
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal free synthesis of ethylene and propylene carbonate from alkylene halohydrin and CO2 at room temperature†
Santosh Govind Khokarale *a and
Jyri-Pekka Mikkola*ab
*a and
Jyri-Pekka Mikkola*ab
aTechnical Chemistry, Department of Chemistry, Chemical-Biological Centre, Umeå University, SE-90187 Umeå, Sweden. E-mail: santosh.khokarale@umu.se
bIndustrial Chemistry & Reaction Engineering, Department of Chemical Engineering, Johan Gadolin Process Chemistry Centre, Åbo Akademi University, FI-20500 Åbo-Turku, Finland
First published on 22nd October 2019
Abstract
Herein we describe a metal free and one-pot pathway for the synthesis of industrially important cyclic carbonates such as ethylene carbonate (EC) and propylene carbonates (PC) from molecular CO2 under mild reaction conditions. In the actual synthesis, the alkylene halohydrins such as alkylene chloro- or bromo or iodohydrin and organic superbase, 1,8-diazabicyclo-[5.4.0]-undec-7-ene (DBU) reacted equivalently with CO2 at room temperature. The syntheses of cyclic carbonates were performed in DMSO as a solvent. Both 1,2 and 1,3 halohydrin precursors were converted into cyclic carbonates except 2-bromo- and iodoethanol, which were reacted equivalently with DBU through n-alkylation and formed corresponding n-alkylated DBU salts instead of forming cyclic carbonates. NMR analysis was used to identify the reaction components in the reaction mixture whereas this technique was also helpful in terms of understanding the reaction mechanism of cyclic carbonate formation. The mechanistic study based on the NMR analysis studies confirmed that prior to the formation of cyclic carbonate, a switchable ionic liquid (SIL) formed in situ from alkylene chlorohydrin, DBU and CO2. As a representative study, the synthesis of cyclic carbonates from 1,2 chlorohydrins was demonstrated where the synthesis was carried out using chlorohydrin as a solvent as well as a reagent. In this case, alkylene chlorohydrin as a solvent not only replaced DMSO in the synthesis but also facilitated an efficient separation of the reaction components from the reaction mixture. The EC or PC, [DBUH][Cl] as well as an excess of the alkylene chlorhydrin were separated from each other following solvent extraction and distillation approaches. In this process, with the applied reaction conditions, >90% yields of EC and PC were achieved. Meanwhile, DBU was recovered from in situ formed [DBUH][Cl] by using NaCl saturated alkaline solution. Most importantly here, we developed a metal free, industrially feasible CO2 capture and utilization approach to obtain EC and PC under mild reaction conditions.
1. Introduction
Cyclic carbonates, especially ethylene and propylene carbonate are industrially important chemicals considering their widespread application.1 Being non-toxic and bio-degradable and having high boiling points and aprotic polar nature, these cyclic carbonates are preferentially replacing other harmful organic solvents such as dimethylformamide (DMF), hexamethylphosphoramide (HMPA), N-methyl-2-pyrrolidone (NMP), dimethylacetamide and acetonitrile (ACN).1–3 Cyclic carbonates have also been successfully implemented as intermediates upon synthesis of fine chemicals such as dialkyl carbonates, glycols, carbamates etc. Further, they find use as electrolytes for batteries, precursors for polymeric materials and as fuel additives.4 The traditional industrial production of cyclic carbonates follows the phosgene route. However, due to the toxic and corrosive nature of phosgene as well as production of large amounts of chlorinated salts and solvents, this process is not found to be sustainable in terms of environmental impact and work safety. As an alternative approach, the synthesis of cyclic carbonates from epoxides and carbon dioxide (CO2) was invented as an elegant, non-toxic and atom-efficient carbon capture and utilization (CCU) pathway (Fig. 1).5 This pathway introduced an opportunity to use CO2 as an inexpensive, nontoxic, and renewable carbon C1 building block for the synthesis of valuable chemicals. However, considering its more oxidized state and hence chemically inert nature, the CO2 molecule usually gives rise to low reactivity. Hence, for the chemical activation of CO2 molecule and its subsequent interaction with epoxides, several homogeneous and heterogeneous catalytic systems including metal based catalysts, organocatalysts, ionic liquids etc. were developed to synthesize cyclic carbonates.4–8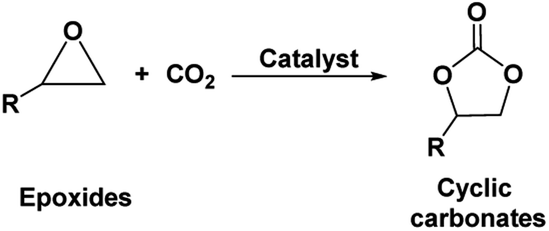 | ||
| Fig. 1 Synthesis of cyclic carbonates from epoxides and CO2. | ||
Apart from the catalytic systems, various types of epoxide substrates were also successfully used. These include both petroleum (for e.g. ethylene oxide, propylene oxide and cyclohexene oxide) as well as bio-based epoxides (for e.g. epichlorohydrine, limonene oxide and vinylcyclohexene oxide).6,9–11 Even though the synthesis of cyclic carbonates from various epoxides is industrially applied process but the synthesis of epoxides accompanied with the use of toxic or costly reagents as well as involves a tedious workup for separation.
Alternative to epoxide as a reagent, the synthesis of cyclic carbonates was also carried out using alkylene halohydrins as a one of the reagent with CO2. The various alkali metal carbonates such as K2CO3, Na2CO3, Cs2CO3 etc. were used as an catalysts for the synthesis of the cyclic carbonates from various alkylene halohydrins. In this case the cyclic carbonates were obtained with high yield (>90%) under mild reaction conditions such as room temperature and atmospheric pressure.12–15 The metal free reaction approach for the synthesis of cyclic carbonates from alkylene halohydrins was also used where amine such as triethylamine was used as a both activating agent and solvent.16 Even though these reaction approaches emerged as an alternative to the epoxide based processes but it is also accompanied with various challenges. In case of metal carbonate based processes involves the use of organic solvents (e.g. dimethylformamide, DMF) and additives (e.g. amines) as well as the generation of equivalent amount of waste such as metal bicarbonates and metal halides. In addition, it was also observed that the desired cyclic carbonates were formed with slow reaction kinetics under applied reaction conditions. Further with triethylamine-involved processes, even though 90% yield of cyclic carbonates was obtained and recovery of amine was achieved, but the process was carried out at high temperature and pressure. Hence, the synthesis of cyclic carbonates from alkylene halohydrins need few improvements where combined reaction approach involving mild reaction conditions along with no metal waste generation can be more preferred.
Herein, we are introducing one-pot and metal free synthesis of ethylene carbonate (EC) and propylene carbonate (PC) from molecular CO2 at room temperature. In this route, the equivalent mixture of various alkylene halohydrin such as 2-chloroethanol or 2-bromoethanol or 2-iodoethanol or 1-chloro-2-propanol or 3-chloro-1-propanol or 3-bromo-1-propanol and organic superbase DBU react with molecular CO2 to form EC or PC (reaction Scheme 1).
 | ||
| Scheme 1 Synthesis of ethylene or propylene carbonate from alkylene halohydrins. | ||
The process described in this report for the synthesis of EC and PC uses equivalent amounts of DBU, alkylene halohydrin and CO2 where synthesis was carried out at room temperature and atmospheric pressure. In addition, as shown in the reaction Scheme 1, the organic superbase DBU was recovered in the form of its halide salt. Further, the cyclic carbonates and salt of DBU were separated in the pure form with mild solvent extraction and vacuum distillation approach. The DBU was also recovered from its halide salt following alkaline solvent's treatment.
The synthesis of EC and PC was initially carried out in DMSO (as a solvent) while in order avoid to use of DMSO, the same process was further developed to yield a DMSO free solvent system where alkylene halohydrins themselves were used as solvents. The progress of the reaction as well as recovery of EC or PC and DBU were monitored by the means of NMR spectroscopic techniques. The numbering and labelling for the chemical species used and formed in this cyclic carbonate synthesis are shown in Fig. 2.
 | ||
| Fig. 2 Chemical species used and formed in the reaction mixture in cyclic carbonate synthesis. | ||
2. Experimental
2.1 Chemicals and methods
2.2. Synthesis and separation of MP and MMA and recovery of DBU
 | (1) |
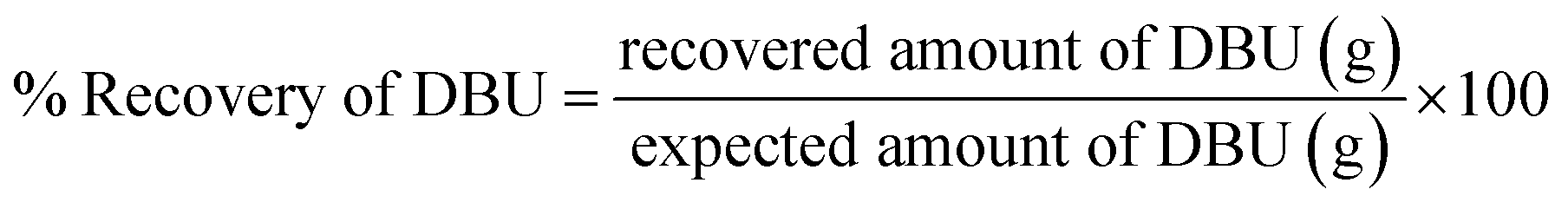 | (2) |
3. Result and discussion
The synthesis of cyclic carbonates such as EC or PC was preceded by the introduction of organic superbase DBU through the interaction between molecular CO2 and alkylene chlorohydrins such ethylene or propylene chlorohydrins, respectively. Initially, the synthesis of cyclic carbonates was performed in a DMSO solvent where equivalent mixture of DBU and alkylene chlorohydrin were dissolved.The primary aim of the use of DBU was to activate the CO2 molecule prior to formation of cyclic carbonates. This was facilitated via the formation of a switchable ionic liquid (SIL) through equivalent interaction between DBU, alkylene chlorohydrin and CO2. The synthesis of organic superbase comprised SIL is a promising approach for the chemical activation of CO2 and its storage under mild reaction conditions.18 In general, being a superbase, DBU is able to abstract the proton of –OH group of an alcohol (low and high molecular weight alcohols as well as biomolecules such as cellulose) and the thus formed alkoxide anion subsequently interacts with CO2 and form SIL i.e. [DBUH][alkyl carbonate] (reaction Scheme 2a).19
 | ||
| Scheme 2 Plausible mechanism for the synthesis of ethylene or propylene carbonate from chlorohydrin and CO2. Formation of (a) switchable ionic liquid (SIL), [DBUH][ClCH(R)CH2CO3] and, (b) ethylene or propylene carbonate. | ||
In this report, as shown in the reaction Scheme 2, the synthesis of cyclic carbonates from DBU, alkylene chlorohydrin and CO2 was initially expected to occur in two steps. The process involves the formation of SIL i.e. [DBUH][ClCH(R)CH2CO3] (R = –H or –CH3) at room temperature, followed by the synthesis of cyclic carbonates through release of [DBUH][Cl] under thermal treatment with or without use of catalysts. However, based on the NMR analysis of the reaction mixture, it was observed that during bubbling of CO2 in the reaction mixture, the synthesis of cyclic carbonate took place in line with a one-pot process. As shown in Fig. 3a and b, the 1H NMR spectra reveals that after interaction of DBU and ethylene chlorohydrin with CO2 at room temperature, the two doublets signals of four proton atoms (3.88–4.10 ppm) as well as singlet for the proton in hydroxyl group (6.59 ppm, broad) in ethylene chlorohydrin disappeared. On the other hand, two new signals with the chemical shifts 4.85 (intense) and 11.11 ppm (broad) were observed. Further, after comparing the 1H NMR spectra of the reaction mixture with the 1H NMR spectra of commercially available EC, the similar intense signal at the 4.85 ppm was also observed for the commercially available EC. The singlet for the 4 proton atoms in the EC appeared as all these protons have identical chemical environment. The signal observed at 11.11 ppm is related to the proton attached to the quaternized and sp2-hybridised nitrogen atom in protonated DBU i.e. [DBUH]+ cation.20 The occurrence of the EC and [DBUH]+ cation were also further confirmed from the 13C NMR analysis.
 | ||
| Fig. 3 1H NMR spectra for (a) equivalent mixture of DBU and ethylene chlorohydrin, (b) reaction mixture after bubbling of CO2 in DBU and ethylene chlorohydrin, and (c) commercially available ethylene carbonate in DMSO solvent. (NMR analysis with D2O capillary). | ||
The 13C NMR spectra for the reaction mixture obtained after reaction of DBU, ethylene chlorohydrin and CO2 in DMSO is shown in Fig. 4. The signals for the non-identical carbon atoms denoted by C-a and C-b in ethylene chlorohydrin were observed at 47.36 and 62.31 ppm, respectively (Fig. 4a and b). After bubbling of CO2 in the DMSO solution of the equivalent mixture of DBU and ethylene chlorohydrin, similar to 1H NMR spectra, the signals for C-a and C-b disappeared and new signals at 65.94 and 156.40 ppm were observed. After comparison between the NMR spectra of the reaction mixture and commercially available EC, it was observed that the signals appearing at 65.94 and 156.40 ppm, respectively, belong to the aliphatic (C-a′ and C-b′) and carbonyl carbon (denoted by star) of the EC molecule, respectively (Fig. 4b and c). Further, after the interaction of DBU and ethylene chlorohydrin with CO2, the carbon atoms in the DBU molecule, at position 6 and 9, got shielded while the carbon atom at position 7 was deshielded which confirmed the formation of [DBUH]+ cation.20 Hence, based on both 1H and 13C NMR spectra, the EC was formed after equivalent interaction between DBU, ethylene chlorohydrin and CO2 molecule in a one-pot process.
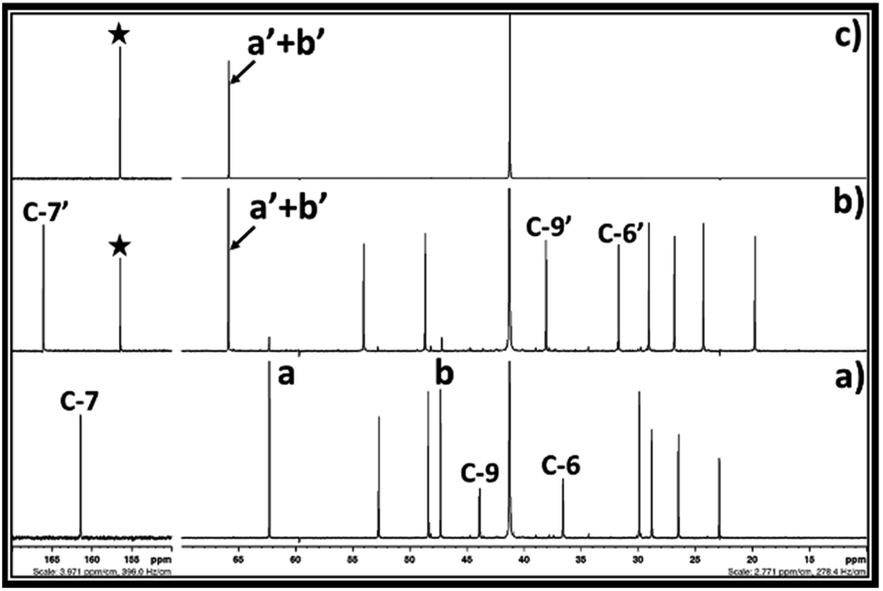 | ||
| Fig. 4 13C NMR spectra for (a) equivalent mixture of DBU and ethylene chlorohydrin, (b) reaction mixture after bubbling of CO2 in DBU and ethylene chlorohydrin, and (c) commercially available ethylene carbonate in a DMSO solvent. (NMR analysis with D2O capillary). | ||
Based on the NMR analysis, even though EC formed in the reaction mixture, it was not confirmed that the synthesis was preceded either through two sub-steps mentioned in the Scheme 2 or other alternative pathways. Therefore, in order to confirm the mechanism of the formation of EC, CO2 was bubbled though equivalent mixture of DBU and ethanol (non-chlorinated analogue of ethylene chlorohydrin). As shown in Fig. S1,† although SIL i.e. [DBUH][CH3CH2CO3] formed after interaction of equivalent mixture of DBU and ethanol with CO2, unlike in the EC synthesis, the isolated signals for the carbon atoms C-a′ and C-b′ in the [CH3CH2CO3]− anion were observed in the 13C NMR spectra. Hence, due to absence of leaving group i.e. Cl atom at the carbon atom C-a in ethanol, the subsequent intermolecular ring closing for EC synthesis after CO2 capture was not observed. However, as [DBUH][CH3CH2CO3] formed in this control experiment and also [DBUH][Cl] formed in the EC synthesis process, we can conclude that the formation of EC proceeds through the two sub-step comprised concerted mechanism shown in reaction Scheme 2. Hence, as shown in the reaction Scheme 2, initially it seems that the SIL forms in the reaction mixture. Further due to presence of Cl atom as a good leaving group, the negatively charged oxygen atom in the carbonate anion induced a nucleophilic attack at the carbon atom to which Cl atom is attached and formation of EC took place through the release of [DBUH][Cl]. Identical mechanism was also proposed previously in case of metal carbonate and amine catalysed processes.12–15
Further, in order to confirm the origin of carbon atom in the EC, the 13C enriched CO2 was used as a CO2 source (tracer technique). The 13C enriched CO2 was bubbled in the equivalent mixture of DBU and ethylene chlorohydrin in DMSO and the one as well as two dimensional NMR analysis of the reaction mixture was carried out. As shown in Fig. 5c, after the use of 13C enriched CO2, a similar peak pattern was observed for the reaction mixture after comparing it with the NMR of the reaction mixture when ‘normal’ (non-13C enriched) CO2 was used. However, in case of 13C enriched CO2, the signal observed for the carbonyl carbon in the EC at the chemical shift 156.40 ppm was having a high intensity compared to the signal for the C-7′ in the [DBUH]+ cation (Fig. 5c). On the contrary, in case of the reaction mixture obtained after use of normal CO2, the intensity of the signal at 156.40 ppm was found comparable with the intensity of the signal for C-7′ in [DBUH]+ cation. In addition, it was also observed that the protons in the EC showed the doublet signal at the chemical shift 4.85 ppm in the 1H NMR spectra when 13C enriched CO2 was used in the synthesis (ESI S2-a†). In this case, the carbonyl carbon atom of EC contained a higher percentage of 13C isotope than 12C and as the protons of EC are in adjutants to this 13C enriched carbonyl carbon atom, these protons showed doublet signal in the NMR spectra.21 On the other hand, the signal for these protons was observed as a singlet at the same chemical shift in the 1H NMR of the reaction mixture when normal CO2 was used (Fig. 2b). So, these observations proved that the EC was formed from the 13C enriched CO2 and ethylene chlorohydrin through the equivalent interaction with DBU.
 | ||
| Fig. 5 13C NMR spectra for (a) equivalent mixture of DBU and ethylene chlorohydrin, reaction mixture after bubbling of (b) CO2, (c) 13C enriched CO2 in DBU and ethylene chlorohydrin, and (d) commercially available ethylene carbonate in DMSO solvent. (NMR analysis with D2O in capillary). | ||
The two dimensional correlation NMR analysis techniques such as 1H–1C HSQC (heteronuclear single quantum coherence) and HMBC (heteronuclear multiple bond correlation) of the reaction mixture was performed when 13C enriched CO2 was used. This analysis was carried out to get more proof about the formation of EC in the reaction mixture. As shown in Fig. S2-b,† the HSQC NMR analysis represents that the signal for the correlation between four equivalent protons and carbon atoms (C-a′ and C-b′) are observed with the chemical shift at 4.85/65.94 ppm. Further, in case of HMBC NMR analysis, the protons in the EC gave rise to long range coupling with the 13C enriched carbonyl carbon atom in the EC and corresponding correlation signal observed at chemical shift 4.85/156.44 ppm (Fig. S2-c†).
Hence, from all these observations based on the both one and two dimensional NMR analysis, it is confirmed that the synthesis of EC proceeds through simultaneous SIL formation followed by intermolecular ring closing reaction shown in the reaction Scheme 2 where chemisorbed CO2 became a part of the structure of thus synthesised EC.
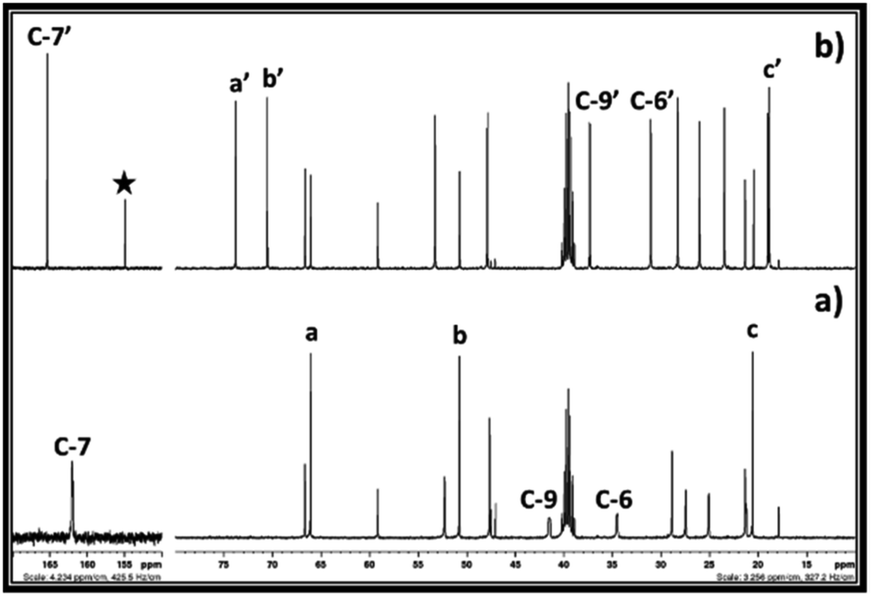
After synthesis of EC from CO2 and ethylene chlorohydrin, the similar reaction approach was further mimicked upon synthesis of PC. In this case, propylene chlorohydrin was used as an alkylene chlorohydrin precursor. However, unlike ethylene chlorohydrin, propylene chlorohydrin purchased from Sigma Aldrich comprised of its two different isomers i.e. 1-chloro-2-propanol (70 wt%) of and 2-chloro-1-propanol (30 wt%). During actual experiment, it was assumed that the one type of isomer of propylene chlorohydrin was present in the purchased sample container and based on the equivalent of DBU, accordingly the amount of the propylene chlorohydrin was taken for the experiment. As shown in Fig. 6a, the 13C NMR spectra also showed that mixed signals for the both types of isomers of propylene chlorohydrin were observed with the signals of DBU. Further CO2 was bubbled in the equivalent mixture of DBU and propylene chlorohydrin in DMSO and the formation of PC was confirmed by NMR analysis. The observed 1H and 13C NMR are shown in the ESI S3† and Fig. 6, respectively. As depicted in Fig. 6a and b, the signals for the both the types of isomers of propylene chlorohydrin disappeared. After comparing the NMR spectra of both the reaction mixture and the commercially available PC sample, it was observed that the PC formed in the reaction mixture (Fig. 6b and c). Also, similar to the EC synthesis, based on the NMR analysis, [DBUH][Cl] also formed in the reaction mixture (Scheme 2 and Fig. 6b). Hence, independent on the type of isomer, it was observed that the PC can be synthesised from mixtures of isomers of propylene chlorohydrin, DBU and CO2 following the reaction Scheme 2.
 | ||
| Fig. 6 13C NMR spectra for (a) equivalent mixture of DBU and propylene chlorohydrin, (b) reaction mixture after bubbling of CO2 in DBU and propylene chlorohydrin, and (c) commercially available propylene carbonate in DMSO solvent. (NMR analysis with D2O in capillary). | ||
Similar to 13C NMR, the 1H NMR spectra of the reaction mixture also gave rise to similar observation where prior to CO2 exposure, the reaction mixture represented characteristic signal for DBU as well as for both the isomers of propylene chlorohydrin (Fig. S3-a†). After the formation of the PC, the signals belonging to the both isomers of the propylene chlorohydrin disappeared and new deshielded signals for the protons in the PC were observed (Fig. S3-b†). Further, it was also observed that the two protons at the carbon atom C-a in the propylene chlorohydrin became non-identical after formation PC and this was further confirmed with commercially available PC.
Hence, based on the NMR analysis, it was preliminary concluded that PC formed form the equivalent interaction between DBU, propylene chlorohydrin and CO2. However, in order to get more confirmation about the origin of the CO2, the 13C enriched CO2 was used in the PC synthesis process and the reaction mixture analysed by one and two dimensional NMR analyses. As shown in Fig. 7c, after the use of 13C enriched CO2, the intensity of the carbonyl carbon of the PC was found higher than the intensity of the carbon atom C-7′ in the [DBUH]+ cation. Meanwhile upon use of normal CO2, the intensities of the carbonyl carbon atoms in PC and C-7′ of [DBUH]+ cation were found nearly identical to each other (Fig. 6b). Further, similar to EC synthesis, upon use of 13C enriched CO2 and normal CO2, the protons at the positions ‘a’ and ‘b’ in the synthesised PCs showed different splitting patterns in their 1H NMR spectra. In case of normal CO2, both protons at the position a′ gave rise to triplets while multiplate signal was observed for the proton at position b′ (Fig. S4-a†). On the other hand, upon use of 13C enriched CO2, the triplet of doublets and broadened singlet were observed for the protons at the positions a′ and b′, respectively (Fig. S4-a†). Hence, the NMR analysis confirmed that the PC was formed from 13C enriched CO2 and isomers of propylene chlorohydrin through their equivalent reaction with DBU.
 | ||
| Fig. 7 13C NMR spectra for (a) equivalent mixture of DBU and propylene chlorohydrin, reaction mixture after bubbling of (b) CO2, (c) 13C enriched CO2 in DBU and propylene chlorohydrin, and (d) commercially available propylene carbonate in DMSO solvent. (NMR analysis with D2O in capillary). | ||
Moreover, two-dimensional NMR analysis such as 1H–13C HSQC and HMBC NMR analysis was also used to highlight better the existence of PC in the reaction mixture. In this case, the two dimensional NMR analysis of the reaction mixture was carried out in which 13C enriched CO2 was used in the PC synthesis. As depicted in Fig. S4-b,† the HSQC NMR analysis reveals the correlation signals between the proton and carbon atom, at the positions ‘a’ and ‘b’ and gave rise to the chemical shifts 4.42 or 4.95/71.33 (for two different protons at C-a′) and 5.26/74.57 ppm, respectively. The HMBC NMR analysis also confirmed the signals for the long range correlation between the protons atom at the position ‘a’ and ‘b’ and 13C enriched carbonyl carbon in PC (Fig. S4-c†).
Hence, similar to EC synthesis, the one and two dimensional NMR analysis confirmed that the PC was formed from the equivalent interaction between DBU, propylene chlorohydrin and CO2 where captured CO2 remained as a part of PC (reaction Scheme 2).
In order to understand the substrate scope for the DBU based synthesis of cyclic carbonates, various halohydrins such as 2-bromoethanol, 2-iodoethanol, 3-chloro-1-propanol and 3-bromo-1-propanol were also used. The formation of the corresponding cyclic carbonates was confirmed by NMR analysis. In case of both 2-bromo- and 2-iodoethanol, it was observed that after their addition to DMSO solution of DBU, the sp2-N atom of the DBU molecule became quaternized (ESI S5 and S6†). This was confirmed by the change in the chemical shifts of carbon atoms of DBU in 13C NMR at positions 6, 7 and 9, respectively. Further, after bubbling CO2 in the reaction mixture, the peak positions did not change and the peak belonging to the carbonyl carbon atom for the carbonate species was also not observed. Hence, these observations confirmed that the sp2-N atom in the DBU molecule become alkylated (n-alkylation) through nucleophilic attack on the carbon atom C-a in 2-bromo- or 2-iodoethanol. Therefore, the sp2-N in DBU molecule did not remain available to abstract the proton from the hydroxyl group in these halohydrins during the CO2 capture process. As Br and I are considered as better leaving groups than Cl, 2-chloroethanol was converted to a cyclic carbonate following the reaction Scheme 2 whereas its bromine and iodine analogues were not able to form cyclic carbonates and converted to n-alkylated DBU salts. Hence, it is proven that unlike 2-chloroethanol, 2-bromo 2-iodoethanol cannot be considered as a substrate for the synthesis of cyclic carbonates following the DBU involving synthesis approach.
In case of 1,3 halohydrin, as shown in 13C NMR (Fig. 8) and 1H (ESI S7†), it is possible to synthesise PC from 3-chloro-1-propanol. However, unlike 2-chloroethanol and 1-chloro-2-propanol, the rate of formation of cyclic carbonate i.e. PC was slow. From the 13C NMR analysis, it was observed that the SIL was formed immediately after the interaction of 3-chloro-1-propanol and DBU with CO2. In this case, after the formation of SIL, the peak for the carbonyl carbon atom in the carbonate species was observed with the chemical shift 156.9 ppm. In addition, the peak for the carbon atom C-7 in DBU with the chemical shift 161.4 ppm disappeared while new peak belonging to the carbon atom at position 7 (C-7′) in protonated DBU was observed at 165.8 ppm. Simultaneously, it was also observed that in 20 min PC was formed in the reaction mixture where the peak for the corresponding carbonyl carbon atom was observed at 148.9 ppm (shown by filled star). Further, with an increase in time, the amount of propylene carbonate increased steadily in the reaction mixture as the peak height at chemical shift 148.9 ppm increased. However, even after 1 h of reaction, complete conversion of SIL to cyclic carbonate was not observed. Hence, these observations confirmed that the immediate formation of SIL between DBU, 1-chloro-3-propanol and CO2 occurred immediately, but the subsequent cyclisation in the anion of SIL to form propylene carbonate was found as a slow step in this synthesis compared 1,2 chlorohydrins. The reason could be a different position of leaving group i.e. Cl atom in the 1-chloro-3-propanol molecule compared other 1, 2 chlorohydrins. Compared to 1,2 chlorohydrins, having one more methylene group at the position ‘b’ in 1-chloro-3-propanol increases the electron density at the carbon atom at position ‘a’ to which Cl atom is attached. This renders the Cl atom as a less good leaving group which reduces further the rate of formation of propylene carbonate compared to the 1,2 chlorohydrins. The use of 3-bromo-1-propanol, on other hand, gave rise to a faster reaction rate than 3-chloro-1-propanol where in 20 minutes it was converted to PC (ESI S8†). In this case, Br is considered as a better leaving group than Cl and, therefore, beyond the structural similarities, the Br containing 1,3 halohydrin showed faster reaction kinetics in the synthesis of PC compared to its Cl containing analogue.
 | ||
| Fig. 8 13C NMR spectra for (a) equivalent mixture of DBU and 3-chloro-1-propanol in DMSO, reaction mixture of DBU and 3-chloro-1-propanol in DMSO after bubbling of CO2 for (b) 20 min, (c) 45 min and, (d) 1 h. (NMR analysis with D2O in capillary). | ||
Hence, similar to previously reported metal based reaction approach, organic superbase DBU was also used in the synthesis of cyclic carbonates using various types of halohydrin substrates. In this case, both 1,2 and 1,3 chloro- and bromohydrin except 2-bromo- and 2-iodoethanol were found as useful substrates in the synthesis following the reaction Scheme 1. In case of 1,3 halohydrins, the previous studies involving metal carbonate have controversial results where Cs2CO3 was able to convert both 1,2 and 1,3 halohydrins to cyclic carbonates. On the other hand, K2CO3 promoted only reactions with 1,2 halohydrins to corresponding cyclic carbonates. In this study, DBU based reaction approach except in the case of 2-bromo- and 2-iodo ethanol, was able to convert various types of halohydrins to cyclic carbonates and with the faster reaction kinetics compared to metal based processes.
Although the synthesis of both EC and PC was successfully demonstrated following mild reaction approach, the separation of both these cyclic carbonates from DMSO solvent is an energy intensive process. In other word, cyclic carbonates as well as DMSO are considered as high boiling liquids and, therefore, their separation from each other is not feasible through distillation. Also, the separation using solvent extraction can also be a difficult as EC or PC and DMSO are soluble in most of the organic solvents as well as in water. Further, DMSO is not a green solvent, especially in bulk scale synthesis, when its toxic nature as well as expensiveness are considered. Hence, rather than searching alternative techniques for the separation of DMSO, we decided to replace the DMSO with an excess of alkylene chlorohydrin in the reaction mixture. We have also previously used a similar reaction approach upon acrylic plastic precursor synthesis where excess of methanol was used as the solvent to replace DMSO.22 Herein we also proposed that the methanol not only performed as a solvent media but also served as a reagent in the process. Considering this, the synthesis of EC or PC was carried out in the alkylene chlorohydrin solvent where the 50 vol% solution of DBU in the ethylene or propylene chlorohydrin was used, respectively.
After exposure of the solution of DBU in alkylene chlorohydrin to CO2, the formation of EC or PC was confirmed by NMR analysis. The 13C and 1H NMR spectra for the synthesis of EC in the ethylene chlorohydrin solution are shown in Fig. 9 and in the ESI Fig. S9,† respectively. Similar to synthesis in DMSO, the NMR analysis confirms that EC was formed when the solution of DBU in ethylene chlorohydrin reacted with CO2. The 13C NMR analysis of the reaction mixture gave rise to isolated signals for both EC and [DBUH][Cl] as well as for the unreacted ethylene chlorohydrin. Similar to synthesis of EC, 1H and 13C NMR analysis also confirmed the formation of PC after interactions between the solution of DBU in propylene chlorohydrin and CO2 (Fig. 10 and S10†). As shown in Fig. 10, the NMR spectra confirm that the signals for the PC, [DBUH][Cl] and unreacted isomers of the propylene chlorohydrin were observed. Hence, it can be concluded that DMSO free synthesis of the EC or PC can proceed where alkylene chlorohydrins can be used as both solvents and reagents in these processes.
 | ||
| Fig. 9 13C NMR spectra for (a) 50 vol% solution of DBU in the ethylene chlorohydrin and, (b) reaction mixture after bubbling of CO2 in 50 vol% solution of DBU in the ethylene chlorohydrin (NMR analysis in DMSO-d6). | ||
 | ||
| Fig. 10 13C NMR spectra for (a) 50 vol% solution of DBU in the propylene chlorohydrin and, (b) reaction mixture after bubbling of CO2 in 50 vol% solution of DBU in the propylene chlorohydrin (NMR analysis in DMSO-d6). | ||
Further, as discussed previously, excess of alkylene chlorohydrins were used in order to facilitate the separation of components in the reaction mixture after the formation of cyclic carbonates. The separation of the EC or PC, [DBUH][Cl] and the excess of the alkylene chlorohydrins was achieved following the separation approaches mentioned in the experimental section. The composition of the [DBUH][Cl] obtained through its precipitation after addition of ethyl acetate in the reaction mixture was confirmed by 1H NMR analysis. As shown in the ESI S12-a,† pure form of [DBUH][Cl] was recovered from the reaction mixture after the synthesis of the cyclic carbonates. After the removal of the [DBUH][Cl], the remaining ethyl acetate solution was further processed for the recovery of the excess of alkylene chlorohydrin and EC or PC. The ethyl acetate was removed using rotation evaporator followed by alkylene chlorohydrins were separated from the EC or PC using high vacuum distillation process at 40 °C. The composition of the recovered EC or PC were analysed by the 1H NMR analysis (Fig. S11†) while their recovery was calculated by eqn (1) (Experimental section). The 1H NMR spectra of the EC and PC showed that pure cyclic carbonates were recovered where recovery levels of 92 and 96%, respectively, were reached. The high recovery as well as purity of these cyclic carbonates was achieved because of their high boiling points compared to respective alkylene chlorohydrins. Therefore, high vacuum distillation at 40 °C allowed efficient separation of alkylene chlorohydrins from cyclic carbonates. Hence, use of alkylene chlorohydrin solution of the DBU instead of DMSO in the cyclic carbonates synthesis facilitated the separation of the reaction fractions such as cyclic carbonates and [DBUH][Cl] from each other.
DBU was recovered from the [DBUH][Cl] applying alkaline solvent treatment and solvent extraction process. The purity and the recovery of the DBU was determined by the 1H NMR analysis and the eqn (2) (Experimental section). The NMR analysis confirmed that pure DBU was recovered while 76% recovery was achieved (Fig. S12-b†). However, as mentioned in a previous report, the recovery of the DBU using alkaline solvent treatment from its cationic form is inconsistent and not totally optimized.17 However, considering the price as well as the importance of DBU in the activation of CO2 under mild reaction conditions, more work needs to be performed in order to reach higher recovery of the DBU from [DBUH][Cl].
Hence, in this report, compared to previous study where metal carbonates were used as an activating agent for cyclic carbonate synthesis from halohydrins, the similar synthesis was performed at room temperature and atmospheric pressure with faster reaction kinetics and no use of the metal based catalysts or co-catalysts. As it was described previously, even though metal based synthesis approach for cyclic carbonate synthesis can be applied for various types of halohydrin substrates, it is also accompanied with the generation of the 2 equivalents of metal salts as a waste per equivalent of cyclic carbonate (MHCO3 and MCl or MBr where M = K, Na or Cs). In this case, authors also did not describe the recovery of these salts as well as regeneration of active metal carbonate catalysts. Unlike these metal based processes, the generation of waste did not occur in DBU based reaction approach except the formation of easily recoverable by-product such as [DBUH][Cl] salt. Besides that, the recovered DBU salt can be converted to DBU with alkaline solvent treatment. Hence, the route proposed in this report for the synthesis of cyclic carbonates from various chlorohydrins can be more preferable compared to the metal based approach.
4. Conclusions
A one-pot and metal free synthesis of the both ethylene carbonate (EC) and propylene carbonates (EC) from molecular CO2 was successfully carried out under mild reaction conditions. Various Cl, Br and I comprised 1,2 and 1,3 alkylene halohydrins and organic superbase DBU reacted with CO2 to form cyclic carbonate and halide salts of DBU. The NMR based mechanistic studies confirmed that the switchable ionic liquid (SIL) was formed in situ from DBU, alkylene halohydrin and CO2 and was further converted to EC or PC through intermolecular ring closing reaction. In the case of 1,2 halohydrins, 2-chloroethanol and 1-chloro-2-propanol were converted to EC and PC, respectively, whereas 2-bromo and 2-iodoethanol reacted equivalently with DBU and formed n-alkylated of DBU salts instead of cyclic carbonates. Both 1,3 halohydrins such as 3-chloro-1-propanol and 3-bromo-1-propanol could be converted to PC. However due to having Br as a good leaving species compared to Cl, 3-bromo-1-propanol gave rise to faster reaction kinetics compared to 3-chloro-1-propanol during the formation of PC. In order to avoid the use of DMSO as a solvent, the EC or PC synthesis from 1,2 chlorohydrins was further carried out using chlorohydrins as a solvent. In this case, the chlorohydrins not only performed as a solvent media but also served as reagents in the process. Further, the use of chlorohydrins as a solvent also enabled efficient separation of the reaction components through mild experimental approaches. In this process, pure forms of EC or PC were obtained where 92 and 96% of recovery of cyclic carbonates was achieved, respectively. The organic superbase DBU was also recovered with high purity from [DBUH][Cl] using alkaline solvent treatment whereupon the recovery was 76%. Hence, in this report DBU superbase involved synthesis of EC and PC from CO2 was carried out using various types of halohydrins. The synthesis was further successfully upgraded using halohydrins as both solvent media and reagent. Being metal free, one pot and mild reaction condition approach as well as having control over the waste generation, this DBU involved reaction approach can be applied in large-scale synthesis of cyclic carbonates from CO2 and halohydrins.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We wish to thanks Dr Tobias Sparrman and Dr Mattias Hedenström for their assistance in the NMR analysis. This work is part of activities of the Technical Chemistry, Department of Chemistry, Chemical-Biological Centre, Umeå University, Sweden as well as the Johan Gadolin Process Chemistry Centre at Åbo Akademi University in Finland. The Swedish Research Council (Drn: 2016-04090), Bio4Energy programme, Kempe Foundations and Wallenberg Wood Science Center under auspices of Alice and Knut Wallenberg Foundation are gratefully acknowledged for funding this project.References
- J. L. Scott and H. F. Sneddon, Green Solvents: Green Techniques for Organic Synthesis and Medicinal Chemistry, 2018, pp. 21–42, DOI:10.1002/9781119288152.ch2.
- H. L. Parker, J. Sherwood, A. J. Hunt and J. H. Clark, ACS Sustainable Chem. Eng., 2014, 2, 1739–1742 CrossRef CAS.
- M. Morcillo, M. North and P. Villuendas, Synthesis, 2012, 12, 918–1925 Search PubMed.
- T. Sakakura and K. Kohnoa, Chem. Commun., 2009, 1312–1330 RSC.
- T. Sakakura, J. C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS.
- A. J. Kamphuis, F. Picchioni and P. P. Pescarmona, Green Chem., 2019, 21, 406–448 RSC.
- B. Schaffner, F. Schaffner, S. P. Verevkin and A. Borner, Chem. Rev., 2010, 110, 4554–4581 CrossRef CAS.
- M. H. Anthofer, PhD thesis, Development, Optimization and Application of Catalysts in Cyclic Carbonate Synthesis and Olefin Epoxidation, 2015, https://mediatum.ub.tum.de/doc/1237026/1237026.pdf.
- F. D. Bobbink, D. Vasilyev, M. Hulla, S. Chamam, F. Menoud, G. Laurenczy, S. Katsyuba and P. J. Dyson, ACS Catal., 2018, 8, 2589–2594 CrossRef CAS.
- C. M. Byrne, S. D. Allen, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2004, 126, 11404–11405 CrossRef CAS.
- M. Taherimehr, J. P. Sert, A. W. Kleij, C. J. Whiteoak and P. P. Pescarmona, ChemSusChem, 2015, 8, 1034–1042 CrossRef CAS.
- T. Hirose, S. Shimizu, S. Qu, H. Shitara, K. Kodama and L. Wang, RSC Adv., 2016, 6, 69040–69044 RSC.
- Z. Xi, Y. Xiangui, C. Tong, Z. Yi and W. Gongying, Chin. J. Catal., 2009, 30, 7–8 CrossRef.
- J.-L. Wang, L.-N. He, X.-Y. Dou and F. Wu, Aust. J. Chem., 2009, 62, 917–920 CrossRef CAS.
- M. R. Reithofer, Y. N. Sum and Y. Zhang, Green Chem., 2013, 15, 2086–2090 RSC.
- J. R. O. Gomez, O. G.-J. Aberasturi, C. A. R. Lopez, J. N. Mestre, B. M. Madurg and M. Belsue, Chem. Eng. J., 2011, 175, 505–511 CrossRef.
- J. Sun, W. Cheng, Z. Yang, J. Wang, T. Xu, J. Xin and S. Zhang, Green Chem., 2014, 16, 3071–3078 RSC.
- D. J. Heldebrant, C. R. Yonker, P. G. Jessop and L. Phan, Energy Environ. Sci., 2008, 3, 487–493 Search PubMed.
- P. G. Jessop, S. M. Mercer and D. J. Heldebrant, Energy Environ. Sci., 2012, 5, 7240–7253 RSC.
- I. Anugwom, P. Maki-Arvela, P. Virtanen, P. Damlin, S. Sjoholm and J.-P. Mikkola, RSC Adv., 2011, 1, 452–457 RSC.
- R. A. de Graaf, D. L. Rothman and K. L. Behar, NMR Biomed., 2011, 24, 958–972 CrossRef CAS.
- S. G. Khokarale and J. P. Mikkola, Green Chem., 2019, 21, 2138–2147 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra06765e |
| This journal is © The Royal Society of Chemistry 2019 |