reaction with oxygen (from dissolved air); (iii) with the amount of Fe(III) in the structure increased, the stability of LDH–GO became weaker and a fraction of the structure collapsed and Fe(III) released into the solution. The released Fe(III) reacted with OH− forming precipitated Fe(OH)3 that subsequently transformed into FeOx;36 (iv) The GO and iron formed strong surface complexes through the Lewis acid–base interaction. As can be seen from Fig. 3(a), the morphologies of H-LDH–GO particles were similar to the LDH–GO reported in other reports.13 The LDH sheets arrays grew on both sides of the GO sheets. The inhomogeneous flakes were smooth and flat. The flakes stacked together to form agglomerates in some areas. When the initial concentration of Fe(II) was 10 mg L−1, the surface of LDH–GO@Fe(II)10 presented a large area continuous porous-flocculent structure, but they did not form separate flakes. When the initial concentration of Fe(II) increased to 50 mg L−1, the flakes formed the basic substrate and almost every flake was upright, and some flakes clustered together on the top of the substrate.37 This may be due to the Fe(II) transferred into parallel lamellae and stretched the distance between the flakes. Moreover, the surface functional groups of the LDH–GO possibly formed intensive electrostatic and chemical interactions with metal ions. The interactions could drag Fe(II) into inner space, which lead parallel flakes more loosely and become upright orientation. The results showed that the LDH–GO@Fe(II) particles had porous structures, which provided effective adsorption sites.
The N2 adsorption and desorption isotherms are used to measure the surface area and pore structure of the obtained particles. The BET and BJH methods are applied to determine the surface area and the pore size distribution considering the desorption and adsorption branch of the N2 isotherms, respectively. The nitrogen adsorption–desorption isotherms are showed in Fig. S3.† All the isotherms were type III-like adsorption isotherms with a H3-type hysteresis loop for the desorption isotherms. The results revealed the particles exhibited mesoporous structures. The obtained specific surface areas were 73.3, 84.4 and 88.3 m2 g−1 for the H-LDH–GO, LDH–GO@Fe(II)10 and LDH–GO@Fe(II)50, respectively. The average pore sizes of the H-LDH–GO, LDH–GO@Fe(II)10 and LDH–GO@Fe(II)50 were 13.11, 11.76 and 11.73 nm. The pore volume of the H-LDH–GO, LDH–GO@Fe(II)10 and LDH–GO@Fe(II)50 were 0.23, 0.24 and 0.26 cm3 g−1, respectively. The modification lead to a change in specific surface area and pore size while no significant difference in pore specific volume. The modified LDH–GO (LDH–GO@Fe(II)10 and LDH–GO@Fe(II)50) had specific surface areas larger than that of the unmodified LDH–GO and thus, and its average pore sizes were smaller. A change in the average pore size range was likely due to the iron formations coating. The iron formations possibly blocked certain pores of the LDH–GO support, in particular small diameter pores.38 The results indicated that Fe(II)-decoration affected the pore structure of the LDH–GO, which increased its specific surface area. The increase in specific surface area was due to the presence of non-crystalline Fe formations located at the surface of the LDH/GO crystal. Generally, a poorly crystalline phase is desirable for adsorption because the lack of a 3D crystalline structure results in high specific surface area.21 The enlarged surface area and pore size of LDH–GO@Fe(II)10 and LDH–GO@Fe(II)50 particles, made Cd(II) more freely diffuse and moved into the larger meso-channels, and readily be removed.
3.2. Cd(II) removal by LDH–GO@Fe(II) composites
It is shown in Fig. 2 that the Cd(II) adsorption performance depended on the initial Fe(II) concentrations. The removal rate of Cd(II) increased from 66.8% of the H-LDH–GO to 77.2% of the LDH–GO@Fe(II)10. It is interesting to observe that with the further increase of Fe content in the modified LDH–GO, the removal capacity for Cd(II) removal had a decline. The LDH–GO@Fe(II)10 composites showed the highest removal capacity for Cd(II).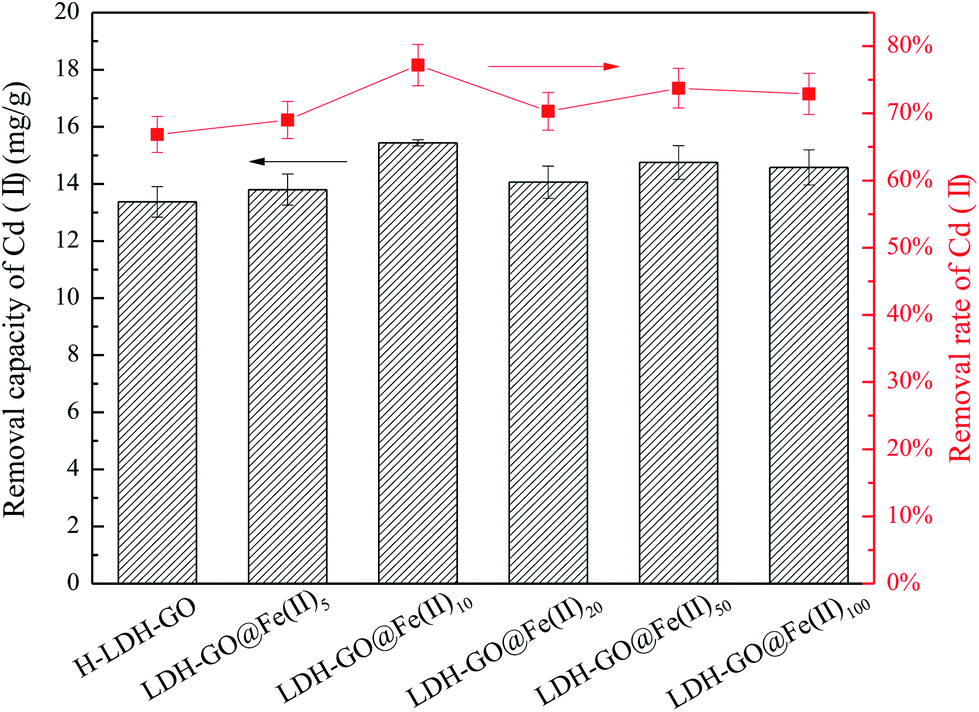 | ||
| Fig. 2 The removal performance of Cd(II) on LDH–GO@Fe(II)x. | ||
Generally speaking, adsorbents own large surface area and porosity have great adsorption capacity for adsorbates. The LDH–GO@Fe(II)10 composites had smaller surface area and pore volume than LDH–GO@Fe(II)50, but showed higher removal capacities for Cd(II). This phenomenon demonstrated that doping a certain amount of Fe(II) into LDH–GO particles can enlarge the surface area and provide more adsorption sites. But, excessive Fe(II) will restrain the adsorption for Cd(II). When the composites exposed to Fe(II)-containing solution, isomorphic substitution of Mg(II) took place. Replacement ions must have the same total ionic charge and approximately the same size as those replace.39 Fe(II) substituted part of Mg(II) through isomorphous, and then Fe(II) were oxidized to Fe(III) on the surface of the LDH–GO. The results of XPS spectrums also confirmed the existence of Fe(III) in the composites. The Fe(III) in the structure of LDH–GO made the brucite-like sheets more positively charged, thus formed an electrostatic repulsion with positively charged Cd(II). On the other hand, the delocalized π electron systems of graphene layer can act as Lewis base to form electron donor–acceptor complexes with metal ions.40 During the decoration of Fe(II) on the LDH–GO, strong surface complexation between the GO and Fe(II) and Fe(III) occurs through the Lewis acid–base interaction, which also occupied part of the adsorption sites on GO sheets. As a result, 10 mg L−1 was the optimum precursor concentration for LDH–GO modification by Fe(II).
Adsorption kinetic tests for Cd(II) (Fig. 3) were performed to evaluate the contact time needed for sorption equilibrium, with an initial Cd(II) concentration of 20 mg L−1, the sorbent dosage of 1.0 g L−1, the pH value of 5.0 and the temperature of 25 °C. The equilibrium solution pH after sorption was about 6. It is possible that the increase in pH was due to the alkalinity of the adsorbent. The results are shown in Fig. 3, the adsorption amount significantly increased within the first 4 h, followed by a slow increase until equilibrium was reached. The necessary time to reach equilibrium was about 12 h. To ensure adsorption equilibrium, a contact time of 24 h was chosen.
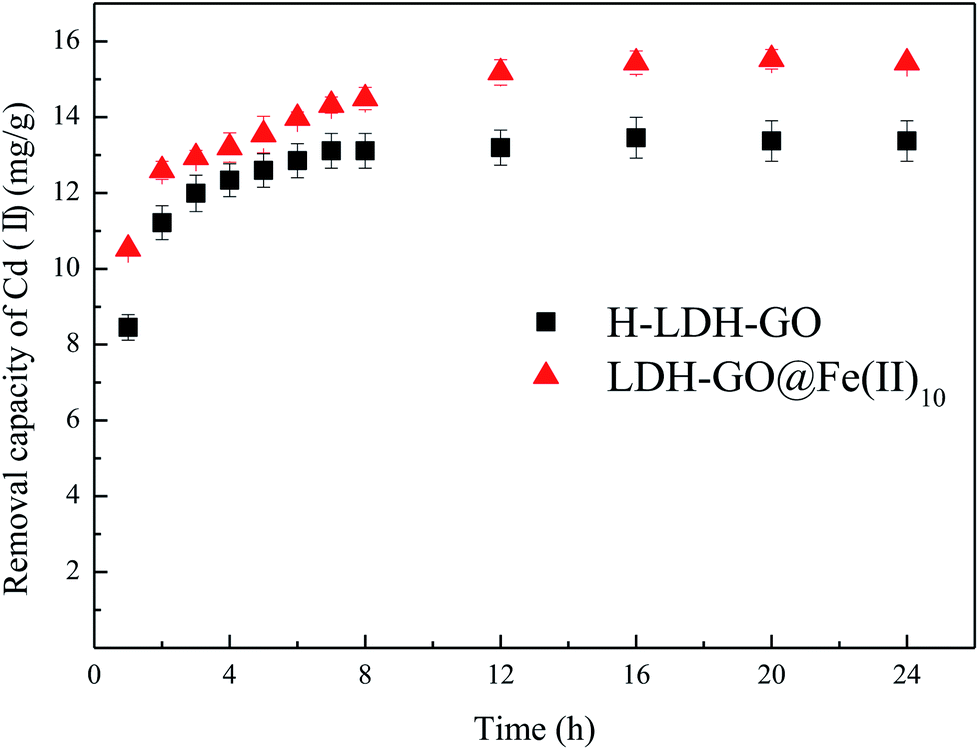 | ||
| Fig. 3 Effect of contact time on the adsorption of Cd(II) onto H-LDH–GO and LDH–GO@Fe(II)10. | ||
The suitable results obtained from different models are summarized in Table 1. It can be seen that the correlation coefficient of the pseudo-second-order model (R2 = 0.999) was much higher than the pseudo-first order. Therefore, the adsorption kinetics followed the pseudo-second-order model, suggesting a chemisorption process. The chemisorption mechanism involved formation of chemical bonds between the adsorbents function groups and the metal ions.41,42
| Adsorbents | qe,exp (mg g−1) | Pseudo-first order | Pseudo-second order | ||||
|---|---|---|---|---|---|---|---|
| qe,cal (mg g−1) | k1 (min−1) | R2 | qe,cal (mg g−1) | k2 (g mg−1 min−1) | R2 | ||
| H-LDH–GO | 13.37 | 13.02 | 1.0092 | 0.9681 | 13.68 | 0.1718 | 0.9998 |
| LDH–GO@Fe(II)10 | 15.43 | 14.69 | 1.2357 | 0.8995 | 15.97 | 0.0871 | 0.9996 |
The adsorption isotherm is shown in Fig. 4 and the relative parameters calculated from the two models are listed in Table 2. Based on the Langmuir parameters, LDH–GO@Fe(II)10 exhibited a higher affinity for Cd(II) than H-LDH–GO and LDH–GO@Fe(II)50, as reflected by its higher adsorption capacity. However, the Fe content of LDH–GO@Fe(II)10 was much lower than that of LDH–GO@Fe(II)50, which indicated that the Fe species in LDH–GO@Fe(II)10 were more efficient. The sorption isotherms were better fitted by the Langmuir model than by the Freundlich model. The increased sorption capacities can be explained by the increased specific surface area and the high affinity of amorphous Fe oxides with Cd(II) on the surface of LDH–GO@Fe(II)10. The previous study inferred that the FeMnOx phases of FMBC contributed to the sorption of Cd(II) and Cu(II), the increased uptake was mainly due to the formation of strong mono- or multidentate inner-sphere complexes (e.g., COO–M (M = Cu or Cd) and Fe–Mn–O–M).34 Previous studies showed the oxygenic functional groups on GO can bind with heavy metal ions and form complexes.43,44 It can be speculated that the adsorption of Cd(II) was own to the formation complexes between graphene oxide (e.g., COO–Cd and O–Cd) and Fe species (FeOx–Cd). Excess Fe species in LDH–GO@Fe(II)50 occupied the binding sites for Cd(II) and Fe(III) in LDH–GO@Fe(II)50 brought positive charge to adsorbent which hindered Cd(II) adsorption via electrostatic repulsion.
 | ||
| Fig. 4 Adsorption isotherm of Cd(II) onto H-LDH–GO, LDH–GO@Fe(II)10 and LDH–GO@Fe(II)50. | ||
| Adsorbents | qe,exp (mg g−1) | Langmuir | Freundlich | ||||
|---|---|---|---|---|---|---|---|
| qe,cal (mg g−1) | KL (L mg−1) | R2 | KF (mg1−n Ln g−1) | n | R2 | ||
| GO | 6.88 | 6.91 | 0.74 | 0.9971 | 2.73 | 3.53 | 0.9074 |
| LDH | 16.56 | 16.39 | 0.95 | 0.9918 | 6.84 | 3.51 | 0.7428 |
| LDH–GO | 19.80 | 19.96 | 1.28 | 0.9972 | 8.10 | 3.25 | 0.7447 |
| H-LDH–GO | 19.50 | 19.68 | 0.95 | 0.9934 | 7.50 | 3.19 | 0.7872 |
| LDH–GO@Fe(II)10 | 28.98 | 29.41 | 1.10 | 0.9944 | 10.60 | 2.82 | 0.7917 |
| LDH–GO@Fe(II)50 | 24.01 | 24.45 | 0.96 | 0.9917 | 8.80 | 2.93 | 0.8025 |
3.3. Removal mechanism
To further investigate the Cd(II) removal mechanisms on the Fe-decorated LDH–GO particles, the samples after adsorption of Cd(II) were characterized by XRD, SEM/EDS, and XPS techniques.Fig. 5(a) illustrates the XRD patterns of the final solid sample after treating 20 mg L−1 Cd(II). The peaks attributed to the LDH–GO@Fe(II)10 were impaired. The main phases were indexed Cd(OH)2 or/and Cd(OH)Cl,45 which indicated the precipitation form of cadmium on the surface of LDH–GO@Fe(II)10.
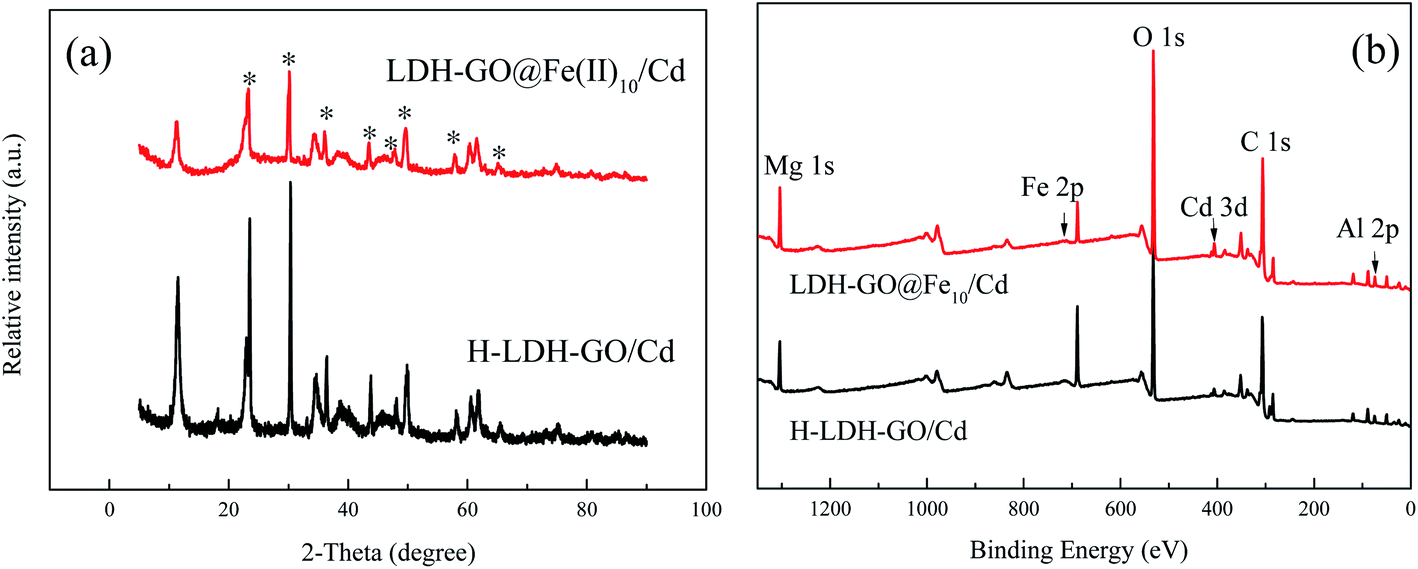 | ||
| Fig. 5 (a) The XRD patterns of H-LDH–GO and LDH–GO@Fe(II)10 after adsorption of Cd(II) (the asterisk signs indicate Cd(OH)2 or Cd(OH)Cl); (b) XPS survey scan of H-LDH–GO and LDH–GO@Fe(II)10 after removal of Cd(II). | ||
In order to further prove the presence of Cd on LDH–GO@Fe(II)10, the content of various elements on the surface of LDH–GO@Fe10 after Cd(II) adsorption was measured by EDS (Fig. S3†). The EDS spectrum showed 0.80% (atomic%) of Cd was loaded on the surface of LDH–GO@Fe(II)10. The content of Fe on the surface of LDH–GO@Fe(II)10 dramatically declined to 0.79% (atomic%), compared to the original LDH–GO@Fe(II)10 (6.10%). The content of O on the surface of LDH–GO@Fe(II)10 increased from 46.12% (atomic%) to 54.53% (atomic%) after Cd(II) adsorption. This was due to the adsorbed Cd(II) formed Cd hydroxides and Cd-complex with oxygen-containing groups on the surface of LDH–GO@Fe(II)10. The SEM results (Fig. S3†) showed that the porous surface was substituted by lumpy structure. The Cd hydroxides cover part of the original porous structure. This study inferred that the LDH–GO@Fe(II)x contributed to the sorption of Cd(II) through surface complexation as well as precipitation.
The XPS survey scan of the H-LDH–GO and LDH–GO@Fe(II)10 after cadmium adsorption is shown in Fig. 5(b). The result displayed the remarkable presentation of Cd 3d spectra after cadmium adsorption, which signified the presence of Cd on the surface of the adsorbents. In Fig. S4(a),† the doublet characteristics of Cd appeared at 406 and 412 eV were in line with those of Cd(OH)2, which indicated the precipitation form of cadmium.20,46 Combined with XRD pattern, it is clear that in this study, the removal of Cd(II) from aqueous solution was mainly conducted by surface-induced precipitation as hydroxides. Notably, the peaks of Fe 2p were impaired after Cd(II) adsorption (Fig. S4(b)†), which indicated the Cd(II) hydroxides covered the surface of LDH–GO@Fe(II)10 and Fe atoms were wrapped in the inner structure.
Combined with the XRD and XPS results, the deposition of Cd(II) played a significant role in the adsorption process. Complexation between Cd(II) and FeOx as well as O-containing groups should also be considered. The O content on the surface of the adsorbents increased after Cd(II) adsorption, which might be due to the formation of complex with oxygen-containing groups (such as COO–Cd, O–Cd, FeOx–Cd). Su et al.47 demonstrated that heavy metal sorption on amorphous hydrous manganese dioxide usually related to the inner-sphere complex formation. Thus, complex formation can be included as another contributor for Cd(II) adsorption on the Fe-decorated LDH–GO. Cd(II) removal by the LDH–GO@Fe(II) composites were controlled by chemical adsorption via isomorphic surface-induced precipitation and complexes formation, which was constant with results of pseudo-second-order model. The decoration of Fe(II) on LDH–GO facilitated the Cd(II) adsorption process. With the modification of Fe(II), non-crystalline Fe formations located at the surface of the LDH/GO crystal. Iron oxides nanoparticles exhibited favorable sorption to heavy metals in terms of high capacity and selectivity. Uheida et al.48 demonstrated Co(II) was effectively removed by iron oxides nanoparticles, and ion exchange and surface complexation were considered as the possible uptake mechanisms. The LDH–GO@Fe(II)50 particles had higher Fe loading than that of LDH–GO@Fe(II)10, but a lower removal capacity for Cd(II). This revealed that the iron oxides formed on the LDH–GO played a role in the adsorption of Cd(II), but not a major role. The amorphous Fe-phases formed on LDH/GO surface resulted in increasement of specific surface area and pore volume. The porous structure can be attributed to the enhancement for Cd(II) removal. While, excessive Fe(II) would restrain the adsorption for Cd(II). The Fe(II) in the structure substituted part of Mg(II), and then Fe(II) was oxidized to Fe(III), which made the brucite-like sheets more positively charged. Thus, an electrostatic repulsion formed with positively charged Cd(II).
4. Conclusions
In summary, Fe-decorated LDH–GO composites with high active surface area was successfully synthesized. The loading of Fe on LDH–GO was achieved by contacting the powdered LDH–GO with Fe(II)-containing solution for 1 h. The LDH–GO@Fe(II)10 composites had the highest removal rate for Cd(II). With further increase of Fe content, electrostatic repulsion and amorphous Fe oxides would block the adsorption for Cd(II). The results of XPS confirmed Fe(III) in the composites after modification with Fe(II). The XRD and XPS results revealed Cd(OH)2 formed after the adsorption for Cd(II). The adsorption followed pseudo-second-order kinetics, indicating chemisorption. The equilibrium data were fitted well with the Langmuir isotherm model. Cd(II) removal by the LDH–GO@Fe(II) composites was controlled by chemical adsorption via isomorphic surface-induced precipitation and complexes formation. It was noteworthy that Fe-decorated LDH–GO composites had a favorable adsorption capacity and chemical stability for the adsorption of metal ions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the International Scientific and Technological Innovation and Cooperation Project of Sichuan (No. 2019YFH0170).References
- S. Arghavani-Beydokhti, M. Rajabi and A. Asghari, Anal. Methods, 2018, 10, 1305–1314 RSC.
- M. A. González, I. Pavlovic and C. Barriga, Chem. Eng. J., 2015, 269, 221–228 CrossRef.
- X. Lu, X. Huangfu and J. Ma, J. Hazard. Mater., 2014, 280, 71–78 CrossRef CAS PubMed.
- M. K. Uddin, Chem. Eng. J., 2017, 308, 438–462 CrossRef CAS.
- E. Gutierrez-Segura, M. Solache-Rios, A. Colin-Cruz and C. Fall, J. Environ. Manage., 2012, 97, 6–13 CrossRef CAS PubMed.
- D. Xu, X. Tan, C. Chen and X. Wang, J. Hazard. Mater., 2008, 154, 407–416 CrossRef CAS PubMed.
- N. Fiol, I. Villaescusa, M. Martínez, N. Miralles, J. Poch and J. Serarols, Sep. Purif. Technol., 2006, 50, 132–140 CrossRef CAS.
- C. Bertagnolli, S. J. Kleinübing and M. G. C. D. Silva, Appl. Clay Sci., 2011, 53, 73–79 CrossRef CAS.
- Y. Cao, G. Li and X. Li, Chem. Eng. J., 2016, 292, 207–223 CrossRef CAS.
- M. Q. Zhao, Q. Zhang, J. Q. Huang and F. Wei, Adv. Funct. Mater., 2012, 22, 675–694 CrossRef CAS.
- X. Ge, J. Liu, X. Song, G. Wang, H. Zhang, Y. Zhang and H. Zhao, Chem. Eng. J., 2016, 301, 139–148 CrossRef CAS.
- H. N. Tran, D. T. Nguyen, G. T. Le, F. Tomul, E. C. Lima, S. H. Woo, A. K. Sarmah, H. Q. Nguyen, P. T. Nguyen, D. D. Nguyen, T. V. Nguyen, S. Vigneswaran, D. N. Vo and H. P. Chao, J. Hazard. Mater., 2019, 373, 258–270 CrossRef CAS PubMed.
- V. Gbb, O. A. Oyetade, S. Rana, B. S. Martincigh, S. B. Jonnalagadda and V. O. Nyamori, ACS Appl. Mater. Interfaces, 2017, 9, 17290 CrossRef PubMed.
- L. Tan, Y. Wang, Q. Liu, J. Wang, X. Jing, L. Liu, J. Liu and D. Song, Chem. Eng. J., 2015, 259, 752–760 CrossRef CAS.
- X. Yuan, Y. Wang, J. Wang, C. Zhou, Q. Tang and X. Rao, Chem. Eng. J., 2013, 221, 204–213 CrossRef CAS.
- W. Liao, H. Wang, H.-q. Li and P. Yang, Environ. Eng. Sci., 2019 DOI:10.1089/ees.2019.0179.
- W. M. Gitari, T. Ngulube, V. Masindi and J. R. Gumbo, Desalin. Water Treat., 2013, 53, 1578–1590 CrossRef.
- C. Luengo, V. Puccia and M. Avena, J. Hazard. Mater., 2011, 186, 1713–1719 CrossRef CAS PubMed.
- X. Sun, C. Hu and J. Qu, Desalin. Water Treat., 2012, 8, 139–145 CrossRef.
- S. He, Y. Li, L. Weng, J. Wang, J. He, Y. Liu, K. Zhang, Q. Wu, Y. Zhang and Z. Zhang, Sci. Total Environ., 2018, 637–638, 69–78 CrossRef CAS PubMed.
- M. K. Doula, Chemosphere, 2007, 67, 731–740 CrossRef CAS PubMed.
- F. Yang, S. Zhang, Y. Sun, K. Cheng, J. Li and D. C. W. Tsang, Bioresour. Technol., 2018, 265, 490–497 CrossRef CAS PubMed.
- S. H. William Jr and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef.
- F. Shemirani and B. T. S. Akhavi, Anal. Lett., 2001, 34, 2179–2188 CrossRef CAS.
- W. Yao, S. Yu, J. Wang, Y. Zou, S. Lu, Y. Ai, N. S. Alharbi, A. Alsaedi, T. Hayat and X. Wang, Chem. Eng. J., 2017, 307, 476–486 CrossRef CAS.
- C. Meng, W. Zhikun, L. Qiang, L. Chunling, S. Shuangqing and H. Songqing, J. Hazard. Mater., 2018, 341, 198–206 CrossRef PubMed.
- Y. Zheng, B. Cheng, W. You, J. Yu and W. Ho, J. Hazard. Mater., 2019, 369, 214–225 CrossRef CAS PubMed.
- M. Park, C. L. Choi, Y. J. Seo, S. K. Yeo, J. Choi, S. Komarneni and J. H. Lee, Appl. Clay Sci., 2007, 37, 143–148 CrossRef CAS.
- F. Zhang, Y. Song, S. Song, R. Zhang and W. Hou, ACS Appl. Mater. Interfaces, 2015, 7, 7251–7263 CrossRef CAS PubMed.
- J. Li, X. Dou, H. Qin, Y. Sun, D. Yin and X. Guan, Water Res., 2019, 148, 70–85 CrossRef CAS PubMed.
- K. Volgmann, F. Voigts and W. Maus-Friedrichs, Surf. Sci., 2012, 606, 858–864 CrossRef CAS.
- H. Li, J. Wan, Y. Ma, M. Huang, Y. Wang and Y. Chen, Chem. Eng. J., 2014, 250, 137–147 CrossRef CAS.
- X. Yin, W. Liu and J. Ni, Chem. Eng. J., 2014, 248, 89–97 CrossRef CAS.
- Q. Zhou, B. Liao, L. Lin, W. Qiu and Z. Song, Sci. Total Environ., 2018, 615, 115–122 CrossRef CAS PubMed.
- M. Liu, Y. Wang, L. Chen, Y. Zhang and Z. Lin, ACS Appl. Mater. Interfaces, 2015, 7, 7961–7969 CrossRef CAS PubMed.
- X. He, X. Qiu and J. Chen, Colloids Surf., A, 2017, 516, 362–374 CrossRef CAS.
- Y. Zhou, X. Li, K. Wang, F. Hu, C. Zhai, X. Wang and J. Bai, J. Alloys Compd., 2019, 770, 6–16 CrossRef CAS.
- F. Unob, B. Wongsiri, N. Phaeon, M. Puanngam and J. Shiowatana, J. Hazard. Mater., 2007, 142, 455–462 CrossRef CAS PubMed.
- X. Liang, Y. Zang, Y. Xu, X. Tan, W. Hou, L. Wang and Y. Sun, Colloids Surf., A, 2013, 433, 122–131 CrossRef CAS.
- L. Li, L. Fan, M. Sun, H. Qiu, X. Li, H. Duan and C. Luo, Colloids Surf., B, 2013, 107, 76–83 CrossRef CAS PubMed.
- A. A. Bakr, N. A. Sayed, T. M. Salama, I. O. Ali, R. R. Abdel Gayed and N. A. Negm, Egypt. J. Pet., 2018, 27(4), 1215–1220 CrossRef.
- M. A. Badawi, N. A. Negm, M. T. H. Abou Kana, H. H. Hefni and M. M. Abdel Moneem, Int. J. Biol. Macromol., 2017, 99, 465–476 CrossRef CAS PubMed.
- C. J. Madadrang, H. Y. Kim, G. Gao, N. Wang, J. Zhu, H. Feng, M. Gorring, M. L. Kasner and S. Hou, ACS Appl. Mater. Interfaces, 2012, 4, 1186–1193 CrossRef CAS PubMed.
- L. Fan, C. Luo, M. Sun, X. Li and H. Qiu, Colloids Surf., B, 2013, 103, 523–529 CrossRef CAS PubMed.
- H. Zhou, Z. Jiang, S. Wei and J. Liang, Water, Air, Soil Pollut., 2018, 229, 78 CrossRef.
- W. Wang, R. He, T. Yang, Y. Hu, N. Zhang and C. Yang, RSC Adv., 2018, 8, 25754–25766 RSC.
- Q. Su, B. Pan, S. Wan, W. Zhang and L. Lv, J. Colloid Interface Sci., 2010, 349, 607–612 CrossRef CAS PubMed.
- A. Uheida, G. Salazar-Alvarez, E. Bjorkman, Z. Yu and M. Muhammed, J. Colloid Interface Sci., 2006, 298, 501–507 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra07305a |
| This journal is © The Royal Society of Chemistry 2019 |