
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of uniform Prussian blue nanoparticles by a polyol process using a polyethylene glycol aqueous solution†
Fumiyuki Shiba *a,
Ushio Mameudaa,
Seitarou Tatejimab and
Yusuke Okawaa
*a,
Ushio Mameudaa,
Seitarou Tatejimab and
Yusuke Okawaa
aDepartment of Materials Science, Chiba University, 1-33 Yayoicho, Inageku, Chiba 263-8522, Japan. E-mail: shiba@faculty.chiba-u.jp
bDepartment of Image and Materials Science, Chiba University, 1-33 Yayoicho, Inageku, Chiba 263-8522, Japan
First published on 28th October 2019
Abstract
A polyol process was applied to the synthesis of Prussian blue nanoparticles that have a narrow size distribution. Potassium hexacyanidoferrate(III) and iron(III) nitrate aqueous solutions were introduced into a 50% polyethylene glycol (PEG) aqueous solution under magnetic stirring at 50 °C and reacted for 48 h. The shape of the so-obtained particles was cubic with somewhat rounded edges and the mean size was 70 nm. In the formation process, nanoparticles of Prussian green, which is a partially oxidized state of Prussian blue, were firstly generated via reduction of the precursors by PEG molecules. The Prussian green nanoparticles were then reduced subsequently to Prussian blue nanoparticles. Rate constants for both the reduction steps have been estimated using the time evolution of absorbance.
1. Introduction
Prussian blue (PB) is known as a synthetic inorganic pigment that consists of Fe3+ and [FeII(CN)6]4−. The blue color is caused by the intervalence charge transition (IVCT) absorption between the Fe(III) and Fe(II).1 Two types of PB, KFeIII[FeII(CN)6] and FeIII4[FeII(CN)6]3, have been described in the literature, and sometimes referred to as soluble and insoluble forms of PB, respectively, even though both forms are almost insoluble (or sparingly soluble).2In both the so-called soluble and insoluble forms, PB has cubic crystal structure in which Fe3+ and [FeII(CN)6]4− are alternatively arranged in a similar way to Na+ and Cl− in NaCl crystals.3 In the soluble form, cations, typically K+, are inserted in the interstitial position to balance the positive and negative charges in the crystal. In the insoluble form, on the other hand, the charges are balanced by forming vacant sites at 1/4 of the [FeII(CN)6]4− positions. The intermediate Fe3+/[FeII(CN)6]4− ratio could be possible with insertion of corresponding number of K+. Hence, it might be rather suitable to consider that the soluble and insoluble forms of PB are of the stoichiometric compositions of PB: PB may be formulated more generally as KxFeIII[FeII(CN)6]{(3+x)/4} (x = 0 − 1).
In 1978, Neff reported that PB films indicated electrochromism (EC), or reversible color changes associated with electrochemical redox reactions.4 Assuming the soluble form, the reaction can be expressed as
| KFeIII[FeII(CN)6] + e− + K+ ⇄ K2FeII[FeII(CN)6] | (1) |
The reduced form is colorless, since both irons are divalent so that the ICVT absorption is vanished (Everitt's salt, or Prussian white, PW). Another EC process is also possible by oxidation from PB, KFeIII[FeII(CN)6], to Prussian green, K(1−x)FeIII[FeII(CN)6](1−x)[FeIII(CN)6]x (PG), and finally to Prussian yellow, FeIII[FeIII(CN)6] (PY).5 It should be noted that the crystal lattice, or the framework structure of Fe–[Fe(CN)6] bindings, is unchanged by the redox processes; K+ compensating the charge balance is inserted into, or excluded from, the interstitial position of the crystal lattice.
After the Neff's report, PB has been widely studied as a functional material not only as the EC materials6 but also as chemical sensors,7 batteries/capacitors,8 cation storage materials such as for H+ or Cs+,9 catalysts,10 etc. Nowadays, PB nanoparticles are also expected to apply to medical uses including photothermal therapy, in which cancer cells are killed by exotherm of PB by absorption of near-infrared light that is passed into the internal body through the near-infrared window in biological tissue.11,12 Especially on the medical uses, nanoparticles with a narrow size distribution would be one of the appropriate forms of PB to be delivered to the target tissues of human body.
Prussian blue nanoparticles can be precipitated by mixing Fe3+ with [FeII(CN)6]4− or Fe2+ with [FeIII(CN)6]3−: the latter precipitates was called Turnbull's blue historically but it is practically same to the former (PB) due to the nature of IVCT.13 The reaction are instantaneous in both reactant pairs, and usually forms quite small particles. In other words, appropriate ways of storing and supplying the reactants are required to obtain well-controlled PB nanoparticles.
The synthesis procedures for PB nanoparticles may be classified by number of reactant species used, i.e. using a soluble hexacyanidoferrate salt singly or using a pair of soluble iron and hexacyanidoferrate salts. In the former system,14–17 hexacyanidoferrate ions are decomposed generating iron ions under a certain condition, to react with the undecomposed ones. For example, Wu et al. proposed sonochemical synthesis from K4[FeII(CN)6], in which Fe2+ was produced by the decomposition of [FeII(CN)6]4− by ultrasonic irradiation in hydrochloric acid; the Fe2+ was oxidized to Fe3+ to react with remaining [FeII(CN)6]4− ions.14 Alternatively, Shen et al. employed K3[FeIII(CN)6] to supply Fe3+, followed by reduction by polyvinylpyrrolidone (PVP) that was contained as a protective colloid.15 Ming et al. also reported that the size and morphology were affected by both the concentrations of K3[FeIII(CN)6] and PVP.16 These formation processes are controlled simply through the controlling the decomposition process, although free CN− ions are possibly liberated as a by-product.18
In the latter system,19–24 on the other hand, the reaction between the pair of reactants should be prevented at first and then triggered to start the reaction by a suitable procedure. One of the strategies is using photoreduction of photosensitive iron salts such as (NH4)3[Fe(C2O4)3],19 in which Fe2+ ions liberated by light irradiation react with surrounding [FeIII(CN)6]3− ions, as similar to a historical photographic process called cyanotype.2 A pair of Fe3+ and [FeIII(CN)6]3− salts, which forms soluble Fe3+–[FeIII(CN)6]3− complex but hardly precipitates hexacyanidoferrate solid,25 is also applicable; monodisperse PB nanoparticles have been prepared by reducing the complex with citric acid.20,21
Preventing coagulation among nanoparticles is another important factor for nanoparticle synthesis. In addition to the typical protective colloids such as PVP15,16,22 and gelatin,20,21 other biocompatible polymers such as poly(diallyldimethylammonium chloride),22 soluble starch26 and chitosan17,23 have been applied to preparation of PB nanoparticles. Recently, Qian et al. have used low concentration of polyethylene glycol (PEG) to a hydrothermal synthesis of PB/Fe3O4 composite particles in an ethylene glycol medium.24 The surface of PB nanoparticles are sometimes modified with more suitable molecules after the precipitation process. For example, the PEGylation of PB surface is achieved by adsorbing PEG molecules via oleylamine that is preliminarily adsorbed on PB surface.27
The authors have proposed a synthesis procedure of PB nanoparticles using gelatin as a protective colloid, in which an aqueous solution containing Fe3+ and [FeIII(CN)6]3− ions was reduced by citric acid (CA) to form PB.20,21 This procedure enabled us to prepare monodisperse PB nanoparticles of about 70 nm in the average size.20 In addition, their size was controllable by applying additional growth or enforced-nucleation methods.21 Although gelatin is an effective protective colloid, however, it has some difficulties on storing, i.e. rot of gelatin. To elude the problem, in the present study, so-called polyol process is applied to the synthesis of PB nanoparticles.
Polyol process is a synthesis method that employs polyols such as ethylene glycol and polyethylene glycol (PEG) as a dispersing medium and/or reducing agent during formation of nanoparticles, and has been applied to wide variety of metal nanoparticles.28 Even Fe nanoparticles have been synthesized from iron salts.29 In the present study, we employ PEG to the polyol synthesis of PB nanoparticles under a simple and mild reaction condition. The formation process is also evaluated.
2. Experimental
2.1. Materials
All reagents were purchased from Wako Pure Chemicals (Osaka, Japan) and used as-received: Fe(NO3)3·9H2O and K3[FeIII(CN)6] were of analytical grade whereas PEG was of practical grade.2.2. Preparation of PB nanoparticles
In a glass vessel, 50% PEG aqueous solution was prepared by dissolving 10 g of PEG (#20000) in 10 g of distilled water and was kept at 50 °C in a water bath. K3[FeIII(CN)6] aqueous solution (0.4 mol L−1, 0.5 mL) and then Fe(NO3)3 aqueous solution (0.54 mol L−1, 0.5 mL) were introduced into the PEG solution under magnetic stirring to start the reaction. The reacting solution was then moved into an air-oven at 50 °C and magnetic stirred for 48 h.The PB particles formed were separated from the supernatant solution by centrifugation (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, 15 min). Then the nanoparticles were re-dispersed in distilled water and centrifuged again: this process was repeated 4 times to wash the particles. So-obtained particles were freeze-dried.
000 rpm, 15 min). Then the nanoparticles were re-dispersed in distilled water and centrifuged again: this process was repeated 4 times to wash the particles. So-obtained particles were freeze-dried.
The crystal structure of nanoparticles was confirmed by powder XRD analysis using Bruker D8 advance (Cu Kα radiation, λ = 1.5418 Å). The shape of particles was observed with a field-emission scanning electron microscope (FE-SEM; JEOL JSM-6700F, operated at 15 kV; the specimens were coated with Pt) and the size was evaluated with a transmission electron microscope (TEM; Hitachi H-7650, operated at 100 kV).
2.3. Evaluation of formation process
Time changes of particle size, Fe content remaining in the solution phase, and the color of dispersion were evaluated to inspect the formation process of the PB nanoparticles. The PB dispersion was prepared in the same manner written in Section 2.2. A portion of the reacting sol (1.5 mL) was sampled and introduced into 28.5 mL of distilled water to dilute to 1/20. For UV-Vis measurement, 4 mL of the 1/20-diluted sample was diluted again to 20 mL, giving a 1/100-diluted sample of the dispersion.The remained 1/20-diluted sample was then centrifuged (10000 rpm, 15 min) to separate the precipitate from the supernatant solution. 5 mL of the supernatant solution was diluted to 20 mL (0.1 mol L−1 of HNO3 was also contained) for the determination of Fe content in the solution phase by atomic absorption (AA) spectrometry (Varian SpectrAA55 at λ = 248.3 nm with C2H2/air flame). For TEM observations, on the other hand, the precipitate was re-dispersed in distilled water and centrifuged again as described above.
3. Results and discussion
Fig. 1 shows the XRD pattern of nanoparticles formed in the polyol system using 50% PEG #20000 aqueous solution at 50 °C for 48 h. The pattern is matched with that for a face-centered cubic lattice, to which PB belongs. The lattice parameter, 10.13 Å, is estimated from the peak positions and in good agreement with the standard data of PB (ICDD PDF2 01-073-0687). The half-width at 2θ = 17.5° (the 200 reflection) enables us to obtain 70 nm of the crystallite size by using Scherrer's equation, D = Kλ/βcosθ, where λ is the wavelength of X-ray, β is the half-width at the diffraction angle θ, and K is the constant (0.9 is assumed).
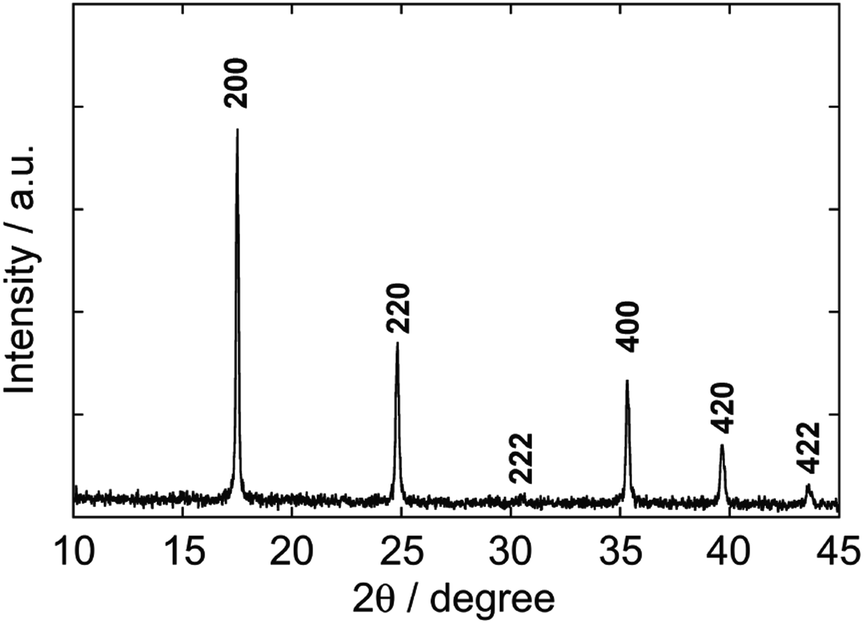 | ||
| Fig. 1 XRD pattern of Prussian blue nanoparticles formed by the polyol process via reduction from Fe(NO3)3 and K3[FeIII(CN)6] in a PEG aqueous solution (#20000, 50%) at 50 °C for 48 h. All peaks are matched with standard ones (ICDD PDF2 01-073-0687). | ||
Electron micrographs of the PB nanoparticles are shown in Fig. 2. As seen in the FE-SEM image in Fig. 2(a), the PB nanoparticles are in cubic shape with somewhat rounded edges. Mean ± one standard deviation of the size was estimated as 77.1 nm ± 13.4 nm from the TEM image in Fig. 2(b). The size is similar to the previous CA/gelatin system20 so that the present PEG system can be an alternative way of PB nanoparticle synthesis. The mean size is close to the crystallite size, suggesting the particles are nearly single-crystalline. This is supported by the selected area electron diffraction (SAED) pattern from a single particle, as shown in Fig. 3. HR-TEM image in Fig. 4 shows fringes whose separation is 0.5 nm, which corresponds to separation of adjacent {200} faces of PB. The fringes are basically continuous although there seems to be some imperfections.
 | ||
| Fig. 2 (a) FE-SEM and (b) TEM images of Prussian blue nanoparticles obtained by the polyol process with PEG #20000 (50%) at 50 °C for 48 h. | ||
 | ||
| Fig. 3 Selected area electron diffraction from a single PB nanoparticle obtained in PEG #20000 aqueous solution (50%) at 50 °C for 48 h. | ||
 | ||
| Fig. 4 HR-TEM image of a PB particle obtained in PEG #20000 aqueous solution (50%) at 50 °C for 48 h. | ||
Fig. 5 shows TEM images of PB nanoparticles at different reaction time during the polyol process. It seems that the size of particles increases and reaches to the final value at about 3 h; this is quantitatively indicated in Fig. 6, where the error bars indicate ± one standard deviation of the size (histograms that indicate the size distributions are shown in Fig. S1(a) in the ESI†).
 | ||
| Fig. 5 TEM images of PB particles at different reaction time during the formation process in PEG #20000 aqueous solution (50%) at 50 °C. | ||
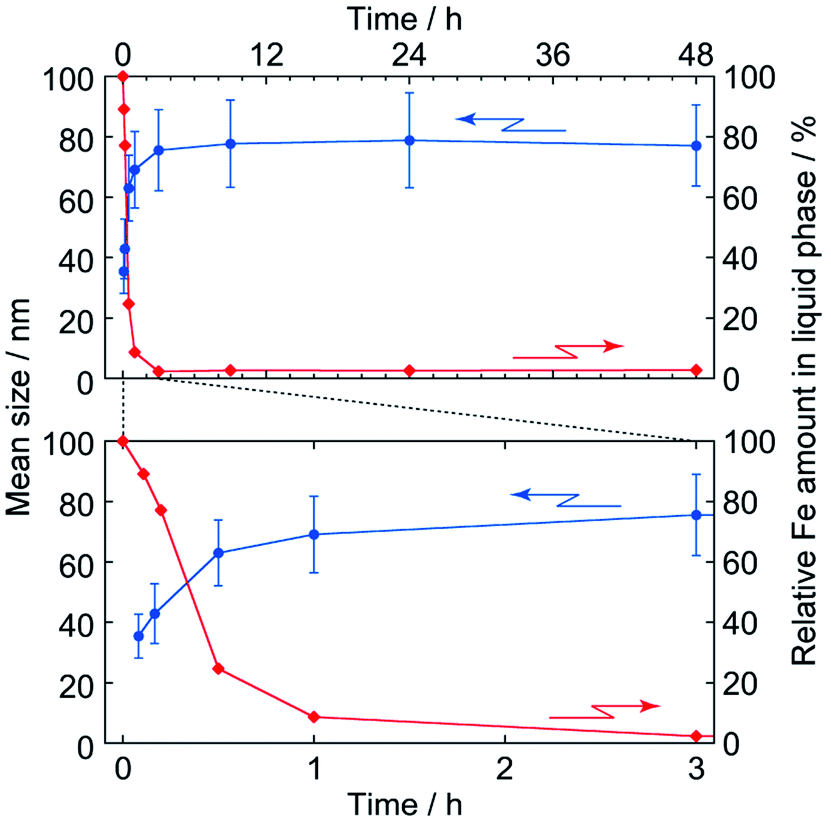 | ||
| Fig. 6 Time evolutions of particle size (●) and Fe amount remaining in the liquid phase (◆). Error bars for the mean size indicate ±one-standard deviation of the size distribution, where the top and bottom figures indicate the whole and early stages of formation, respectively. The Fe amount, relative to initial one, was evaluated by atomic absorption spectrometry. | ||
In addition, it seems to be at about 3 h that the reactants are ran out, as also shown in Fig. 6 by time evolution of the total Fe content (i.e. both Fe3+ and [FeIII(CN)6]3−) remaining in the liquid phase, where the value is estimated by AA spectroscopy and normalized to the initial content. Thus it is strongly suggested the particle formation itself completes in 3 h in this condition.
The color change of sol was, meanwhile, more slowly than the particle growth. The absorption spectra of the 1/100-diluted samples at different times are shown in Fig. 7. The initial brownish color was gradually turns to green, and finally to blue. At 3 h, at which particle growth completes, however, the color was still green; about 48 h is needed to reach the blue color. It should be noted that after at least 3 h, color change of the reacting sol can be considered by color change of the nanoparticles, since almost all reactants are consumed at 3 h.
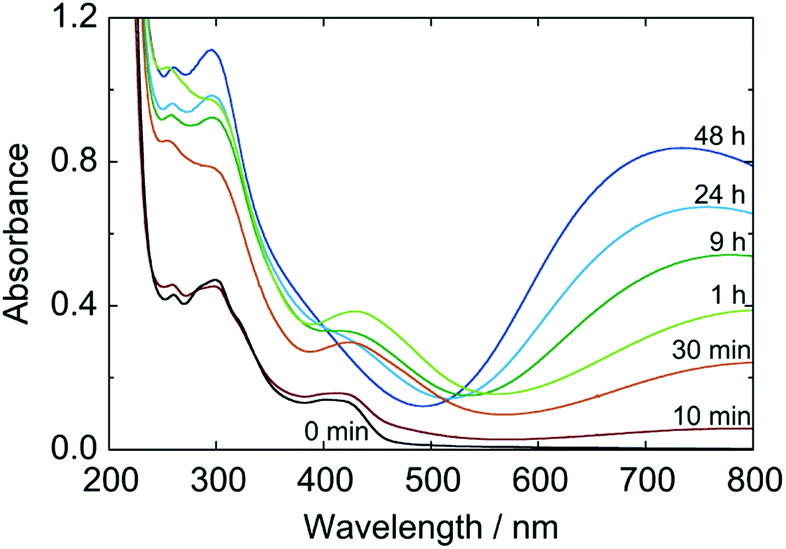 | ||
| Fig. 7 Time change of absorption spectrum during formation process of PB nanoparticles in a 50% PEG #20000 aqueous solution at 50 °C, where the spectra are of 1/100-diluted samples of the reacting sol. | ||
These results imply that the particles firstly formed is Prussian green (PG), the partially-oxidized state of PB containing [FeII(CN)6]4− and [FeIII(CN)6]3− simultaneously with Fe3+ and having the same crystal lattice of PB. In fact, the shape of spectrum at 3 h is very close to that for PG reported in literature.5 Hence the formation process of the PB nanoparticles in the present polyol system would be as follows. At very early stage of the reaction, the reactants exist presumably in the form of soluble Fe3+–[FeIII(CN)6]3−complex as a precursor.25 Once Fe3+–[FeII(CN)6]4− units are produced by reduction, they “coprecipitate” with unreduced Fe3+–[FeIII(CN)6]3− precursors (and some free ions) to generate PG nanoparticles, KxFeIII[FeII(CN)6]y[FeIII(CN)6]z. The PG nanoparticle is then reduced subsequently to PB, K(x+z)FeIII[FeII(CN)6](y+z), also by PEG.
Here, the value (y + z) should be in the range of for the insoluble and soluble forms, 3/4 ≤ (y + z) ≤ 1, and the charge must be balanced as x + 3 = 4y + 3z. The molar ratio of K to Fe for the present PB nanoparticle at 48 h was estimated as 0.10 by TEM-EDX analysis. If one assume there are no contributions of PG and PW, this composition corresponds to K0.18FeIII[FeII(CN)6]0.8 (or K0.72FeIII4[FeII(CN)6]3.2). The reduction reaction from PG to PB involves insertion of K+ so that the incorporation of K+ in the present PB nanoparticles is natural.
Increase of the ICVT absorption would enable us to evaluate the reduction kinetics of Fe3+–[FeIII(CN)6]3− unit, regardless as the precursor in liquid phase or as the component in PG nanoparticles. Fig. 8 shows the time evolution of absorbance at 730 nm of the 1/100-diluted samples. Assuming first order reaction of Fe3+–[FeIII(CN)6]3− concentration, the reduction rate constants are estimated as k1 = 1.8 × 10−4 s−1 and k2 = 1.1 × 10−5 s−1 from the slopes in Fig. S3 in ESI,† where k1 and k2 are for the soluble complex form and for the PG nanoparticles component, respectively. The reduction of PG nanoparticle to PB ones is much slower than of the soluble precursor to PG nanoparticles (k2/k1 = 0.061).
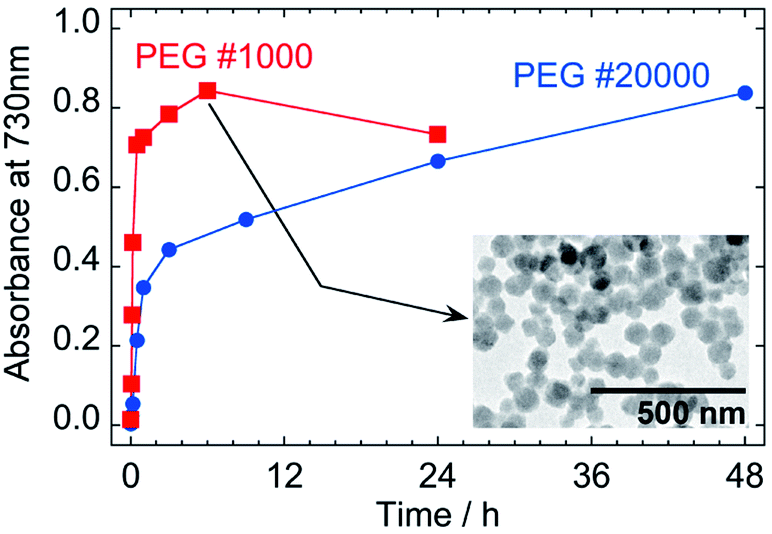 | ||
| Fig. 8 Effect of molecular weight of PEG on the time evolution of absorbance at 730 nm as an indicator of color of the nanoparticles prepared with 50% aqueous solution of PEG at 50 °C. The values of absorbance are of 1/100-diluted samples and the inset is a TEM image of PB nanoparticles at 6 h prepared with PEG #1000. | ||
PB nanoparticles were also synthesized with PEG #1000 for comparison of the reaction rates. Even though increase of the ICVT absorption continued until 6 h, as indicated in Fig. 8, the growth of nanoparticle was completed more quickly about in 30 min, as shown in Fig. S2 in ESI† (the size distributions are also indicated in Fig. S1(b) in ESI†). The particle size at 6 h for PEG #1000 was 67.0 nm ± 12.7 nm and the shape seems to have rounder edges than those by PEG #20000, as shown by the TEM image in the inset in Fig. 8. The decrease of absorbance for PEG #1000 at 24 h from 6 h would reflect further reduction of PB to PW.
The rate constants for PEG #1000 system are estimated as k1 = 1.0 × 10−3 s−1 and k2 = 9.4 × 10−5 s−1 (k2/k1 = 0.094) from the slopes in Fig. S3 in ESI.† Although the reasons causing the differences in rate constants have not been clear, the much faster rate constants for PEG #1000 than for PEG #20000 could be result of the difference in viscosity that affects the mobility of molecules and/or of the difference in the content of reducing site of PEG at the terminal position, –CH2OH.30 On the other hand, it is interesting that the k2/k1 ratios are similar as 0.061 for PEG #20000 and 0.094 for #1000, implying that the formation mechanism is intrinsically same.
The great difference between k1 and k2 could be due to the difference in the state of Fe3+–[FeIII(CN)6]3− unit that is to be reduced. For k1, the Fe3+–[FeIII(CN)6]3− units exist uniformly in the liquid phase as soluble complex, whereas, for k2, the units are localized in the nanoparticles. Thus the frequency meeting it to the reducing site of PEG would be quite different. In addition, adsorption of PEG molecules on the nanoparticle surface31 possibly retard approach of other PEG molecules.
Even in the previous CA/gelatin system,20 PB nanoparticles seem to be formed via PG state as the spectrum has the absorption band around 430 nm, which is attributed to Fe3+–[FeIII(CN)6]3− absorption5 (see Fig. 1 in ref. 20). The increase in IVCT absorption around 730 nm was, however, almost corresponds to the increase in size of nanoparticles (Fig. 4 in ref. 20). In addition, the absorption band around 430 nm was smaller than the PEG system and was quickly disappeared just after the completion of size increase. The smaller molecule size of citric acid than PEG would result in the comparable reduction rates of reactants to PG nanoparticles and PG nanoparticles to PB ones.
The redox potential is another possible reason for the difference among rate constants. According to a potentiostatic deposition study, formation of a PG film from the soluble complex occurs in the range of +0.75 to +0.88 V vs. SCE.32 This range locates, however, at almost the same potential to the redox peak pair for the PB ⇄ PG conversion at around +0.8 V, which is observed in cyclic voltammetric measurements for PB thin films.33 Therefore the effect of reduction potential would not be dominant on the problem of small k2/k1 ratio. On the other hand, reductions of PG nanoparticles to PB ones and PB nanoparticles to PW ones, as observed for PEG #1000, are likely to be discussed in terms of reduction potential since the redox peak pair for the PB ⇄ PW conversion locates at about +0.2 V.33
In the present study, PEG #20000 has been mainly employed because of formation of the sharper edged nanoparticles, in addition to advantages in inspection of the formation process with the enhanced reduction rate difference. The slow rate of the second reduction step enables us to control the color of particles in the range from green to blue. On the other hand, PEG of lower molecular weight would be preferred to prepare PB nanoparticles for application usages, due to shorter reaction time required. In the future study, the reduction process should be investigated more precisely, since the present estimations are based on some assumptions with limited numbers of absorbance data.
4. Conclusions
In the present study, we have shown that the polyol process using PEG aqueous solution enable us to obtain uniform PB nanoparticles, where PEG molecules act as a dispersing agent for the nanoparticles formed, in addition to as a reducing agent for the precursor, presumably soluble Fe3+–[FeIII(CN)6]3− complex. The PB nanoparticles formed at 50 °C with PEG #20000 were in almost single crystalline and in cubic shape with about 77 nm in the mean size. The IVCT absorption continued increasing until at 48 h even though the precipitation of nanoparticles was completed until at 3 h, suggesting that PG nanoparticles, a partially oxidized state of PB, are firstly formed by reduction of the precursor, followed by further reduction of the PG nanoparticles to PB ones. Molecular weight of PEG affects both the reduction rates. The reduction rate constants have been estimated on the basis of time evolution of the IVCT absorption.Conflicts of interest
There are no conflicts to declare.Acknowledgements
XRD measurements were carried out at the Centre for Analytical Instrumentation, Chiba University.Notes and references
- M. B. Robin and P. Day, Adv. Inorg. Chem. Radiochem., 1967, 10, 247 CrossRef CAS.
- M. Ware, J. Chem. Educ., 2008, 85, 612 CrossRef CAS.
- A. Paolella, C. Faure, V. Timoshevskii, S. Marras, G. Bertoni, A. Guerfi, A. Vijh, M. Armand and K. Zaghib, J. Mater. Chem. A, 2017, 5, 18919 RSC.
- V. D. Neff, J. Electrochem. Soc., 1978, 125, 886 CrossRef CAS.
- R. J. Mortimer and D. R. Rosseinsky, J. Chem. Soc., Dalton Trans., 1984, 2059 RSC.
- A. Gotoh, H. Uchida, M. Ishizaki, T. Satoh, S. Kaga, S. Okamoto, M. Ohta, M. Sakamoto, T. Kawamoto, H. Tanaka, M. Tokumoto, S. Hara, H. Shiozaki, M. Yamada, M. Miyake and M. Kurihara, Nanotechnology, 2007, 18, 345609 CrossRef CAS; C. G. Granqvist, Thin Solid Films, 2014, 564, 1 CrossRef.
- R. Koncki, T. Lenarczuk and S. Głąb, Anal. Chim. Acta, 2000, 424, 27 CrossRef CAS.
- H. Minowa, Y. Yui, Y. Ono, M. Hayashi, K. Hayashi, R. Kobayashi and K. I. Takahashi, Solid State Ionics, 2014, 262, 216 CrossRef CAS.
- S. S. Kaye and J. R. Long, Catal. Today, 2007, 120, 311 CrossRef CAS; T. Yasutaka, T. Kawamoto, Y. Kawabe, T. Sato, M. Sato, Y. Suzuki, K. Nakamura and T. Komai, J. Nucl. Sci. Technol., 2013, 50, 674 CrossRef.
- S.-Q. Liu, S. Cheng, L.-R. Feng, X.-M. Wang and Z.-G. Chen, J. Hazard. Mater., 2010, 182, 665 CrossRef CAS; L. I. Doumic, G. Salierno, C. Ramos, P. M. Haure, M. C. Cassanello and M. A. Ayude, RSC Adv., 2016, 6, 46625 RSC.
- M. Gautam, K. Poudel, C. S. Yong and J. O. Kim, Int. J. Pharm., 2018, 549, 31 CrossRef CAS; M. Shokouhimehr, E. S. Soehnlen, J. Hao, M. Griswold, C. Flask, X. Fan, J. P. Basilion, S. Basu and S. D. Huang, J. Mater. Chem., 2010, 20, 5251 RSC.
- G. Fu, W. Liu, S. Feng and X. Yue, Chem. Commun., 2012, 48, 11567 RSC.
- A. Ito, M. Suenaga and K. Ōno, J. Chem. Phys., 1968, 48, 3597 CrossRef CAS.
- X. Wu, M. Cao, C. Hu and X. He, Cryst. Growth Des., 2006, 6, 26 CrossRef CAS.
- X. Shen, S. Wu, Y. Liu, K. Wang, Z. Xu and W. Liu, J. Colloid Interface Sci., 2009, 329, 188 CrossRef CAS.
- H. Ming, N. L. K. Torad, Y.-D. Chiang, K. C.-W. Wu and Y. Yamauchi, CrystEngComm, 2012, 14, 3387 RSC.
- Y. Ding, Y.-L. Hu, G. Gu and X.-H. Xia, J. Phys. Chem. C, 2009, 113, 14838 CrossRef CAS.
- D. D. Kuhn and T. C. Young, Chemosphere, 2005, 60, 1222 CrossRef CAS.
- S. Vaucher, M. Li and S. Mann, Angew. Chem., Int. Ed., 2000, 39, 1793 CrossRef CAS.
- F. Shiba, Colloids Surf., A, 2010, 366, 178 CrossRef CAS.
- F. Shiba, M. Nito, K. Kawakita and Y. Okawa, Part. Sci. Technol., 2015, 33, 671 CrossRef CAS.
- T. Uemura, M. Ohba and S. Kitagawa, Inorg. Chem., 2004, 43, 7339 CrossRef CAS.
- Q. Zhang, L. Zhang and J. Li, Electrochim. Acta, 2008, 53, 3050 CrossRef CAS.
- J. Qian, J. Xu, L. Kuang and D. Hua, ChemPlusChem, 2017, 82, 888 CrossRef CAS.
- J. A. Ibres and N. Davidson, J. Am. Chem. Soc., 1951, 73, 476 CrossRef CAS; D. L. Singleton and J. H. Swinehart, Inorg. Chem., 1967, 6, 1536 CrossRef.
- Q. Zhang, L. Zhang and J. Li, J. Nanosci. Nanotechnol., 2007, 7, 4557 CrossRef CAS.
- H. Chen, Y. Ma, X. Wang, X. Wu and Z. Zha, RSC Adv., 2017, 7, 248 RSC; P. Xue, L. Sun, Q. Li, L. Zhang, Z. Xu, C. M. Li and Y. Kang, J. Colloid Interface Sci., 2018, 509, 384 CrossRef CAS.
- F. Fiévet, in Fine Particles–Synthesis, Characterization, and Mechanisms of Growth, ed. T. Sugimoto, Marcel Dekker, New York, 2000, p. 460 Search PubMed.
- R. J. Joseyphus, D. Kodama, T. Matsumoto, Y. Sato, B. Jeyadevan and K. Toji, J. Magn. Magn. Mater., 2007, 310, 2393 CrossRef CAS; R. J. Joseyphus, K. Shinoda, D. Kodama and B. Jeyadevan, Mater. Chem. Phys., 2010, 123, 487 CrossRef.
- F. Fiévet, J. P. Lagier, B. Blin, B. Beaudoin and M. Figlarz, Solid State Ionics, 1989, 32–33, 198 CrossRef.
- S. Liufu, H. Xiao and Y. Li, Powder Technol., 2004, 145, 20 CrossRef CAS.
- R. J. Mortimer and D. R. Rosseinsky, J. Electroanal. Chem., 1983, 151, 133 CrossRef CAS.
- A. Roig, J. Navarro, J. J. Garcia and F. Vicente, Electrochim. Acta, 1994, 39, 437 CrossRef CAS; D. Ellis, M. Eckhoff and V. D. Neff, J. Phys. Chem., 1981, 85, 1225 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Histograms of particle size, time evolutions of particle size and Fe concentration for PEG #1000, analysis of reduction kinetics. See DOI: 10.1039/c9ra07080j |
| This journal is © The Royal Society of Chemistry 2019 |