DOI:
10.1039/C9RA06629B
(Paper)
RSC Adv., 2019,
9, 35939-35947
Defect-related luminescent nanostructured hydroxyapatite promotes mineralization through both intracellular and extracellular pathways
Received
22nd August 2019
, Accepted 10th October 2019
First published on 5th November 2019
Abstract
Hydroxyapatite (HAP) is a widely used biomaterial for bone tissue substitution due to its chemical similarity with the natural bone. Defect-related luminescent HAP materials have the same chemical composition as normal HAP and excellent biocompatibility. However, only few works have focused on the defect-related luminescent HAP materials on bone regeneration. In this work, we systematically investigated the bone regeneration pathway induced by nanostructured particles using defect-related luminescent hydroxyapatite (S2) materials. We monitored the subcellular distribution and location of S2 during osteoblast differentiation with the property of defect-related luminescence. Nano-scale S2 could be internalized by osteoblasts (OBs) via caveolae-mediated endocytosis and macropinocytosis. S2 incorporated into the lysosomes dissolved and released calcium ions for the formation of mineralized nodules. Extracellular S2 also promoted bone regeneration as a nucleation site. Taken together, the physical properties of hydroxyapatite control the bone regeneration pathway in osteoblasts.
Introduction
Hydroxyapatite (HAP) is the major mineral component of calcified tissues. Synthetic HAP (Ca10(PO4)6(OH)2) has been extensively used as an implant material for bone substitutes due to its biocompatibility, bioactivity and osteoconduction properties.1–3 To monitor the biological relevant testing of HAP, it is desirable to develop fluorescent labeled nanoparticles for tracing. However, traditional HAP generally exhibits no luminescence, which seriously limits its applications. To overcome this limitation, fluorescent HAP nanoparticles doped with rare earth ions (e.g., Eu3+ and Tb3+) and organic dyes have been developed and applied in the bioimaging field.4–6 However, the introduction of these doped ions and organic dyes may affect the mineralization of bone cells, which is unsuitable for studying the effects of HAP on bone regeneration. Thus, it is necessary to develop novel luminescent HAP without introducing rare earth ions and fluorescent dyes.
Defect-related luminescent materials do not contain any transition metal or rare earth activators, and the emission arises from special defects.7 These materials were earlier reported in 19868 and have achieved remarkable development since the synthesis of white phosphors from a silicate-carboxylate sol–gel precursor.9 The defect-related luminescent materials can be used in various electronics-related applications due to the advantages of low toxicity and stability.10,11 In our previous work, three defect-related luminescent hydroxyapatite particles with different morphologies and sizes were synthesized. The defect-related luminescent HAP materials in the nano-scale show excellent biocompatibility in biomedical applications.12 The defect-related luminescent materials are nontoxic for osteoblasts. Previous studies have reported that the materials modified with HAP tend to improve the mineralization of bone cells.13 Thus, we want to know whether the defect-related luminescent HAP materials have any effects on bone regeneration.
In this current work, by taking advantage of the defect-related luminescence properties, an evaluation of the HAP nanoparticles on the subcellular location and their influence on the bone regeneration of osteoblasts was carried out systematically. This work is quite different from our previous work, and it is an extension of the last work.
Materials and methods
Materials
Newborn (1–2 days old) ICR mice were purchased from the Experimental Animal Center of Hebei Medical University. DMEM was purchased from Gibco-BRL (Grand Island, NY, USA). Neonatal bovine serum (NBS) was obtained from Hangzhou Sijiqing Biological Engineering Materials Co., Ltd. (Hangzhou, China). Trypsin, benzylpenicillin, streptomycin, β-glycerphosphate, ascorbic acid, dexamethasone, sirius red, picric acid, alizarin red S (ARS), cetylpyridinium-chloride, propidium iodide (PI) and acridine orange (AO) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Mito-Tracker Green, Lyso-Tracker Red, and wortmannin were purchased from Beyotime Institute of Biotechnology (Haimen, China). Nystatin was purchased from Aladdin (Shanghai, China). Chlorpromazine hydrochloride was purchased from TCI (Tokyo, Japan). An ALP activity kit and a micro-protein assay kit were obtained from Nanjing Jiancheng Biological Engineering Institute (Nanjing, China). TRIzol reagent was purchased from Invitrogen (Carlsbad, CA, USA) The Revertaid First Strand cDNA Synthesis Kit was purchased from Thermo Scientific (Vilnius, Lithuania). UltraSYBR mixture was purchased from Beijing ComWin Biotech Co., Ltd. (Beijing, China). All the other chemicals were of analytical grade and used without further purification.
Synthesis of HAP
A defect-related luminescent HAP sample was synthesized as previously reported.12 Ca(NO3)2·4H2O and hexadecyltrimethylammonium bromide (CTAB) were dissolved in deionized water, and the pH was adjusted to 7.0 using HNO3 or NH3·H2O solutions. Subsequently, a “mixing solution” containing trisodium citrate and (NH4)2HPO4 was added to the above solution. After stirring for 20 min, the mixture was transferred into an autoclave, sealed, and maintained at 180 °C for 24 h. Precipitates were washed with deionized water and absolute ethanol and then air-dried at room temperature. The HAP sample prepared at pH 7.0 was labeled as S2.
Characterization
The morphology of S2 was determined using a JSM-7500 F cold field scanning electron microscope (SEM) (JEOL, Japan) and an FEI Tecnai G2 S-Twin transmission electron microscope (TEM) (FEI, America). The chemical structure was characterized by a Bruker Advance D8 X-Ray diffractometer (XRD) (Bruker, Germany). Photoluminescence (PL) spectra of S2 were tested by a Hitachi F-7000 spectrophotometer.
Isolation and culture of primary OBs
Primary OBs were isolated from neonatal mouse calvaria by sequential digestion with trypsin and collagenase as described previously.12 All studies involving the use of animals were conducted in conformity with NIH guidelines (NIH Publications No. 8023, revised 1978) and were approved by Animal Care and Use Committee of Hebei Medical University. Briefly, calvaria were isolated aseptically from neonatal ICR mice. After eliminating all soft tissues, the calvaria were digested once with 0.25% trypsin for 25 min and twice with 0.1% collagenase II for 1 h each. The cells were collected and cultured in a 75 cm2 flask with complete medium (DMEM containing 10% NBS, 100 U mL−1 penicillin, and 100 μg mL−1 streptomycin) at 37 °C under a humidified atmosphere with 5% CO2. The medium was changed every 3 days.
In Vitro cell uptake
The uptake of S2 was performed according to the method described by Amornwachirabodee et al.14 In brief, OBs were seeded in a 35 mm glass bottom dish at the density of 4 × 104 cells per well and allowed to adhere overnight. Cells were incubated with S2 at 37 °C under a humidified atmosphere with 5% CO2 for 24 h. The media were removed, and the cells were washed thrice with PBS; then, the cells were stained with 6 μg mL−1 acridine orange (AO) for 30 min in the dark. Then, the media were removed, and the cells were washed thrice with PBS before being subjected to confocal laser scanning microscopy (CLSM) (Olympus FV1000, Japan). Excitation at 488 nm and fluorescence signals at 580–680 nm were collected as AO fluorescence signals. Excitation at 405 nm and fluorescence signals at 415–515 nm were collected as S2 fluorescence signals. Images indicating the locations of S2 were then constructed using the overlap signals.
Inhibition of endocytosis
The endocytotic pathways were performed according to the method described by Greulich et al.15 In brief, OBs were seeded in 6-well plates at the density of 1 × 105 cells per well and allowed to adhere overnight. OBs were treated with inhibitors of endocytosis (3.5 μM chlorpromazine hydrochloride, 5 μM nystatin or 200 nM wortmannin) for 30 min at 37 °C under cell culture conditions. Then, OBs were washed twice with PBS and changed for a new complete medium. After this pretreatment, S2 was added to the culture (final concentration: 80 μg mL−1) and the cells were incubated for a further 1 h. Cells treated without inhibitors were set as the control. OBs incubated with S2 were set as a positive control, and the cells incubated without S2 were set as a negative control. Subsequently, the cells were harvested and resuspended in PBS. The uptake of particles into OBs was analyzed by a flow cytometer (FACSCalibur, Becton Dickinson). The side scatter data were analyzed using the CellQuest software. Calibration reagents and solutions for flow cytometry were obtained from Becton Dickinson. After achieving the appropriate instrument settings and compensations, the instrument setup was not changed throughout the study. Ten thousand cells were acquired for each measurement.
Subcellular location of S2
The co-localization studies were performed according to the method described by Greulich et al.15 OBs were cultured as described above and exposed to 80 μg ml−1 S2 particles for 24 h. Following incubation, the OBs were labeled with specific cell organelle fluorescent probes (Beyotime). In order to label the lysosomes and mitochondrion, the cells were incubated with 75 nM Lyso-Tracker Red and 100 nM Mito-Tracker Green in complete medium for 45 min at 37 °C. CLSM was taken (FV1000, Olympus) and processed using the FV10-ASW software. To label the cell nucleus, the cells were fixed with 4% paraformaldehyde for 10 min, followed by permeabilization with 0.2% Triton X-100 in PBS for 2 min. Then, the cells were incubated with 100 μg mL−1 PI in complete medium for 30 min at 37 °C. After three rinses in PBS, the cells were viewed under CLSM (Olympus).
Collagen production analysis
The Sirius red staining–based colorimetric assay was used to quantify collagen deposition. Collagen production analysis was performed according to the method described by Walter et al.16 OBs were seeded in 48-well tissue culture plates at the density of 1 × 104 cells per well with OS. S2 at a final concentration of 80 μg mL−1 was added to complete medium and incubated for 14 d. OBs treated with only OS were used as the control group. The plates were washed three times with ice-cold PBS and fixed for 1 h with 10% formaldehyde and washed with triple-distilled water. Then, 200 μL of dye solution (1 mg Sirius red was dissolved in 1 mL saturated solution of picric acid) per well was added and incubated for 1 h. After that, plates were washed with 0.01 M HCl to remove the excess of the dye solution. The collagen fixed with dye was extracted with 200 μL of 0.1 M NaOH solution per well. OD was measured by a microplate spectrophotometer at 550 nm. All results were normalized by the protein content. The collagen production rate (%) was calculated using the following formula: (ODtreated − ODblank)/(ODcontrol − ODblank) × 100.
Mineralized matrix formation assay
An ARS staining assay was used to evaluate mineralized matrix formation, as described in detail elsewhere.17 OBs were seeded in 48-well culture plates at the density of 1 × 104 cells per well with the osteogenic induction supplement (OS) containing 0.1 μM dexamethasone, 5.0 mM β-glycerophosphate and 50 μg mL−1 ascorbic acid. The medium was changed to differentiation medium OS in the presence or absence of S2 at the concentrations of 20, 40 and 80 μg mL−1 for 17 d. OBs treated with only OS were used as the control group. After incubation, the cells were fixed in 95% ethanol for 10 min at room temperature. The fixed cells were washed with PBS and stained with ARS (40 mM) for 30 min at 37 °C. Quantitation of ARS staining was performed by elution with 10% (w/v) cetylpyridinium chloride 200 μL per well at room temperature for 20 min. OD was measured by a microplate spectrophotometer at 570 nm. All results were normalized by the protein content. The mineralized rate (%) was calculated using the following formula: (ODtreated − ODblank)/(ODcontrol − ODblank) × 100.
Real-time RT-PCR
The quantitative messenger RNA (mRNA) expression of runt-related transcription factor 2 (RUNX2) and osteocalcin (OCN) was characterized by real-time reverse transcriptase polymerase chain reaction (RT-PCR) (ABI Stepone Plus, USA). After 14 days of culture, total RNA was extracted using the TRIzol reagent according to the manufacturer's instructions. The quality and concentration of RNA samples were detected using a spectrophotometer (Bio-Rad Smartspec Plus, USA) at 260, 280 nm absorbance and calibrated by DEPC water. cDNA was reverse transcribed using the Revertaid First Strand cDNA Synthesis Kit. The resultant cDNA was used for the PCR reaction using the UltraSYBR mixture. Reaction conditions were 95 °C for 15 s, 60 °C for 30 s and 72 °C for 30 s. Three independent cultures were analyzed. Gene expression was normalized by glyceraldehyde 3-phosphate dehydrogenase (GAPDH) using a comparative cycle threshold (ΔΔCt) method. Primer pairs used were as follows: GAPDH forward: 5′-GAC TTC AAC AGC AAC TCC CAC-3′, reverse: 5′-TCC ACC ACC CTG TTG CTG TA-3′; OCN forward: 5′-GAA CAG ACT CCG GCG CTA-3′, reverse: 5′-AGG GAG GAT CAA GTC CCG-3′; RUNX2 forward: 5′-TTC TCC AAC CCA CGA ATG CAC-3′, reverse: 5′-CAG GTA CGT GTG GTA GTG AGT-3′.
Nucleation role of S2 in the formation of the mineralized matrix
ARS staining assay was used to stain the mineralized matrix, as described above. In brief, cells were seeded in a glass coverslip at 2 × 104 cells per mL and cultured for 24 h. After treatment with OS in the presence or absence of S2 (80 μg mL−1) for 17 d, cells were stained with ARS as described above. The cells were washed three times with PBS. The mineralized matrix was detected by a fluorescence microscope (Olympus BX53, Japan) with an excitation of 395–410 nm and an emission of 450–470 nm for S2 to trace the fate of HAP.
Statistical analysis
Data were collected from three separate experiments. The results were expressed as mean ± standard deviation (SD). The statistical differences were analyzed using SPSS′ t-test. P < 0.05 was considered to indicate statistical differences.
Results and discussion
Characterization of S2
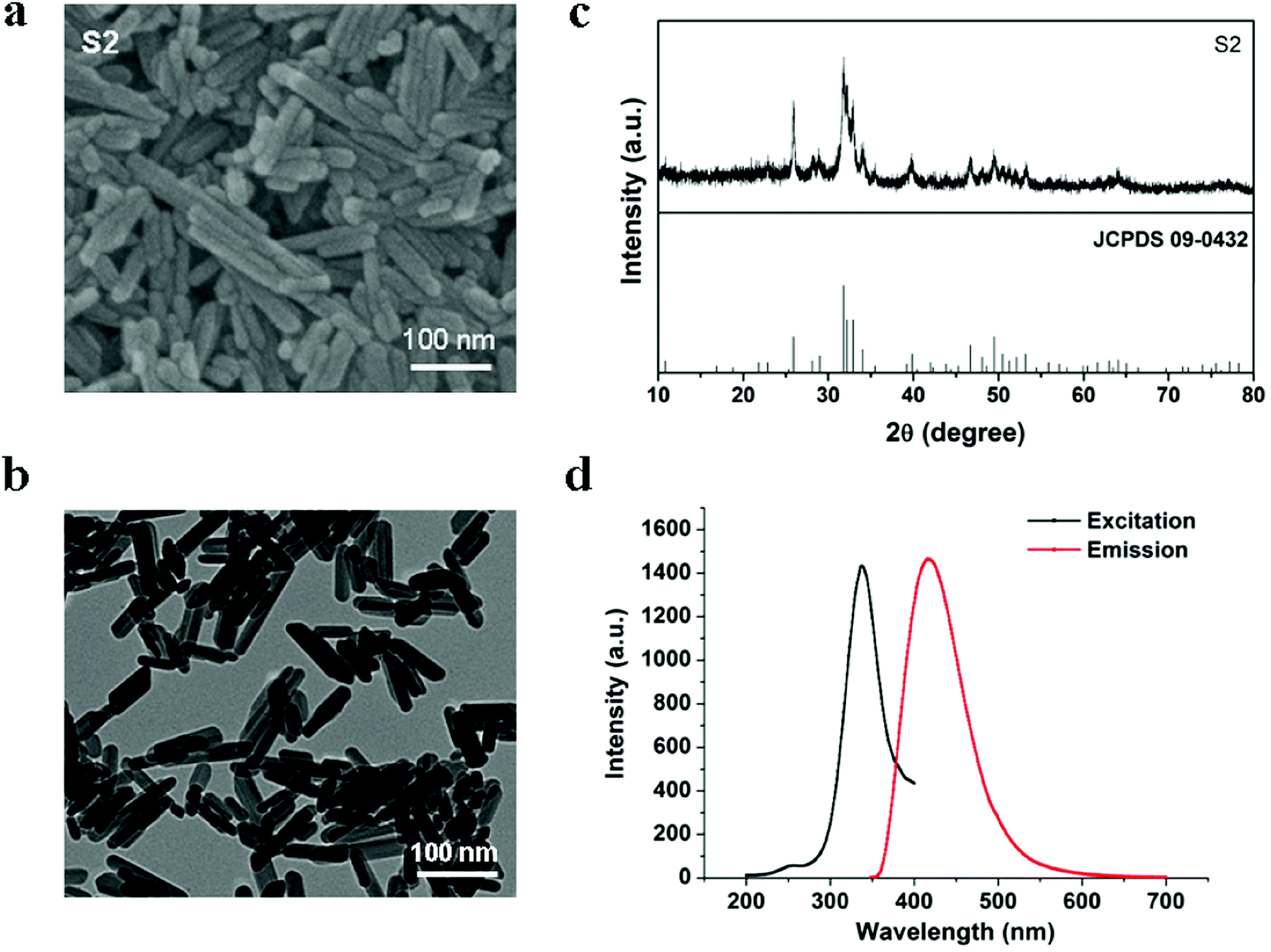
In our experiments, defect-related luminescent hydroxyapatite particles S2 were synthesized according to our previously reported method, in which the fabrication process has been described in detail. Sample S2 particles were formed as nanorods (Fig. 1a and b). The diameter and length of S2 were approximately 25 nm and 100 nm, respectively. Fig. 1c shows that the diffraction peaks of S2 can be well indexed to the hexagonal phase HAP (JCPDS card no. 09–0432). The PL excitation and emission spectra (Fig. 1d) of the S2 nanorods display broad bands centered at 339 and 418 nm, respectively.
 |
| | Fig. 1 (a) SEM image of S2. (b) TEM image of S2. (c) XRD pattern of S2. (d) PL excitation and emission spectra of S2. | |
Uptake and mechanism of S2 in OBs
Nanophase HAP possesses unique properties and many researchers have found that the HAP nanoparticles can easily enter many types of cells.18,19 Strong blue fluorescence was observed inside the OBs incubated with nano-scale S2 for 24 h (Fig. 2). Furthermore, most of the fluorescence signals were within the cytoplasm. It could be confirmed that the S2 nanorods entered OBs.
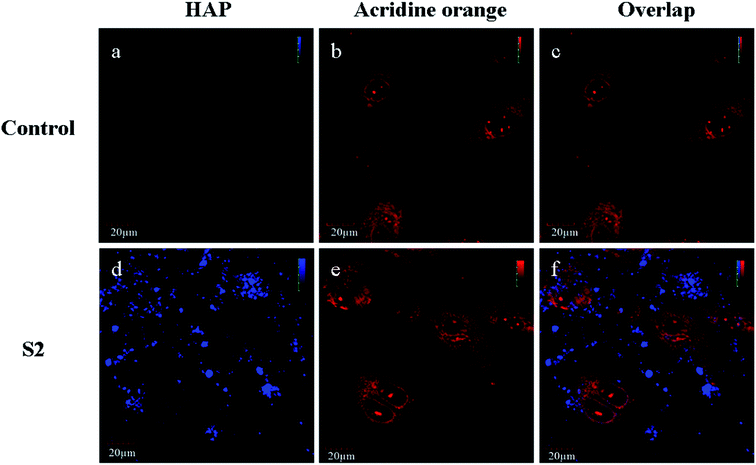 |
| | Fig. 2 Evaluation of the uptake of S2 by OBs. (a–c) Control, (d–f) S2. (a and d) Blue fluorescence (425–475 nm) images from S2 by excitation at 405 nm with a semiconductor laser beam. (b and e) Red fluorescence (617–717 nm) images from AO stained at 488 nm with an argon laser beam. (c and f) The overlap images are nanorods and cells; scale bar is 20 μm. | |
In order to analyze the mechanism of the cellular uptake of nano-scale HAP, selective inhibitors of the specific routes of endocytosis were applied. OBs were preincubated for 30 min with individual inhibitors; then, S2 (80 μg mL−1) was added and cell culture was continued for 1 h (Fig. 3). The quantification of the uptake of nanoparticles was performed by flow cytometry using side scatter analysis. Compared to the result for the negative control, the scatter signal significantly increased in the treatment of S2. Furthermore, the addition of inhibitors via clathrin-mediated endocytosis OBs treated with S2 and a specific clathrin-mediated endocytosis inhibitor (chlorpromazine) showed no significant decrease in the scatter signal. In the presence of a specific caveolae inhibitor (nystatin) and macropinocytosis inhibitor (wortmannin), the scatter signal decreased (6.64% and 5.07%, respectively) in comparison to that for OBs that were not treated with the inhibitor. Nevertheless, the inhibition of caveolae-mediated endocytosis and macropinocytosis did not result in the complete suppression of endocytosis. Caveolae-mediated endocytosis and micropinocytosis were highly involved in the uptake of S2.
 |
| | Fig. 3 Uptake of S2 into OBs in the presence of different endocytosis inhibitors. OBs were preincubated for 30 min with the individual inhibitors and subsequently, the S2 (80 μg mL−1) uptake over 1 h was analyzed by flow cytometric light scatter analysis; the representative scattergrams are shown. | |
Intracellular localization of internalized S2
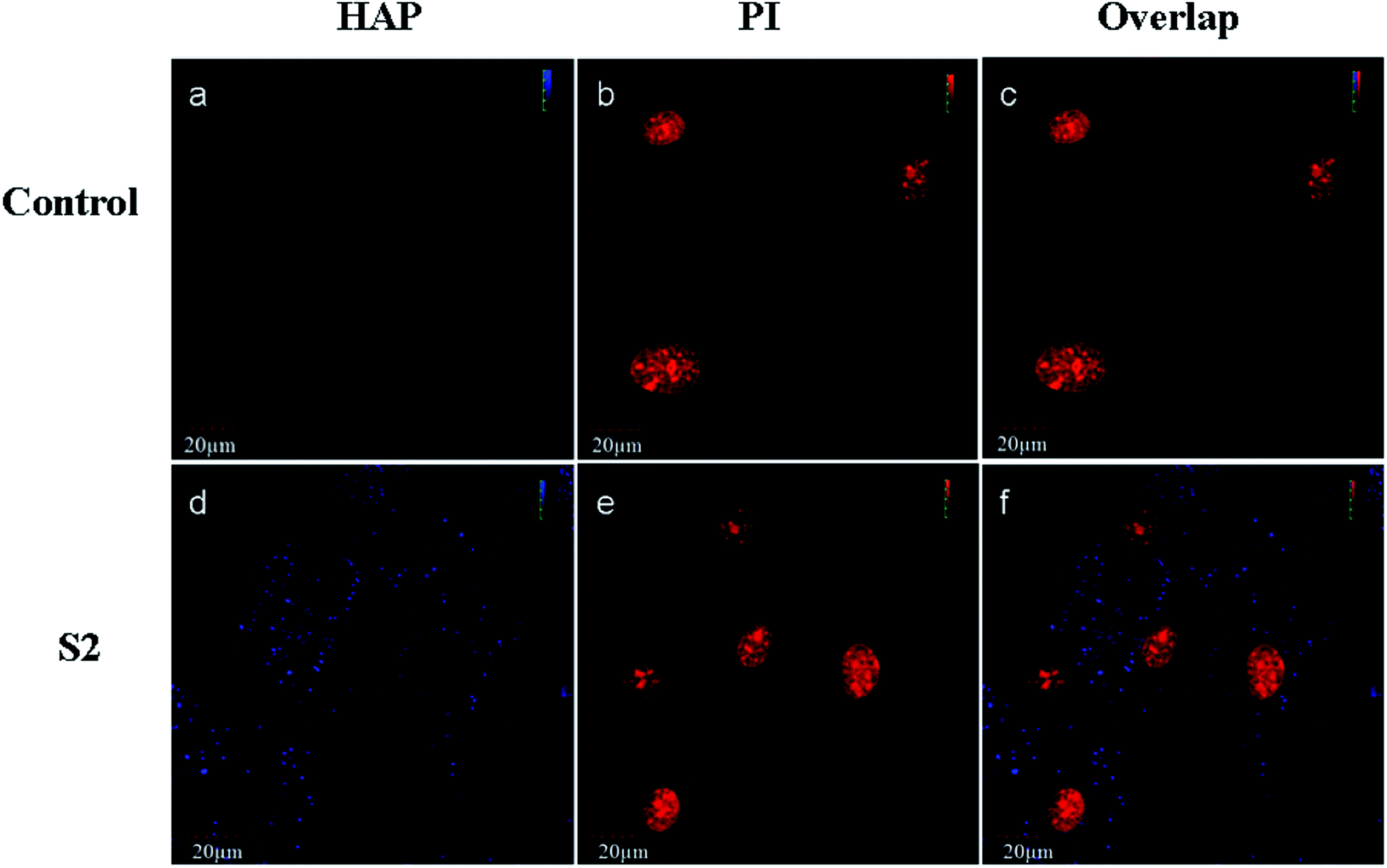
Schmidt et al.20 found that CaP nanoshells were internalized by cells and localized around the cell nucleus. Although research has confirmed that the nanoparticles may internalize, the subcellular distribution is still not clearly understood. To determine the intracellular distribution of internalized S2, the lysosomes and mitochondrion were labeled with a panel of specific organelle markers. In order to demonstrate the uptake of S2, OBs were cultured with S2 at 37 °C for 24 h under cell culture conditions. CLSM was performed on the same cell areas (Fig. 4 and 5). The S2 nanoparticles were mainly incorporated into the cytoplasm and not into the nucleus. As shown in Fig. 4 and 5, the S2 nanoparticles are mainly found to be associated with lysosomes areas (Fig. 4) but not inside the mitochondrion and the nucleus (Fig. 4 and 5). The HAP particles can be efficiently dissolved in the acidic intracellular compartments and release calcium ions.21 The Ca2+ ions promote cell attachment, growth, and differentiation.22 Higher Ca2+ concentrations promote osteoblast differentiation, leading to bone mineralization in both growth and osteogenic conditions.23,24 The HAP particles internalized in the cells release Ca2+ and promote mineralization through an intracellular pathway.
 |
| | Fig. 4 Fluorescence images taken using CLSM. The cells were treated with S2 nanoparticles (80 μg mL−1) for 24 h. (a) Blue fluorescence (425–475 nm) from S2 on excitation at 405 nm with a semiconductor laser beam; (b) green fluorescence (500–545 nm) from mitochondria with Mito-Tracker Green on excitation at 488 nm with an argon laser beam; (c) merged images of (a) and (c); (d) red fluorescence (575–675 nm) from lysosomes stained with Lyso-Tracker Red on excitation at 559 nm with a helium-neon laser beam; (e) merged images of (a) and (d); (f) bright field. Scale bar is 20 μm. | |
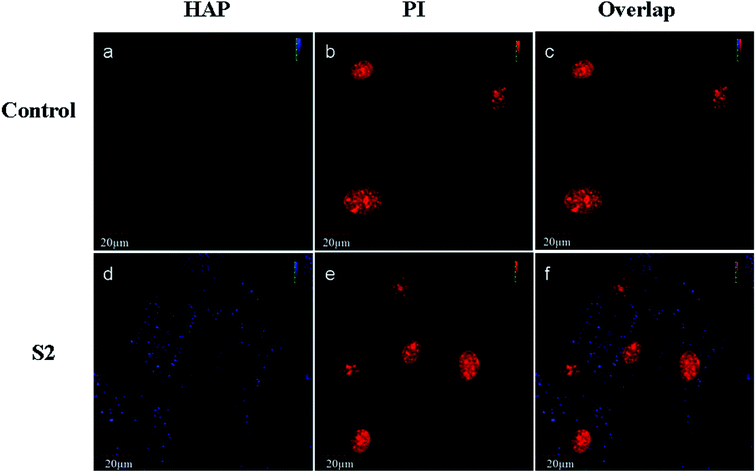 |
| | Fig. 5 Fluorescence images taken under CLSM. The cells were treated with S2 nanoparticles (80 μg mL−1) for 24 h. (a and d) Blue fluorescence (425–475 nm) from S2 on excitation at 405 nm with a semiconductor laser beam; (b and e) red fluorescence (575–675 nm) from nucleus stained with PI on excitation at 559 nm with an argon laser beam; (c and f) overlap. Scale bar is 20 μm. | |
Effect of S2 on the formation of mineralized nodules
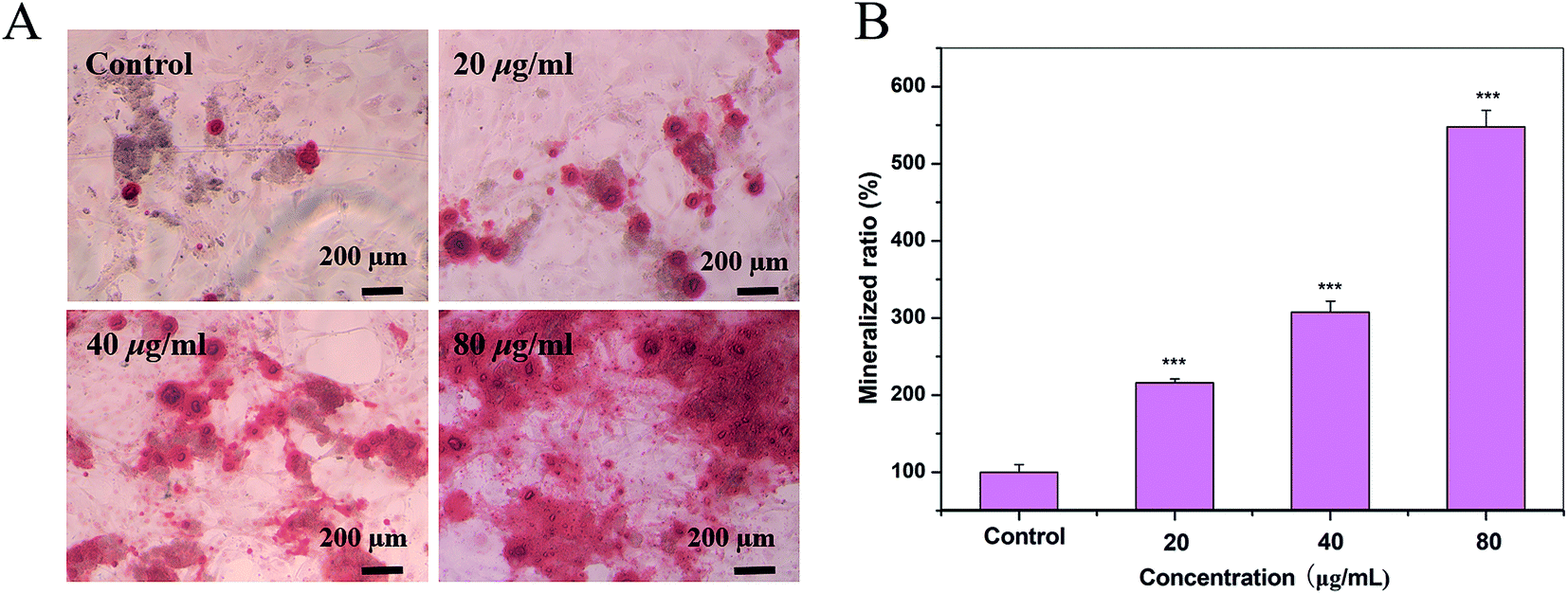
A previous work has demonstrated that HAP enhances bone regeneration.25 On day 17, S2 promoted the formation of the mineralized nodules of OBs at all concentrations in a dose-dependent manner (Fig. 6).
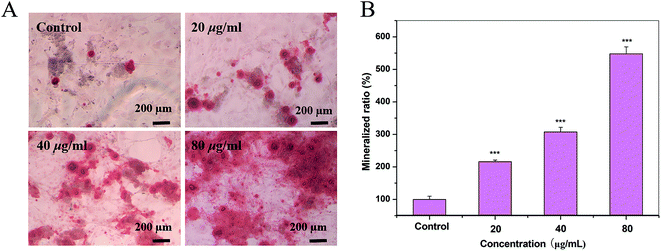 |
| | Fig. 6 Quantification of the mineralization in OBs cultured with S2. (A) Cells incubated with S2 nanoparticles were stained with alizarin red (ARS) after 17 days of culture. (B) The quantification (OD) of the destained ARS. Scale bar is 200 μm. | |
Effect of S2 on the synthesis of collagen of OBs
Collagen I is widely used to evaluate osteogenic differentiation.26 As shown in Fig. 7, after incubating for 14 d, S2 promotes the synthesis of collagen of OBs at the concentration of 80 μg mL−1. The morphologic observation was in accordance with the results.
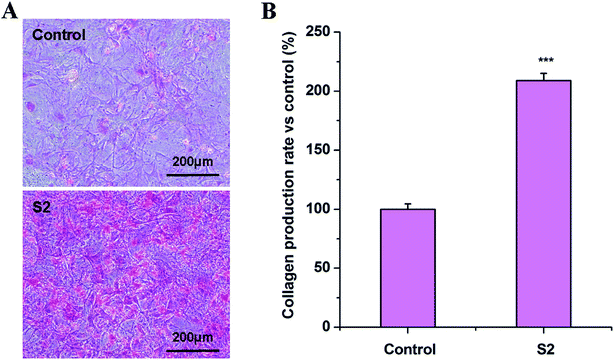 |
| | Fig. 7 Effect of S2 on the synthesis of collagen of OBs. OBs were treated with S2 (80 μg mL−1) for 14 d. The collagen was dyed by a Sirius red solution and quantified by a microplate spectrophotometer at a wavelength of 550 nm. All results were normalized by the protein content. Data are presented as mean ± SD from a representative of three separate experiments performed in sextuplicate. ***P < 0.001 compared with the control; the scale bar is 200 μm. | |
Quantitative osteocalcin gene expression (RT-PCR analysis)
The results for the OCN and RUNX2 gene expression over time are presented in Fig. 8. After 14 days of culture, the mRNA levels of RUNX2 and OCN were about 1.05- and 1.15-fold higher than that of control group, respectively. These results indicated that S2 up-regulated the expression of OCN, while it could not change the expression of RUNX2 under standard culture conditions. OCN is believed to be crucial in regulating osteoblast activity and binding of HAP.27 Combined with our Alizarin staining and Sirius red results, these results suggest that the higher expression of OCN in the HAP nanoparticle-treated osteoblast cells stimulates more mineralization in the culture.28
 |
| | Fig. 8 Quantitative OCN and RUNX2 gene expressions after being cultured with S2. Data are presented as a fold ratio after being normalized to GAPDH values. *P < 0.05, **P < 0.01, ***P < 0.001 vs. control, n = 3. | |
Nucleation role of S2 in the formation of the mineralized matrix
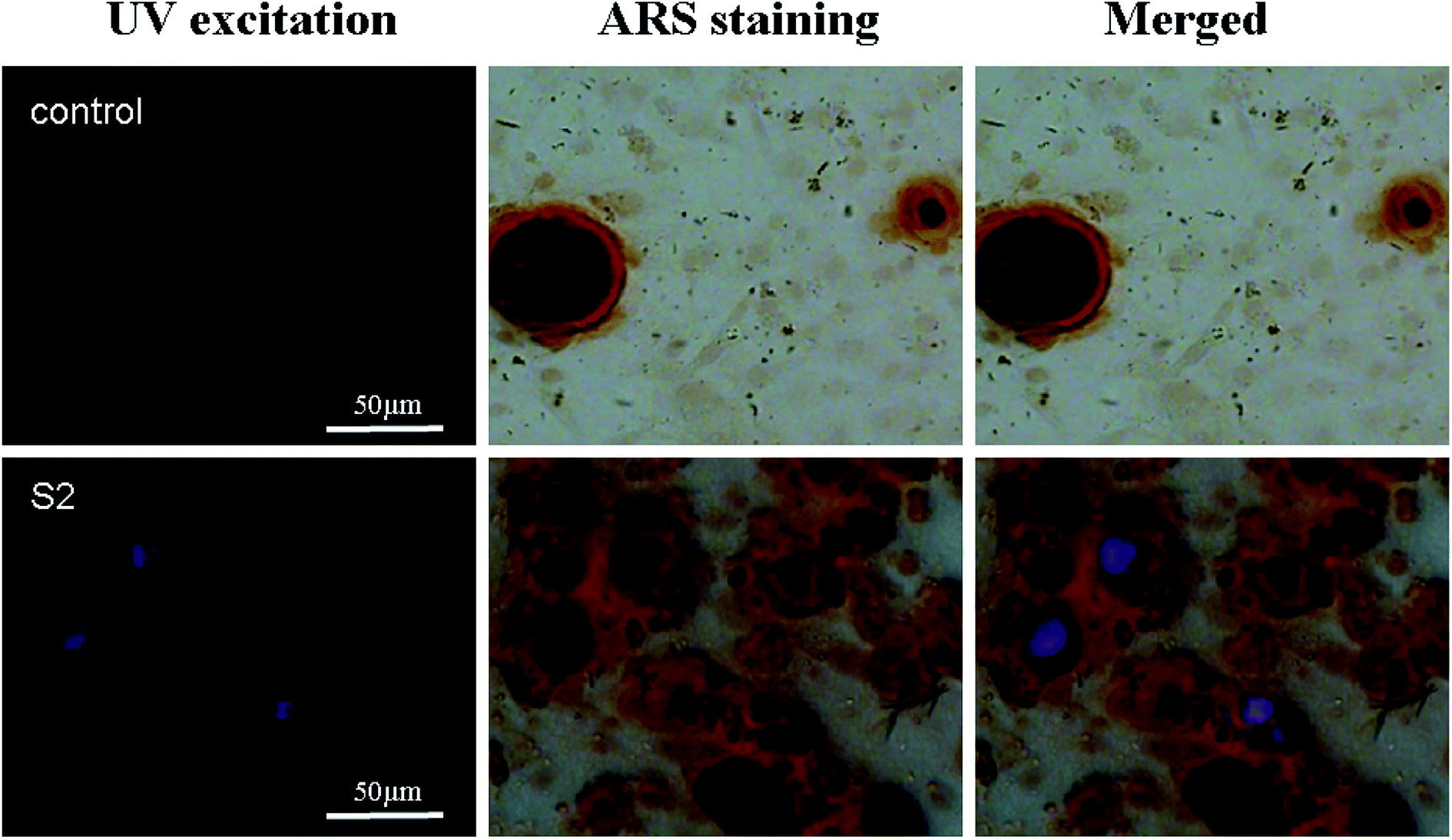
Previous reports have demonstrated that a newly formed bone can be correlated with the presence of HAP nanoparticles, which can be associated with nucleating events.29 To date, there are no reports on whether the HAP particles promote bone regeneration intracellularly or extracellularly and whether they act as a mineral source or a nucleation site for mineral growth.20 The distribution of S2 in the mineralized nodule is shown in Fig. 9. The mineralized nodules produced by OBs have no luminescence, while S2 has defect-related luminescence. By taking advantage of the fluorescence of S2, we found that extracellular S2 is located at the center of the mineralized nodules (Fig. 9). This result was consistent with previous results: the presence of HAP promoted the formation of mineralized nodules. The HAP particles can also promote mineralization through an extracellular pathway.
 |
| | Fig. 9 Fluorescence imaging of mineralized nodules stained with alizarin red (ARS) after 17 days of culture. Blue, the blue light from S2 excited by UV; red, the bright image of the mineralized nodule; merged, the distribution of S2 in the mineralized nodule, the scale bar is 50 μm. | |
In summary, the S2 nanorods were uptaken by OBs through caveolae-mediated endocytosis and macropinocytosis and showed a positive effect in regulating the expression of OCN, the synthesis of collagen and the formation of mineralized nodules. Intracellular S2 could be distributed in lysosomes and dissolved and released calcium ions for the formation of mineralized nodules. The extracellular S2 as a nucleation site promoted the formation of mineralized nodules. The S2 nanorods promoted bone regeneration through both intracellular and extracellular pathways (Fig. 10).
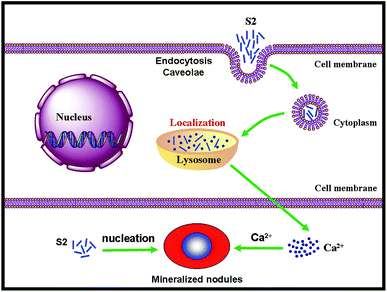 |
| | Fig. 10 The mechanism of S2 nanorods promoting OB mineralization through both intracellular and extracellular pathways. | |
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
This research was supported by the Key Research and Development Plan of Hainan Province (Grant No. ZDYF2018160), National Natural Science Foundation of China (Grant No. 21766007), the Program for Innovative Research Team in University (Grant No. IRT-16R19), Hainan Natural Science Foundation of China (Grant No. 217114), and the financial support from China Scholarship Council (CSC, Grant No. 201808460066, and 201808460067).
References
- H.-W. Kim, J. C. Knowles, V. Salih and H.-E. Kim, J. Biomed. Mater. Res., Part B, 2004, 71, 66–76 CrossRef.
- R. Pallela, J. Venkatesan, V. R. Janapala and S.-K. Kim, J. Biomed. Mater. Res., Part A, 2012, 100, 486–495 CrossRef.
- S. Mirza, I. Zia, R. Jolly, S. Kazmi, M. Owais and M. Shakir, Int. J. Biol. Macromol., 2018, 119, 215–224 CrossRef CAS.
- A. Ashokan, D. Menon, S. Nair and M. Koyakutty, Biomaterials, 2010, 31, 2606–2616 CrossRef CAS.
- D. Li, Z. Liang, J. Chen, J. Yu and R. Xu, Dalton Trans., 2013, 42, 9877–9883 RSC.
- Y. Li, X. Li, D. Wang, C. Shen and M. Yang, Microchim. Acta, 2018, 185, 435 CrossRef PubMed.
- C. Zhang and J. Lin, Chem. Soc. Rev., 2012, 7938–7961 RSC.
- S. Angelov, R. Stoyanova, R. Dafinova and K. Kabasanov, J. Phys. Chem. Solids, 1986, 47, 409–412 CrossRef CAS.
- W. H. Green, Science, 1997, 276, 1826–1828 CrossRef CAS.
- T. Brankova, V. Bekiari and P. Lianos, Chem. Mater., 2003, 15, 1855–1859 CrossRef CAS.
- W.-N. Wang, T. Ogi, Y. Kaihatsu, F. Iskandar and K. Okuyama, J. Mater. Chem., 2011, 21, 5183–5189 RSC.
- C. Dai, J. Duan, L. Zhang, G. Jia, C. Zhang and J. Zhang, Biol. Trace Elem. Res., 2014, 162, 158–167 CrossRef CAS.
- H.-S. Yu, J.-H. Jang, T.-I. Kim, H.-H. Lee and H.-W. Kim, J. Biomed. Mater. Res., Part A, 2009, 88, 747–754 CrossRef.
- K. Amornwachirabodee, K. Chiablaem, S. Wacharasindhu, K. Lirdprapamongkol, J. Svasti, V. Vchirawongkwin and S. P. Wanichwecharungruang, J. Pharm. Sci., 2012, 101, 3779–3786 CrossRef CAS.
- C. Greulich, J. Diendorf, T. Simon, G. Eggeler, M. Epple and M. Köller, Acta Biomater., 2011, 7, 347–354 CrossRef CAS.
- M. N. M. Walter, A. Dehsorkhi, I. W. Hamley and C. J. Connon, Biomater. Sci., 2016, 4, 346–354 RSC.
- T. Kaur and A. Thirugnanam, J. Mater. Sci. Technol., 2017, 33, 734–743 CrossRef.
- I. W. Bauer, S.-P. Li, Y.-C. Han, L. Yuan and M.-Z. Yin, J. Mater. Sci.: Mater. Med., 2008, 19, 1091–1095 CrossRef CAS.
- Y. Cai, Y. Liu, W. Yan, Q. Hu, J. Tao, M. Zhang, Z. Shi and R. Tang, J. Mater. Chem., 2007, 17, 3780–3787 RSC.
- S. M. Schmidt, K. A. Moran, A. M. T. Kent, J. L. Slosar, M. J. Webber, M. J. McCready, C. Deering, J. M. Veranth and A. Ostafin, J. Biomed. Mater. Res., Part A, 2008, 87, 418–428 CrossRef.
- R. D. Bloebaum, G. A. Lundeen, K. N. Bachus, I. Ison and A. A. Hofmann, J. Biomed. Mater. Res., 1998, 40, 104–114 CrossRef CAS.
- A. Bernhardt, A. Lode, C. Mietrach, U. Hempel, T. Hanke and M. Gelinsky, J. Biomed. Mater. Res., Part A, 2009, 90, 852–862 CrossRef CAS.
- S. Maeno, Y. Niki, H. Matsumoto, H. Morioka, T. Yatabe, A. Funayama, Y. Toyama, T. Taguchi and J. Tanaka, Biomaterials, 2005, 26, 4847–4855 CrossRef CAS.
- M. N. Lee, H. S. Hwang, S. H. Oh, A. Roshanzadeh, J. W. Kim, J. H. Song, E. S. Kim and J. T. Koh, Exp. Mol. Med., 2018, 50, 142 CrossRef.
- S. Itoh, S. Nakamura, M. Nakamura, K. Shinomiya and K. Yamashita, Artif. Organs, 2006, 30, 863–869 CrossRef.
- I. Bilic-Curcic, M. Kronenberg, X. Jiang, J. Bellizzi, M. Mina, I. Marijanovic, E. M. Gardiner and D. W. Rowe, Genesis, 2005, 43, 87–98 CrossRef CAS.
- S. Rammelt, M. Neumann, U. Hanisch, A. Reinstorf, W. Pompe, H. Zwipp and A. Biewener, J. Biomed. Mater. Res., Part A, 2005, 73, 284–294 CrossRef.
- D. K. Khajuria, V. B. Kumar, D. Gigi, A. Gedanken and D. Karasik, ACS Appl. Mater. Interfaces, 2018, 10, 19373–19385 CrossRef CAS.
- A. L. Rossi, I. C. Barreto, W. Q. Maciel, F. P. Rosa, M. H. Rocha-Leão, J. Werckmann, A. M. Rossi, R. Borojevic and M. Farina, Bone, 2012, 50, 301–310 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Guangying Chen*a and
David M. Haddleton
*ab,
Guangying Chen*a and
David M. Haddleton