DOI:
10.1039/C9RA06621G
(Paper)
RSC Adv., 2019,
9, 29044-29050
Enzymatic synthesis of biobased aliphatic–aromatic oligoesters using 5,5′-bis(hydroxymethyl)furoin as a building block†
Received
22nd August 2019
, Accepted 6th September 2019
First published on 16th September 2019
Abstract
5,5′-Dihydroxymethyl furoin (DHMF) is a novel biobased difuranic polyol scaffold, achievable from the benzoin condensation of 5-hydroxymethylfurfural (HMF), which has recently been employed as a monomer for the preparation of cross-linked polyesters and polyurethane. Its upgrading by means of enzymatic reactions has not yet been reported. Here we demonstrated that Candida antarctica lipase B (CALB) is a suitable biocatalyst for the selective esterification of the primary hydroxyl groups of DHMF. Exploiting this enzymatic activity, DHMF has been reacted with the diethyl esters of succinic and sebacic acids obtaining fully biobased linear oligoesters with number-average molecular weight around 1000 g mol−1 and free hydroxyl groups on the polymer backbone. The structures of the DHMF-diacid ethyl ester dimers and of the oligomers were elucidated by NMR and MS analyses.
Introduction
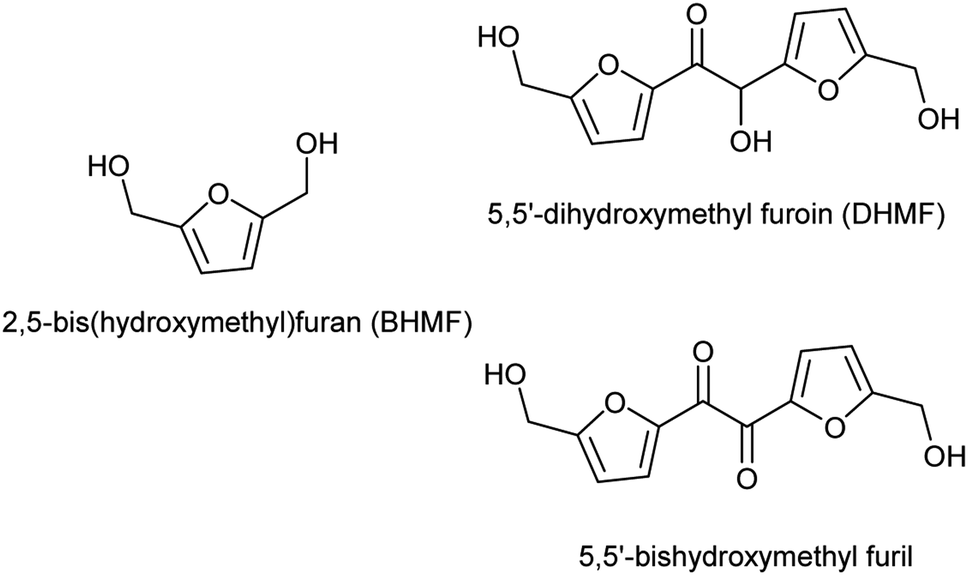
The gradual replacement of fossil resources with renewable biomass-derived feedstocks is imperative for the development of a more sustainable chemical industry. From this perspective, plant oils and sugars (mono- and polysaccharides) are the main carbon sources for the production of primary building blocks, either through chemical or biotechnological processes. In 2004, the US Department of Energy listed the top value-added building blocks achievable starting from sugars.1 In this list, as in its revision published several years later,2 5-hydroxymethylfurfural (HMF), a furan-based building block achievable by acid-catalyzed dehydration of C6 sugars, appeared as one of the most promising platform-chemicals. Indeed, in the last decade, this molecule has received an increasing attention by either academic and industrial researchers.3 A multitude of catalytic approaches has been implemented to produce HMF derivatives with potential uses as fuel additives, solvents or monomers.4–6 Within this last category, 2,5-furandicarboxylic acid (FDCA) and its diester derivatives, have been successfully employed for the production of furanic–aliphatic polyester, which are promising biobased analogues of polyethylene terephthalate7–12 with an increased biodegradability.13,14
Again in the field of monomers for polyester synthesis, HMF is also a valuable precursor for polyols like, 2,5-bis(hydroxymethyl)furan, 2,5-bis(hydroxymethyl)tetrahydrofuran, 1,2,6-hexanetriol, 1,6-hexanediol, achievable through selective catalytic hydrogenation.15–17 Among them, 2,5-bis(hydroxymethyl)furan (BHMF, Fig. 1), is particularly attractive as biobased diol monomer because of the interesting physical and mechanical properties that its rigid heteroaromatic structure could confers to the derived polymers. In spite of this, few studies have been reported on its polycondensation with diacid or diesters17–20 and only one group used an enzyme as catalyst.12,21 Another furan-based polyol scaffold, the 5,5′-dihydroxymethyl furoin (DHMF, Fig. 1), has been recently produced through benzoin-type condensation of HMF promoted by either N-heterocycle carbene (NHC) catalysts22–25 or by the thiamine diphosphate dependent-enzyme BAL.26 This novel HMF derivative has been in principle proposed as a precursor of oxygenated diesel fuels or C12 linear alkane22 which can be obtained through its catalytic hydrogenation or hydrodeoxygenation, respectively. Lately, however, by virtue of the polyhydroxylated rigid aromatic structure, DHMF has attracted the attention of polymer chemists who adopted it for the synthesis of bio-based polyesters27 and polyurethane.28 In particular, Chen and coworkers obtained cross-linked polyesters by reacting DHMF with various linear diacylchlorides while, in order to synthetize linear polyesters, they employed the diol 5,5′-bishydroxymethyl furil (Fig. 1) obtained by selective oxidation of DHMF.27 Until today, a polycondensation procedure selective for the primary alcoholic groups of the triol DHMF has not yet been reported. Such a strategy would allow to prepare linear polyesters with inner secondary hydroxyl groups available for further derivatization. We hypothesized that, by analogy with the above mentioned enzymatic synthesis of BHMF-based polyesters,21 the desired DHMF-based linear polyesters could be obtained by exploiting lipases selectivity although, until now, the enzymatic esterification of DHMF has never yet described.29 Herein we reported the unprecedented enzymatic esterification of DHMF. Furthermore, thanks to the selectivity shown by Candida anctartica lipase B (CALB) for the primary alcoholic groups of DHMF, the reaction conducted under appropriate conditions, has been exploited to synthetize fully bio-based aromatic–aliphatic linear oligoesters, bearing free secondary hydroxyl groups on the polymer backbone.
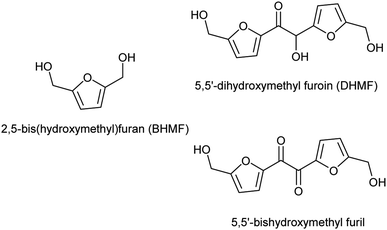 |
| | Fig. 1 Aromatic polyol monomers achievable from HMF. | |
Results and discussion
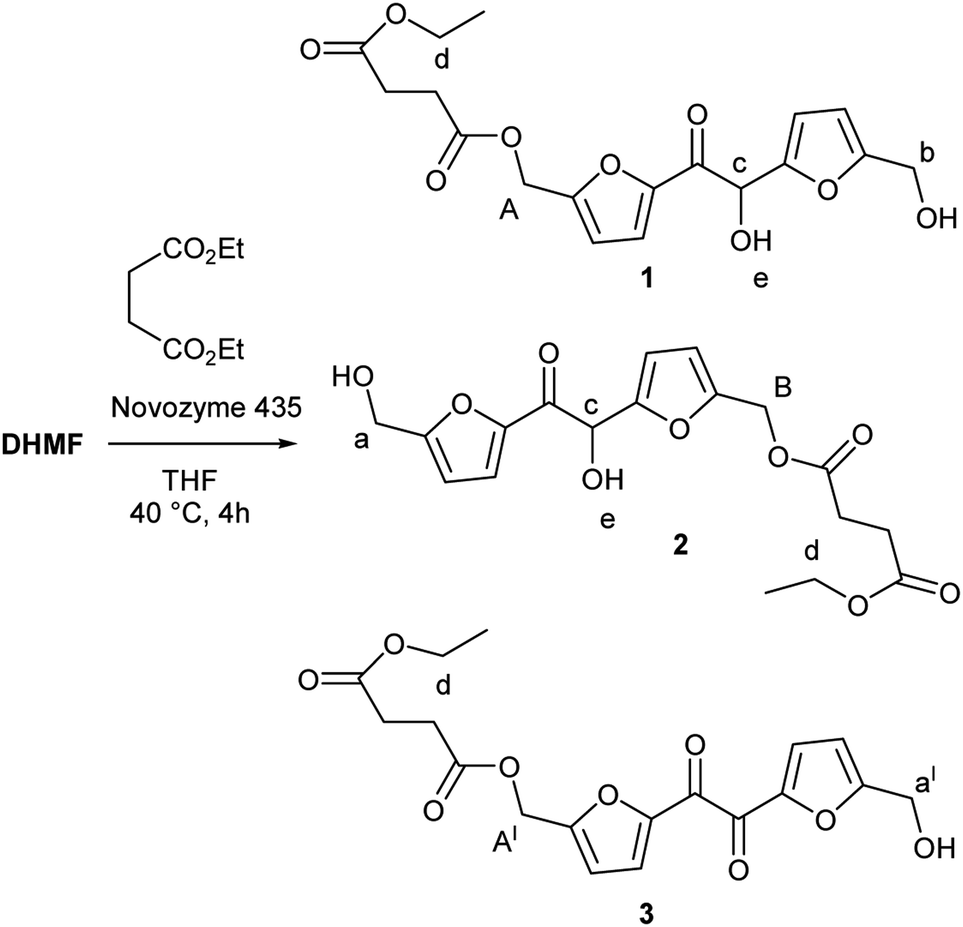
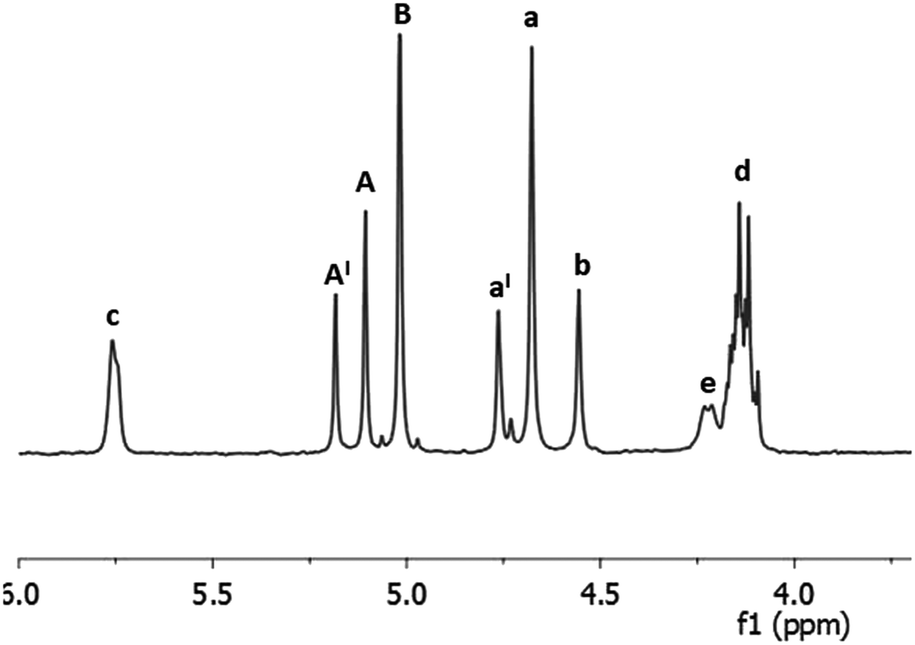
The first issue we addressed in order to evaluate the exploitability of DHMF as polyol monomer for enzymatic polymerization, has been to exploring the affinity of CALB for DHMF. We were encouraged in this attempt by a recent study which reported high yield in the CALB catalyzed esterification of HMF with various acyl donors.30 Since the potential substrate DHMF is not commercially available, we needed to synthetize it. Apart from the enzymatic route recently reported by Chuck and Domínguez de María,26 all the others syntheses of DHMF reported in literature, exploited the umpolung benzoin condensation of HMF catalyzed by NHC catalysts generated from imidazolium,24 benzimidazolium23 or thiazolium25 salts in the presence of a suited base or, in the only case of 1,3,4-triphenyl-4,5-dihydro-lH-1,2,4-triazol-5-ylidene,31 by the thermal decomposition of the methoxylated precursor.22,28 Following this last methodology, we obtained the pure DHMF in 95% yield after simple filtration and washing of the solid product. With the difuran triol in hand, we moved to test the activity of the commercial immobilized CALB Novozym 435, choosing diethyl succinate as the acylating agent. The reaction was performed in dry THF, at 40 °C, with equimolar amounts of the reactants (80 mM) and 50% of enzyme (w/w with respect to DHMF). After four hours, the 1H NMR analysis of the reaction mixture showed the presence of a set of signals attributable to the dimeric products 1 and 2 depicted in Scheme 1. In particular, the two singlets at 5.10 and 5.00 ppm where assigned to the protons of the two kinds of esterified hydroxymethylene groups (Scheme 1, group A of product 1 and group B of product 2, respectively) while, the others two singlets at 4.55 and 4.68 ppm were due to the protons of the corresponding unesterfied hydroxymethylene groups (Scheme 1, group b of product 1 and group a of product 2, respectively). The enzyme was removed from the reaction mixture and after evaporation of the solvent, the residue was chromatographed on silica gel, giving a mixture of products (80% yield) which surprisingly contained, together with 1 and 2, a new product that, on the basis of the additional set of signal appeared in the 1H NMR spectrum (Fig. 2), we identified as the monoesterified diketone 3 (Scheme 1). The results of this preliminary study demonstrated that CALB catalyzes the selective esterification of both the primary hydroxyl groups of DHMF, leaving the secondary one free. Worthy to note is the fact that, in spite of the chiral nature of DHMF, both the enantiomers of the racemic mixture used for the transesterification reactions were accepted as substrate from CALB, as demonstrated by the almost complete conversion of the reactant. In light of this, we moved to investigate the possibility to synthetize linear polyesters using DHMF and diethyl succinate as comonomers by exploiting the catalytic activity of CALB. In doing that, we followed the approach recently reported by Loos and coworkers for the enzymatic synthesis of polyesters with BHMF (Fig. 1) as the diol monomer.21 They described a three-stage method, where CALB catalyzed the formation of oligomers in the first two stages conducted at atmospheric pressure and under a weak vacuum (350 mmHg), respectively. In the third stage, performed under high vacuum (3 mmHg) the above prepolymers undergoes to polycondensation, giving oligomers (MW ranging from 1.300 to 1.500, depending on the number of carbon atoms of the diacid monomer). Using the same conditions, namely DHMF to diethyl succinate molar ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, CALB 20% (w/w, with respect to DHMF) and diphenyl ether as the solvent, after 1.5 hours at atmospheric pressure and 80 °C, the 1H NMR spectrum of the reaction mixture (spectrum a, Fig. 3), showed the presence of the two singlets at 5.10 and 5.00 ppm (A and B) typical of DHMF esters while the signals of the free hydroxymethylene groups (a and b) were negligible. This indicated that the main product was the trimer 4 (Fig. 3) derived from the esterification of both the primary alcoholic groups of DHMF. Furthermore, the ratio between the sum of the integrals A and B and that of the multiplet at 4.15 ppm (d in Fig. 3) which is due to the resonance of the ethyl groups of 4 and of the unreacted diethyl succinate, suggested a low conversion. The reaction was then left at the same temperature and after 3 hours the pressure was reduced to 300 mmHg.
:1, CALB 20% (w/w, with respect to DHMF) and diphenyl ether as the solvent, after 1.5 hours at atmospheric pressure and 80 °C, the 1H NMR spectrum of the reaction mixture (spectrum a, Fig. 3), showed the presence of the two singlets at 5.10 and 5.00 ppm (A and B) typical of DHMF esters while the signals of the free hydroxymethylene groups (a and b) were negligible. This indicated that the main product was the trimer 4 (Fig. 3) derived from the esterification of both the primary alcoholic groups of DHMF. Furthermore, the ratio between the sum of the integrals A and B and that of the multiplet at 4.15 ppm (d in Fig. 3) which is due to the resonance of the ethyl groups of 4 and of the unreacted diethyl succinate, suggested a low conversion. The reaction was then left at the same temperature and after 3 hours the pressure was reduced to 300 mmHg.
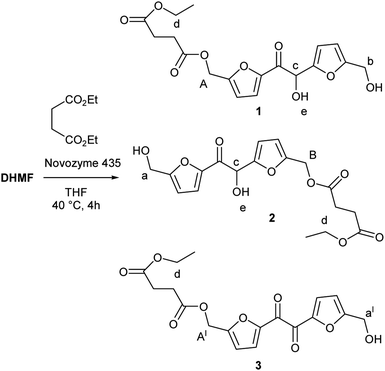 |
| | Scheme 1 CALB catalyzed transesterification of diethyl succinate with DHMF; reaction conditions: anhydrous THF 1.0 mL, DHMF 0.08 M, diethyl succinate 0.08 M, CALB 50% (w/w with respect to DHMF), 4 h, 40 °C. | |
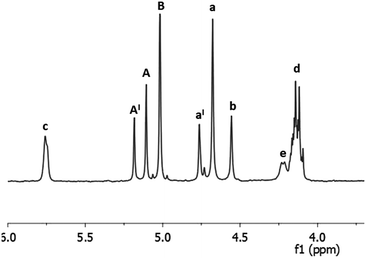 |
| | Fig. 2 1H NMR spectrum (in CDCl3) of the mixture of products 1, 2 and 3 (Scheme 1) obtained from the CALB catalyzed transesterification of diethyl succinate with DHMF after column chromatography. | |
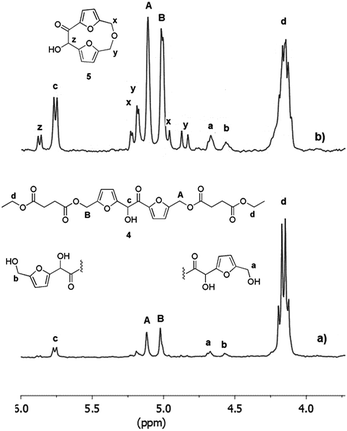 |
| | Fig. 3 1H NMR spectra of the reaction mixture (10 μL in 1 mL of CDCl3) acquired after 1.5 (spectrum a) and 6 hours (spectrum b) of the enzymatic polymerization of DHMF and diethyl succinate conducted at 80 °C. Reaction conditions: diphenyl ether 0.18 mL, DHMF 0.7 M, diethyl succinate 0.7 M, CALB 20% (w/w with respect to DHMF), 80 °C, 3 h under atmospheric pressure followed by 3 h at 300 mmHg. | |
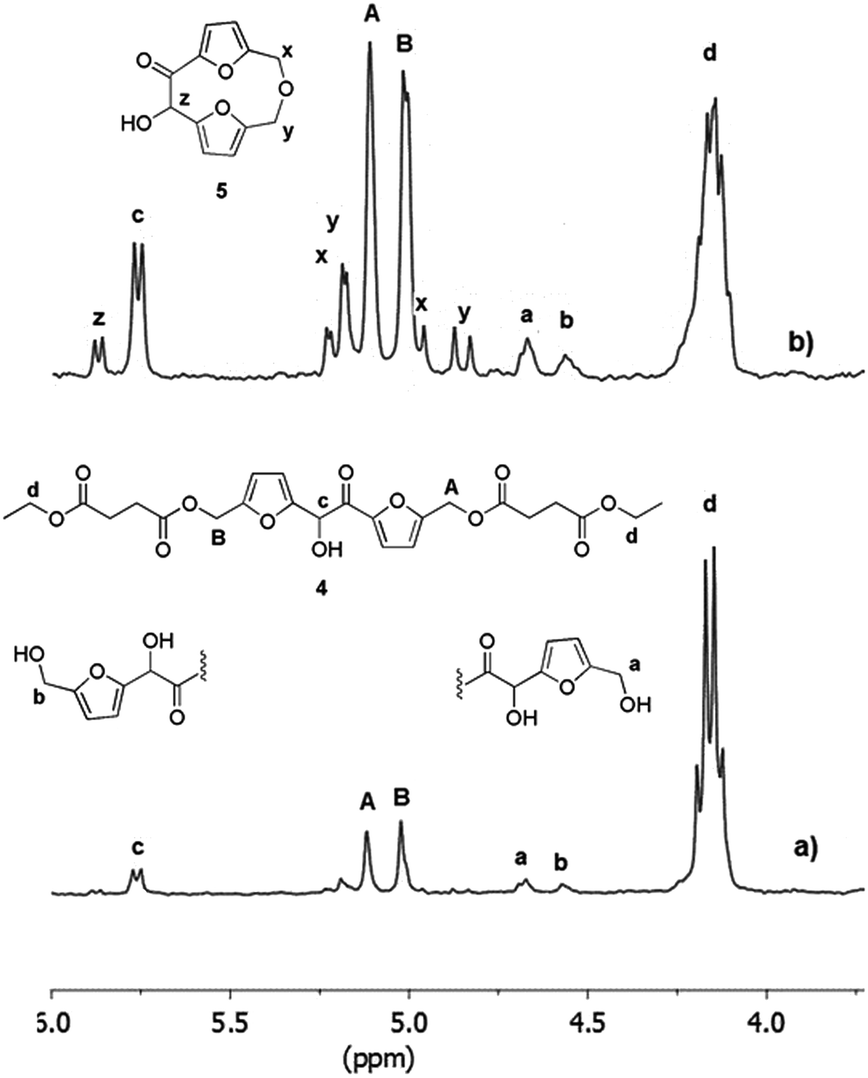
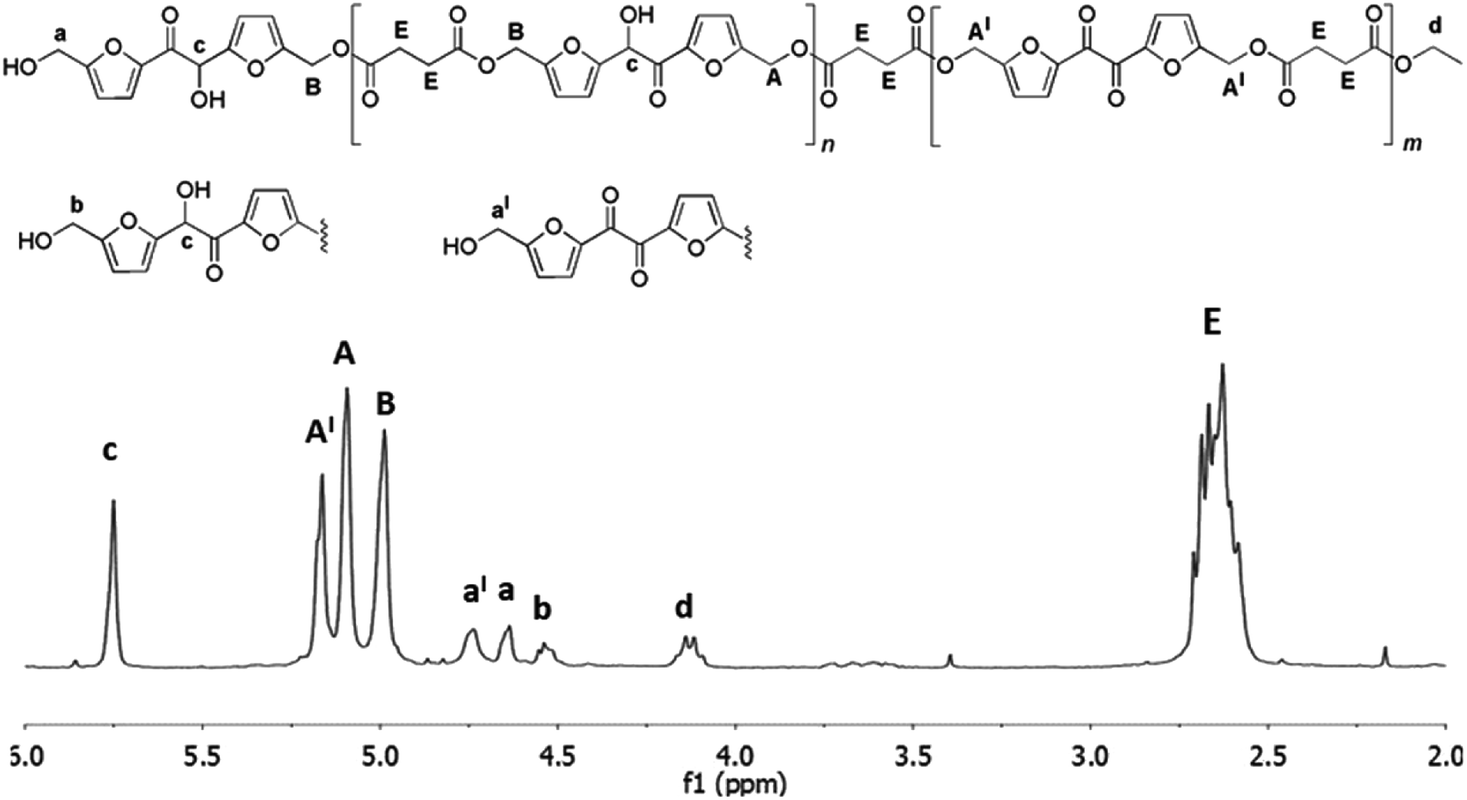
After overall 6 hours, the 1H NMR spectrum of the reaction mixture (spectrum b, Fig. 3) revealed a complete conversion of the diethylsuccinate. The main product of the mixture was the trimer 4 (Fig. 3) accompanied by lower amounts of the esters 1 and 2, as revealed by the presence of the small signals of the free hydroxymethylene groups (a and b). In addition, the spectrum showed the appearance of a byproduct (signals x, y and z, spectrum b, Fig. 3) which formation is probably due to the tendency of the hydroxymethylene groups of DHMF to dehydrate and react with alcoholic functions forming ether linkages. This behavior has been also documented for the BHMF which, under the same conditions, dehydrated and reacted with ethanol.21 On the basis of 2D 1H NMR COSY experiment (ESI S3†) and of HPLC-MS analyses (ESI S11†), we deduced that, in the case of DHMF, after dehydration of one of the hydroxymethylene groups, the cyclic ether 5 (Fig. 3) was formed through an intramolecular mechanism. In order to reduce the formation of such byproduct, we decided to repeat the reaction starting with the lower temperature and the lower pressure of 50 °C and 18 mmHg, respectively. After six hours under these conditions, the conversion determined by 1H NMR was about 28% and the signals of the byproduct 5 were negligible. At this point, the temperature was increased to 60 °C and the reaction was continued for additional four hours. After this period, about 80% of diethyl succinate was converted and the byproduct 5 was present in trace amount (ESI S4†). In order to favor the condensation of the oligomers between them and with the unreacted DHMF the pressure was reduced to 3 mmHg, in this following the procedure described by Loos and coworkers.21 The reaction was maintained at 60 °C under vacuum for additional ten hours and then diluted with chloroform in order to precipitate the residual DHMF. After filtration, the chloroform was evaporated from the organic solution and a yellow solid was precipitated from the diphenyl ether solution by adding dropwise cyclohexane. The solid was recovered by filtration, washed with cyclohexane and dried. The mass of the solid was about 70% of the starting mass of the reactants. The 1H NMR analysis of the solid showed, in addition to signals of the esterified hydroxymethylene groups of the DHMF units (A and B in Fig. 4), also the signal of the corresponding groups of the diketone units (A′ in Fig. 4). From the spectrum it can be deduced that about 30% of the DHMF units incorporated in the oligomers were oxidized to the corresponding diketone form [ratio of integrals A′/(A + B)]. The free hydroxymethylene groups of the DHMF and of the diketone terminal units (signals a, b and a′ in Fig. 4) together with the ethyl groups (signal d in Fig. 4), represented all the possible ending groups. Finally, the spectrum showed an intense multiplet centered at 2.60 ppm generated by the proton's resonance of the succinyl units (signal E in Fig. 4). The 13C NMR spectrum of the solid, allowed to identify all the types of carbon atom present in the oligomer structures (ESI S6†). The number-average molecular weight (Mn) of the oligomers calculated from the 1H NMR was about 1.150 (see Experimental section). Several of the oligomers were recognized also by HPLC-MS analyses (ESI S12†).
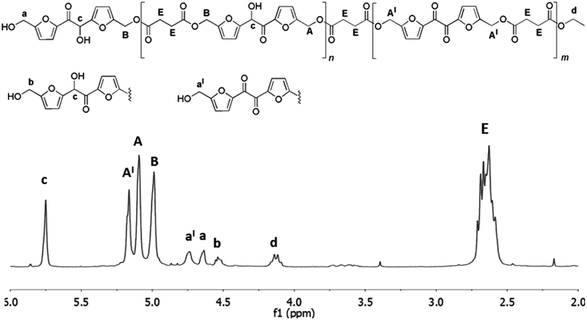 |
| | Fig. 4 1H NMR spectrum (in CDCl3) and products structures of the oligoesters mixture obtained after the three-step enzymatic polymerization of DHMF with diethyl succinate. Reaction conditions: diphenyl ether 0.18 mL, DHMF 0.7 M, diethyl succinate 0.7 M, CALB 20% (w/w with respect to DHMF), 50 °C, 18 mmHg for the first step of six hours, followed by a second step of four hours at 60 °C under the same pressure and finally by ten hours at 60 °C and 3 mmHg. | |
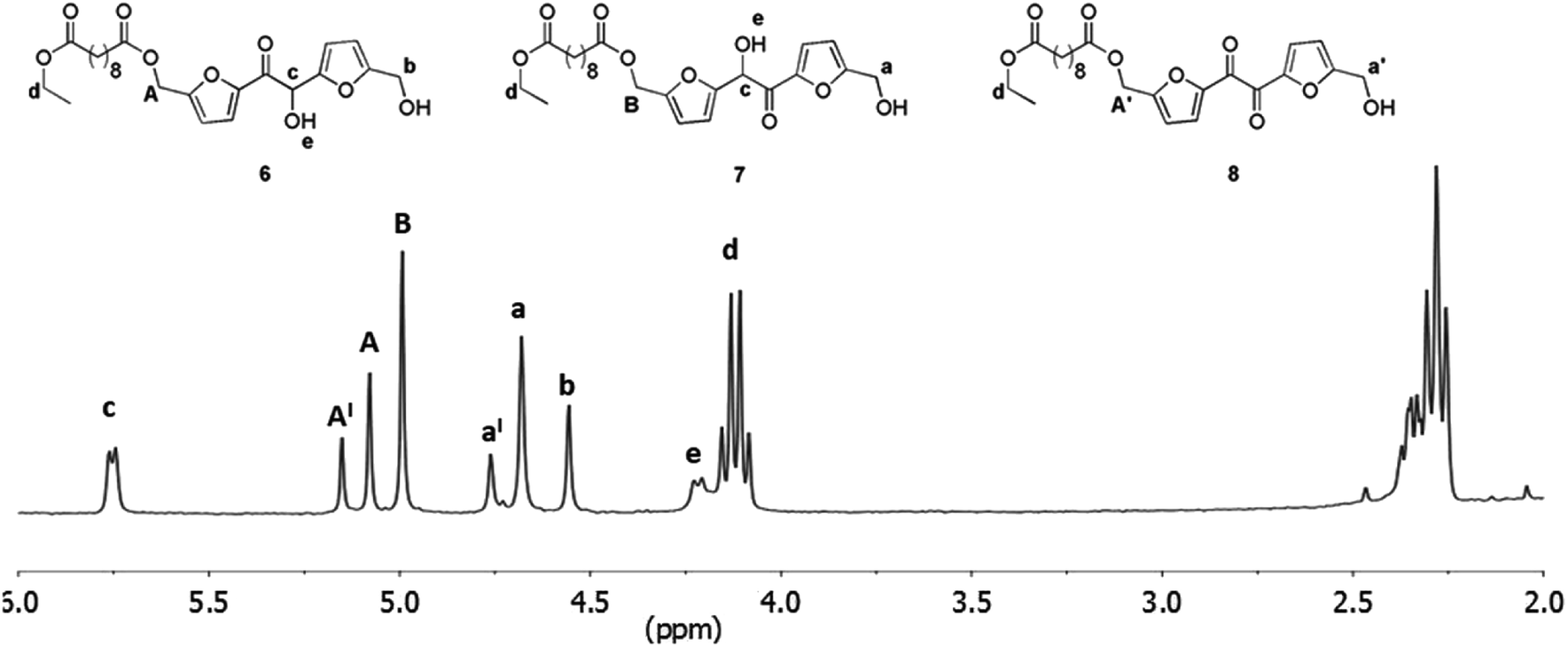
The CALB recovered after the polymerization was washed with THF and dried under vacuum at 40 °C. After this treatment the enzyme was used for further three reaction cycles without significant loss of activity. The number-average molecular weight of the oligomers obtained, is in the same range of that reported for the products obtained from the enzymatic polycondesation of diethyl succinate with BHMF.21 On the other hand, recent studies suggested a beneficial effect of an increase of the diester length on the results of enzymatic polycondensation.12,32,33 For these reasons we moved to verify the extendibility of the approach to longer diacid esters. The sebacic acid diethyl ester was chosen as the model substrate and its transesterification with DHMF was first proven at atmospheric pressure in dry THF, as described for diethyl succinate. Also in this case, after chromatography, we obtained a mixture of the three products 6, 7 and 8 (Fig. 5) analogues to the compounds 1, 2 and 3 produced with diethyl succinate. After that, we moved to study the enzymatic polymerization of DHMF with diethyl sebacate following the same three-step procedure adopted for the synthesis of the DHMF-succinate oligoesters (50 °C, 18 mmHg, 6 h; 60 °C, 18 mmHg, 4 h; 60 °C, 3 mmHg, 10 h).
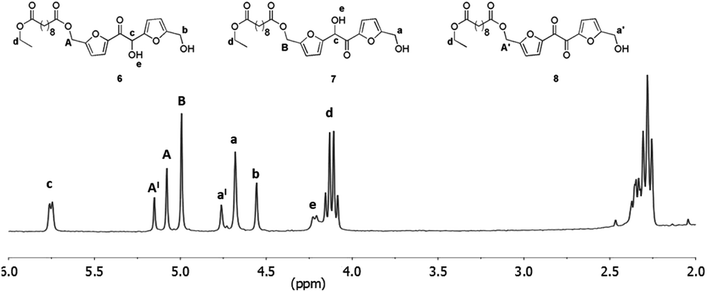 |
| | Fig. 5 1H NMR spectrum (in CDCl3) of the products mixture obtained from the CALB catalyzed transesterification of diethyl sebacate with DHMF after column chromatography; reaction conditions: anhydrous THF 1.0 mL, DHMF 0.08 M, diethyl sebacate 0.08 M, CALB 50% (w/w with respect to DHMF), 4 h, 40 °C. | |
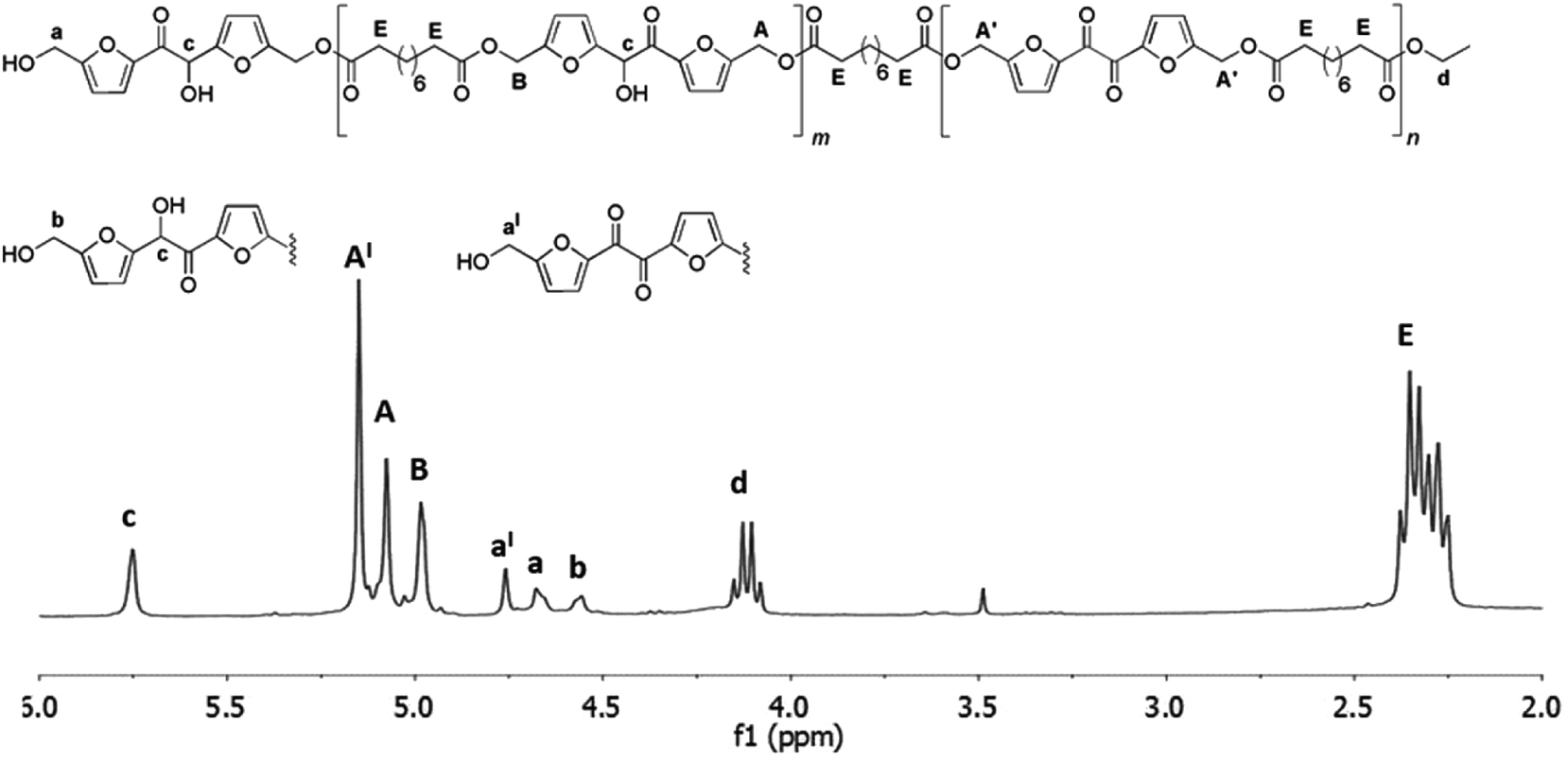
In this case, the 1H NMR analysis of the reaction mixture revealed the formation of oligomers including about 30% of diketone units already after the first stage which increase up to 40% at the end of the second stage. Being the aim of the work the synthesis of polyhydroxylated linear polyesters, we decided to stop the reaction at this stage. The attempt to accelerate the rate of the polymerization by increasing the temperature was not considered because it would favor the formation of the byproduct 5 as well. After the same workup described for the polymerization with diethyl succinate a yellow solid was recovered (about 75% of the initial mass of reactants). This products mixture was analyzed by 1H NMR (Fig. 6) and HPLC-MS analyses (ESI S14†) and a Mn of about 1.000 was calculated from the 1H NMR. The lacking increase of molecular weight by switching from the diethyl succinate to diethyl sebacate is in agree with the results of a precedent study where the enzymatic polymerization of BHMF with both the two diethyl ester furnished oligomers of comparable Mn (about 1500 g mol−1).21
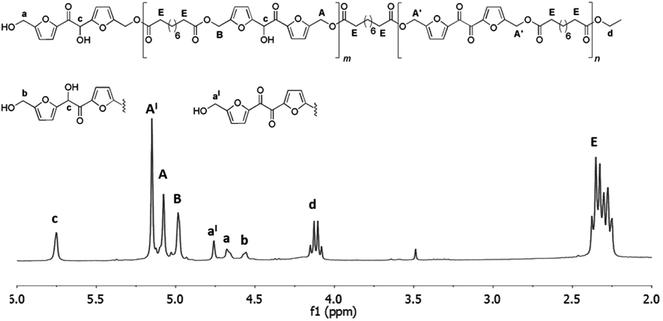 |
| | Fig. 6 1H NMR spectrum (in CDCl3) and structures of the oligoesters mixture obtained after the two-step enzymatic polymerization of DHMF with sebacic acid diethyl ester. Reaction conditions: diphenyl ether 0.18 mL, DHMF 0.7 M, diethyl succinate 0.7 M, CALB 20% (w/w with respect to DHMF), 50 °C, 18 mmHg for the first step of six hours, followed by a second step of four hours at 60 °C under the same pressure. | |
Conclusions
DHMF is an emerging HMF derivative whose synthetic upgrading is receiving increasing attention. In spite of the recently reported enzymatic synthesis, further biocatalyzed elaborations of its polyfunctional structure have not yet documented. In the present study we demonstrated that the primary hydroxyl groups of DHMF can be selectively esterified using commercial CALB as the catalyst. The enzymatic reaction with diethyl succinate or diethyl sebacate as acylating agents, performed under appropriate conditions, afforded oligoesters with number-average molecular weight of about 1000 Da. In view of the growing interest for green technologies devoted to the processing of biobased chemicals, we think that the present study could be an important starting point for further researches addressed to improve the polymerization procedure by exploring the activities of other lipases and/or modifying the reaction conditions. Furthermore, the herein reported results contribute to increase the synthetic tools available to enhance the value of DHMF as multifunctional synthetic scaffold for applications in life and material sciences.
Experimental section
Materials and methods
The lipase B from Candida antarctica (CALB) immobilized on acrylic resin, Novozym 435, was purchased from Novozyme. The enzyme was dried at 60 °C under vacuum (0.8 mmHg) for 8 hours and stored in a desiccator before use. Diphenyl ether (99%) from Acros Organics was distilled under vacuum and stored with activated 4 Å molecular sieves. Tetrahydrofuran (THF) was distilled under nitrogen atmosphere from Na/benzophenone immediately prior to use. Diethyl succinate (≥99%) and diethyl sebacate (98%) were Sigma Aldrich products. The pre-catalyst 5-methoxy-1,3,4-triphenyl-4,5-dihydro-1H-1,2,4-triazoline (TPT-OMe) was synthetized as described in ref. 27, while the dihydroxymethyl furoin (DHMF) was prepared as described in ref. 25. Flash column chromatography was performed on silica gel 60 (230–400 mesh). 1H and 13C NMR spectra were recorded on 300 and 400 MHz spectrometers at room temperature using CDCl3 or DMSO-d6 as solvent. Chemical shifts (δ) are reported in ppm relative to residual solvent signals. Liquid chromatographic separation was performed with a micro HPLC (model Surveyor Plus, Thermo Scientific, USA) equipped with solvent delivery system, column compartment and 4 channels pump. Mobile phases were water and acetonitrile with formic acid 0.1% (v/v). Experimental conditions were: gradient elution from 35% to 95% of acetonitrile in 20 min, then isocratic for 5 min; flow are 100 μL min−1; injection volume 2 μL; column Symmetry (Waters) C18 150 × 2.1 mm, 3.5 μm. HPLC was hyphenated to a LTQ XL mass spectrometer (Thermo Scientific, USA) with linear ion trap mass analyzer by ESI ionization interface. MS experimental conditions were: ESI positive mode; spray voltage 4.00 kV; capillary temperature 275 °C; capillary voltage 25 V; tube lens 95 V.
CALB catalyzed synthesis of ethyl succinate-DHMF monoesters 1, 2 and 3
A 1.5 mL flask was fed with DHMF (20 mg, 0.08 mmol), diethyl succinate (14 mg, 0.08 mmol), freshly distilled THF (1 mL) and CALB (10 mg). The flask was stoppered and gently shaken (100 rpm) at 40 °C for four hours. After that, CALB was filtered out and washed three times with THF (3 × 5 mL). The solutions were combined, concentrated under reduced pressure and the products were dried under vacuum (0.8 mmHg) at room temperature for two hours. The residue was chromatographed on a silica gel column with ethyl acetate/cyclohexane 5:1 as the eluent obtaining 24 mg (80% yield) of a mixture composed by the esters 1 (22%), 2 (44%) and 3 (14%): ester 1: 1H NMR, CDCl3, selected data δ 1.22 (t, 3H, CH3), 2.55–2.70 (m, 4H, C2H4), 4.08–4.18 (m, 2H, O–CH2CH3), 4.55 (s, 2H, CH2OH), 5.00 (s, 2H, CH2C(O)), 5.78 (s, 1H, CHOH). MS, m/z ESI = 381 Da [M + H]+; ester 2: 1H NMR, CDCl3, selected data δ 1.22 (t, 3H, CH3), 2.55–2.70 (m, 4H, C2H4), 4.08–4.18 (m, 2H, O–CH2CH3), 4.68 (s, 2H, CH2OH), 5.10 (s, 2H, CH2C(O)), 5.78 (s, 1H, CHOH). MS, m/z ESI = 381 Da [M + H]+; ester 3: 1H NMR, CDCl3, selected data δ 1.22 (t, 3H, CH3), 2.55–2.70 (m, 4H, C2H4), 4.08–4.18 (m, 2H, O–CH2CH3), 4.76 (s, 2H, CH2OH), 5.20 (s, 2H, CH2C(O)). MS, m/z ESI = 379 Da [M + H]+. Control reactions performed under the same conditions in the absence of the enzyme did not afford any reaction product.
CALB-catalyzed polycondesation of diethy succinate and DHMF
DHMF (30 mg, 0.12 mmol), diphenyl ether (180 μL), diethyl succinate (17 mg, 0.12 mmol) and CALB (6 mg) were fed into a 10 mL flask. The flask was connected to the vacuum pump and the pressure was reduced to 18 mmHg. The reaction mixture was gently shaken at 50 °C for six hours and then the temperature was increased to 60 °C for additional four hours. After this period, the pressure was lowered to 3 mmHg and the mixture was maintained at 60 °C for ten hours. The reaction was stopped by increasing pressure to 760 mmHg, adding chloroform (5 mL) and filtering out CALB. The enzyme was washed with the same solvent (5 mL) three times and the solutions were combined and concentrated under reduced pressure. Cyclohexane (3 mL) was added dropwise to precipitate the products from the diphenyl ether solution. The products were washed with cyclohexane (2 × 2 mL) and the residual solvent was evaporated under vacuum (0.8 mmHg) at room temperature for 3 hours obtaining 37 mg of dried oligomers mixture (90% yield): 1H NMR, CDCl3, selected data δ 1.22 (t, 3H, CH3 ethyl end groups), 2.55–2.70 (m, 4H, CH2–C(O)-succinyl units, signal E in Fig. 5), 4.08–4.18 (m, 2H, CH2 ethyl end groups, signal d in Fig. 5), 4.52 (m, 2H, CH2–OH end group, signal b in Fig. 5), 4.65 (m, 2H, CH2–OH end groups, signal a in Fig. 5), 4.75 (m, 2H, CH2–OH end groups, signal a′ in Fig. 5), 5.00 (m, 2H, CH2–O of DHMF units, signal B in Fig. 5), 5.10 (m, 2H, CH2–O of DHMF unit, signal A in Fig. 5), 5.18 (m, 2H, CH2–O of DHMF diketone units, signal A′ in Fig. 5), 5.75 (m, CH–OH, signal c in Fig. 5); 13C NMR, DMSO-d6, δ 185.12 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, ketones), 172.28, 172.07, 171.98, 171.90 (CO esters), 155.00, 153.41, 149.84, 149.72 (C furanic), 121.95, 113.86, 113.07, 112.34, 110.08, 109.79 (CH furanic), 69.94 (CHOH), 60.51 (CH2 ethyl end group), 58.17, 58.06, (furyl-
O, ketones), 172.28, 172.07, 171.98, 171.90 (CO esters), 155.00, 153.41, 149.84, 149.72 (C furanic), 121.95, 113.86, 113.07, 112.34, 110.08, 109.79 (CH furanic), 69.94 (CHOH), 60.51 (CH2 ethyl end group), 58.17, 58.06, (furyl-![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H2–O–C(O)), 56.29, 56.04 (CH2OH end groups), 28.95, 28.73 (C2H4), 14.46 (CH3 ethyl end group). Control reactions performed under the same conditions in the absence of the enzyme did not afford any reaction product.
H2–O–C(O)), 56.29, 56.04 (CH2OH end groups), 28.95, 28.73 (C2H4), 14.46 (CH3 ethyl end group). Control reactions performed under the same conditions in the absence of the enzyme did not afford any reaction product.
CALB catalyzed synthesis of ethyl sebacate-DHMF monoesters 6, 7 and 8
The enzymatic synthesis of the ethyl sebacate-DHMF monoesters was performed in anhydrous THF following the same procedure described for the esters 1, 2 and 3 in the Section 2.2, by using 21 mg (0.08 mmol) of diethyl sebacate in place of the diethyl succinate. After the same workup, the residue was chromatographed on a silica gel column with ethyl acetate/cyclohexane 2:1 as the eluent obtaining 28 mg (83% yield) of a mixture composed by the esters 6 (29%), 7 (52%) and 8 (19%): ester 6: 1H NMR, CDCl3, selected data δ 1.15–1.40 (m, 9H, CH2 and CH3), 1.40–1.70 (m, 6H, CH2), 2.20–2.40 (m, 4H, CH2–CO), 4.08–4.18 (m, 2H, O–CH2–CH3), 4.55 (s, 2H, CH2OH), 5.00 (s, 2H, CH2C(O)), 5.78 (s, 1H, CHOH). MS, m/z ESI = 465 Da [M + H]+; ester 7: 1H NMR, CDCl3, selected data δ 1.15–1.40 (m, 9H, CH2 and CH3), 1.40–1.70 (m, 6H, CH2), 2.20–2.40 (m, 4H, CH2–CO), 4.08–4.18 (m, 2H, O–CH2–CH3), 4.68 (s, 2H, CH2OH), 5.08 (s, 2H, CH2C(O)), 5.78 (s, 1H, CHOH). MS, m/z ESI = 465 Da [M + H]+; ester 8: 1H NMR, CDCl3, selected data δ 1.15–1.40 (m, 9H, CH2 and CH3), 1.40–1.70 (m, 6H, CH2), 2.20–2.40 (m, 4H, CH2–CO), 4.08–4.18 (m, 2H, O–CH2–CH3), 4.76 (s, 2H, CH2OH), 5.17 (s, 2H, CH2C(O)). MS, m/z ESI = 463 Da [M + H]+. Control reactions performed under the same conditions in the absence of the enzyme did not afford any reaction product.
CALB-catalyzed polycondesation of diethyl sebacate and DHMF
DHMF (30 mg, 0.12 mmol), diphenyl ether (180 μL), diethyl sebacate (31 mg, 0.12 mmol) and CALB (6 mg) were fed into a 10 mL flask. The flask was connected to the vacuum pump and the pressure was reduced to 18 mmHg. The reaction mixture was gently shaken at 50 °C for six hours and then was warmed up to 60 °C for additional four hours. After that the reaction was stopped and worked up as reported for the DHMF-succinate oligomers (Section 2.3), obtaining 45 mg of dried solid (89% yield): 1H NMR, CDCl3, selected data δ 2.20–2.40 (m, 4H, CH2–C(O) of sebacic units, signal E in Fig. 6), 4.08–4.18 (m, 2H, CH2 ethyl end groups, signal d in Fig. 6), 4.58 (m, 2H, CH2OH end groups, signal b in Fig. 6), 4.65 (m, 2H, CH2OH end groups, signal a in Fig. 6), 4.75 (m, 2H, CH2OH end groups, signal a′ in Fig. 6), 5.00 (m, 2H, CH2–O of DHMF units, signal B in Fig. 6), 5.10 (m, 2H, CH2–O of DHMF units, signal A in Fig. 6), 5.18 (m, 2H, CH2–O of DHMF diketone units, signal A′ in Fig. 6), 5.75 (m, CH–OH, signal c in Fig. 6). 13C NMR, DMSO-d6, δ 185.23 (CO, ketones), 177.82, 174.57, 173.00, 172.91 (CO esters), 156.47, 155.02, 153.81, 149.80, 148.51 (C furanic), 126.52, 122.22, 114.53, 113.15, 112.80, 110.11 (CH furanic), 69.95 (CHOH), 60.01 (CH2 ethyl end group), 57.77 (furyl-H2–O–C(O)), 56.55, 56.23 (CH2OH end groups), 34.21, 27.54, 25.03 (CH2), 14.82 (CH3 ethyl end group).
Calculation of the number-average molecular weight (Mn) of the oligomers
The number-average molecular weight (Mn) was calculated from 1H NMR spectrum using the following equation:
| Mn = [(ΣA′+A+B/4 × MDHMF) + (E/4 × MDA)]/[(Σa′+a+b/2 + d/2) × 0.5] + MF |
where ΣA′+A+B is the sum of the integrals of signals A′, A and B, MDHMF is the mass of the DHMF repeating unit (234), E is the integral of signal E, MDA is the molecular weight of the diacid repeating unit (100 for succinic acid and 184 for sebacic acid), Σa′+a+b is the sum of the integrals of signal a′, a and b, d is the integral of signal d and MF is the mass of the end group assuming that the all the oligoesters terminated with OH/ethyl group. The protons which resonance generates signals A′, A, B, a′, a, b, c, d and e are indicated in Fig. 2.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by University of Ferrara (grants FAR 2018 and FIR 2017). We gratefully thank Mr Paolo Formaglio for NMR experiments, and Dr Tatiana Bernardi for HPLC-MS analyses.
Notes and references
- T. Werpy and G. R. Petersen, Top value added chemicals from biomass Volume I - Results of screening for potential candidates from sugars and synthesis gas, US department of Energy (DOE), Oak Ridge, TN, USA, 2004 Search PubMed.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539 RSC.
- R. J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499 CrossRef CAS PubMed.
- H. Zang, K. Wang, M. Zhang, R. Xie, L. Wang and E. Y.-X. Chen, Catal. Sci. Technol., 2018, 8, 1777 RSC.
- N. K. Gupta, A. Fukuoka and K. Nakajima, ACS Sustainable Chem. Eng., 2018, 6, 3434 CrossRef CAS.
- A. Brandolese, D. Ragno, G. Di Carmine, T. Bernardi, O. Bortolini, P. P. Giovannini, O. G. Pandoli, A. Altomare and A. Massi, Org. Biomol. Chem., 2018, 16, 8955 RSC.
- C. Moreau, M. N. Belgacem and A. Gandini, Top. Catal., 2004, 27, 11 CrossRef CAS.
- Y. Jiang and K. Loos, Polymers, 2016, 8, 243 CrossRef PubMed.
- E. de Jong, M. A. Dam, L. Sipos and G. J. M. Gruter, ACS Symp. Ser., 2012, 1105, 1 CrossRef CAS.
- A. F. Sousa, C. Vilela, A. C. Fonseca, M. Matos, C. S. R. Freire, G. J. M. Gruter, J. F. J. Coelho and A. J. D. Silvestre, Polym. Chem., 2015, 6, 5961 RSC.
- Y. Jiang, A. J. J. Woortman, G. O. R. Alberda van Ekensteina and K. Loos, Polym. Chem., 2015, 6, 5198 RSC.
- D. Maniar, Y. Jiang, A. J. J. Woortman, J. van Dijken and K. Loos, ChemSusChem, 2019, 12, 990 CrossRef CAS PubMed.
- A. Pellis, K. Haernvall, C. M. Pichler, G. Ghazaryan, R. Breinbauer and G. M. Guebitz, J. Biotechnol., 2016, 235, 47 CrossRef CAS PubMed.
- S. Weinberger, K. Haernvall, D. Scaini, G. Ghazaryan, M. T. Zumstein, M. Sander, A. Pellis and G. M. Guebitz, Green Chem., 2017, 19, 5381 RSC.
- X. Tang, J. Wei, N. Ding, Y. Sun, X. Zeng, L. Hu, S. Liu, T. Lei and L. Lin, Renewable Sustainable Energy Rev., 2017, 77, 287 CrossRef CAS.
- A. Gandini, T. M. Lacerda, A. J. F. Carvalho and E. Trovatti, Chem. Rev., 2016, 116, 1637 CrossRef CAS PubMed.
- L. Hu, J. Xu, S. Zhou, A. He, X. Tang, L. Lin, J. Xu and Y. Zhao, ACS Catal., 2018, 8, 2959 CrossRef CAS.
- C. Zeng, H. Seino, J. Ren, K. Hatanaka and N. Yoshie, Macromolecules, 2013, 46, 1794 CrossRef CAS.
- T. Ikezaki, R. Matsuoka, K. Hatanaka and N. Yoshie, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 216 CrossRef CAS.
- P. P. Upare, Y. K. Hwang and D. W. Hwang, Green Chem., 2018, 20, 879 RSC.
- Y. Jiang, A. J. J. Woortman, G. O. R. Alberda van Ekenstein, D. M. Petrović and K. Loos, Biomacromolecules, 2014, 15, 2482 CrossRef CAS PubMed.
- D. Liu and E. Y.-X. Chen, ChemSusChem, 2013, 6, 2236 CrossRef CAS PubMed.
- L. Wang and Y. Y.-X. Chen, ACS Catal., 2015, 5, 6907 CrossRef CAS.
- D. Liu, Y. Zhang and E. Y.-X. Chen, Green Chem., 2012, 14, 2738 RSC.
- H. Zang and E. Y.-X. Chen, Int. J. Mol. Sci., 2015, 16, 7143 CrossRef CAS PubMed.
- J. Donnelly, C. R. Müller, L. Wiermans, C. J. Chuck and P. Domínguez de María, Green Chem., 2015, 17, 2714 RSC.
- Z. Mou and E. Y.-X. Chen, ACS Sustainable Chem. Eng., 2016, 4, 7118 CrossRef CAS.
- Z. Mou, S. K. Feng and E. Y.-X. Chen, Polym. Chem., 2016, 7, 1593 RSC.
- L. Hu, A. He, X. Liu, J. Xia, J. Xu, S. Zhou and J. Xu, ACS Sustainable Chem. Eng., 2018, 6, 15915 CrossRef CAS.
- M. Krystof, M. Pérez-Sánchez and P. Domínguez de María, ChemSusChem, 2013, 6, 630 CrossRef CAS PubMed.
- D. Enders, K. Breuer, U. Kallfass and T. Balensiefer, Synthesis, 2003, 1292 CAS.
- D. Juais, A. F. Naves, C. Li, R. A. Gross and L. H. Catalani, Macromolecules, 2010, 43, 10315–10319 CrossRef CAS.
- M. Kanelli, A. Douka, S. Vouyiouka, C. D. Papaspyrides, E. Topakas, L.-M. Papaspyridi and P. Christakopoulos, J. Appl. Polym. Sci., 2014, 131, 40820 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra06621g |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Arianna Brandolese
,
Arianna Brandolese