
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnusual rearrangement of imidazo[1,5-a]imidazoles and imidazo[1,2-b]pyrazoles into imidazo[1,5-a]pyrimidines and pyrazolo[1,5-a]pyrimidines†
Khadija Gambouzabc,
Mohsine Driowya c,
Mohammed Loubidiac,
Zahira Tberac,
Hassan Allouchid,
Saïd El Kazzouli*b,
Mohamed Akssira*a and
Gérald Guillaumet*c
c,
Mohammed Loubidiac,
Zahira Tberac,
Hassan Allouchid,
Saïd El Kazzouli*b,
Mohamed Akssira*a and
Gérald Guillaumet*c
aLaboratoire de Chimie Physique et Chimie Bioorganique, URAC 22 FSTM, Université Hassan II – Casablanca, Morocco. E-mail: mohamed.akssira@fstm.ac.ma
bEuromed Research Center, Euromed Faculty of Enginnering, Euromed University of Fes (UEMF), Route de Meknès, 30000 Fès, Morocco. E-mail: s.elkazzouli@ueuromed.org
cInstitut de Chimie Organique et Analytique, Université d’Orléans, UMR CNRS 7311, 45067 Orléans Cedex 2, BP 6759, France. E-mail: gerald.guillaumet@univ-orleans.fr
dLaboratoire de Chimie Physique, équipe EA 7502 SIMBA, Faculté de Pharmacie, Université de Tours, 31, avenue Monge, 37200 Tours, France
First published on 16th September 2019
Abstract
A multicomponent reaction giving easy and cheap access to a variety of bicyclic 5,5-fused hetero-rings has been developed. Then, an usual rearrangement of imidazo[1,5-a]imidazoles or imidazo[1,2-b]pyrazoles leading to bi-heterocyclic imidazo- and pyrazolo[1,5-a]pyrimidines in the presence of a specific amount of I2 in THF at room temperature has been achieved. This new method enables the hitherto unreported synthesis of functionalized imidazo- and pyrazolo[1,5-a]pyrimidines.
Introduction
Polynitrogenated heterocycles are specific candidates for the design and discovery of new compounds with a large spectrum of biological activities.1–4 Widely known examples are those molecules containing imidazole or pyrazole as frameworks of high interest in medicinal chemistry, such as 5-(2-(3-((2,6-dichlorophenyl)amino)imidazo[1,2-a]pyrimidin-2-yl)-3,5-dimethoxyphenyl)-N-methyl-1,3,4-oxadiazol-2-amine (A) which is used as an SGLT2 inhibitor for type 2 diabetes,5 R′-8-carbamoyl-4-(3-R-phenyl)-2-methyl-1,4-dihydroimidazo[1,5-a]pyrimidine-3-carboxylate (B) as a calcium antagonist,6 2,7-di(1H-indol-3-yl)pyrazolo[1,5-a]pyrimidine-6-carbonitrile (C) which has anti-oxidant activity7 and 3-(((3-bromo-5-(2-fluorophenyl) pyrazolo[1,5-a]pyrimidin-7-yl)amino)methyl)pyridine 1-oxide (D) which exhibited inhibition of CDK2 (ref. 8) (Fig. 1).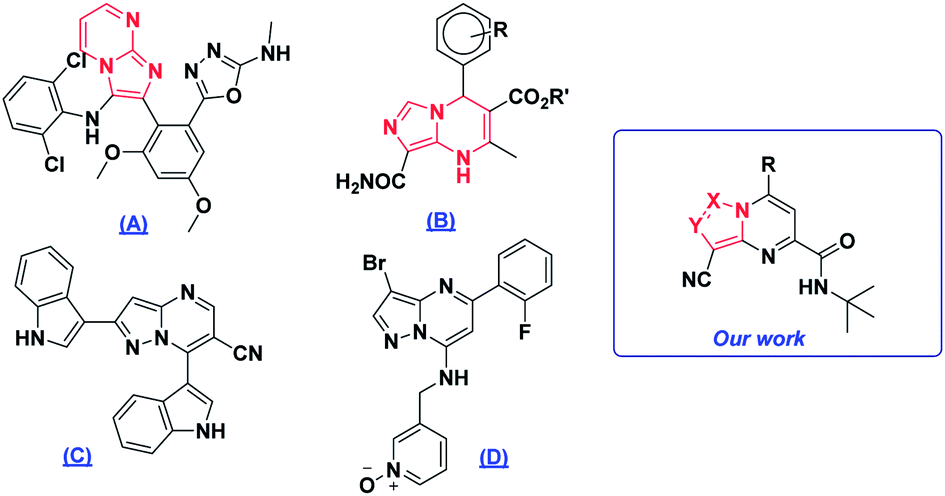 | ||
| Fig. 1 Examples of active imidazolo and pyrazolo-pyrimidine derivatives. | ||
Polycyclisation reaction has been steadily gaining importance in synthetic organic chemistry. It's an interesting way to access structural diversity and new privileged structures.9 The multicomponent reactions (MCR), especially those based on isocyanide called Groebke–Blackburn–Bienaymé reactions,10 are a method of choice to access complex heterocyclic molecules in a single step process from commercially available starting materials. Based on our expertise in the synthesis and functionalization of diverse polynitrogen-containing heterocycles,11 we wish to describe herein the preparation of original imidazo- and pyrazolo-[1,5-a]pyrimidine derivatives from synthesized imidazo[1,5-a]imidazoles and imidazo[1,2-b]pyrazoles using MCR followed by iodine catalyzed intramolecular rearrangement.
Results and discussion
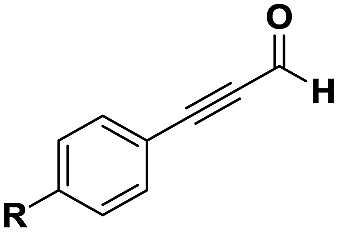
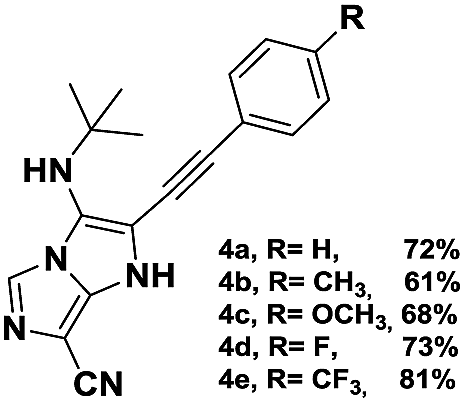
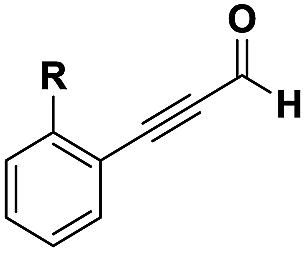
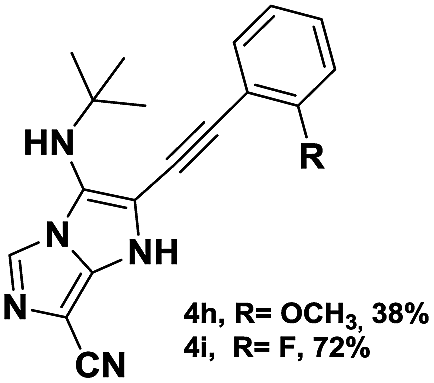
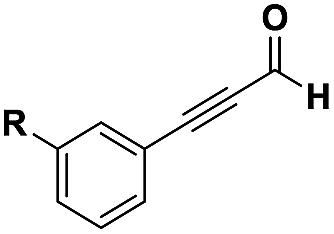
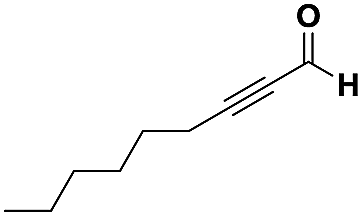
The imidazo-imidazole derivatives were prepared using MCR as described in the literature.12 In this method, we treated 5-aminoimidazole-4-carbonitrile and tert-butyl isocyanide with various commercially available or readily prepared propargylic aldehydes 3a–j (see ESI† for new aldehydes preparation) in the presence of perchloric acid as catalyst in methanol at room temperature for 24 h. The imidazo[1,5-a]imidazole substrates 4a–j were isolated in moderate to good yields. The use of either electron-donating or withdrawing groups on the aromatic ring of 3a–j provided the desired products in acceptable yields (Table 1).
|
|||
|---|---|---|---|
| Aldehyde 3 | Product 4 | Aldehyde 3 | Product 4 |
 |
 |
 |
 |
 |
 |
 |
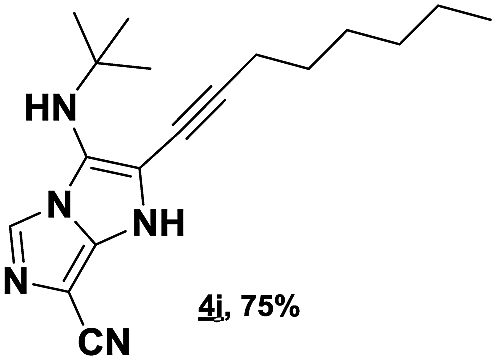 |
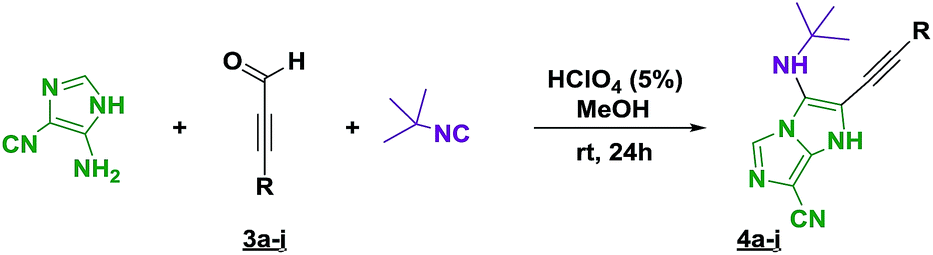
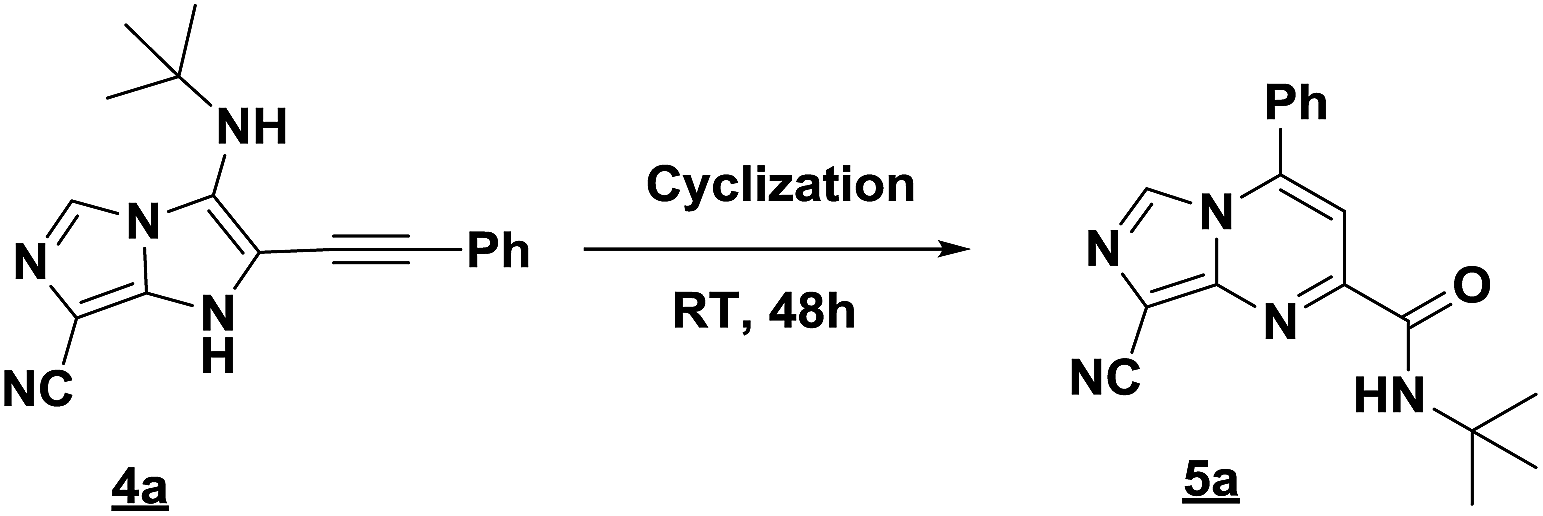
Firstly, the phenylpropargyl aldehyde was examined and the desired compound 4a was isolated in 72% yield. The effect of substituents on the aromatic ring of benzaldehydes was then investigated. The 3-(p-tolyl)propiolaldehyde and 3-(p-methoxyphenyl)propiol-aldehyde, bearing electron-donor groups led to expect products 4b and 4c in 61 and 68% yields, respectively. These yields are slightly lower than those obtained using the electron withdrawing groups with both 3-(p-fluorophenyl)propiolaldehyde and 3-(p-(trifluoromethyl)phenyl)propiolaldehyde, the corresponding compounds 4d and 4e were isolated in 73 and 81% yields, respectively. Importantly, the use of the aliphatic aldehyde 2-octynal was also effective leading to compound 4j in 75% yield. In contrast, we noticed a decrease in reaction yields when using aldehydes containing a methoxy group on the benzoic ring at either ortho or meta- positions. However, no significant decrease was observed when using fluorine group at either ortho or meta- positions on the aromatic ring. Once a variety of 3-(tert-butylamino)-2-(arylethynyl)-1H-imidazo[1,5-a]imidazoles has been synthesized, we decided to expand the utility of these scaffolds by using iodine polycyclisation reaction. According to our previous reports, the formation of tricyclic fused 3-iodo-pyrrolo[3′,2′:4,5]-imidazo[1,5-a]imidazoles could be easy obtained by an iodine intramolecular cyclisation of the multicomponent compounds in the presence of I2 in DCM (Scheme 2).13 So, we first investigated the general reaction conditions using 3-(tert-butylamino)-6-cyano-2-(phenylethynyl)-1H-imidazo[1,5-a]imidazole 4a as model substrate in the presence of 2 equivalents of I2 in DCM at room temperature. Under these reaction conditions, 60% of starting material was recovered (entry 1, Table 2). Surprisingly, the use of THF as solvent for the reaction gave access to imidazo[1,5-a]pyrimidine 5a instead of tricyclic compound 6 (entry 2, Table 2). The structure of 5a was confirmed by a single-crystal X-ray study (Scheme 1).
|
|||
|---|---|---|---|
| Entry | Iodine source | Solvent | Yield (%) |
a Isolated starting material.b Reaction under argon.c 6 equiv. of water was added to the reaction![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :degradation. :degradation. |
|||
| 1 | I2 (2 equiv.) | DCM | Traces(60)a |
| 2 | I2 (2 equiv.) | THF | 15(53)a |
| 3 | I2 (4 equiv.) | THF | 54(31)a |
| 4 | I2 (6 equiv.) | THF | 71 |
| 5 | I2 (8 equiv.) | THF | Traces |
| 6 | I2 (6 equiv.) | THF | Tracesb |
| 7 | I2 (6 equiv.) | THF | 0c |
| 8 | ICl (6 equiv.) | THF | 0(90)a |
| 9 | NIS (6 equiv.) | THF | Traces(60)a |
| 10 | HI (6 equiv.) | THF | 32(30)a |
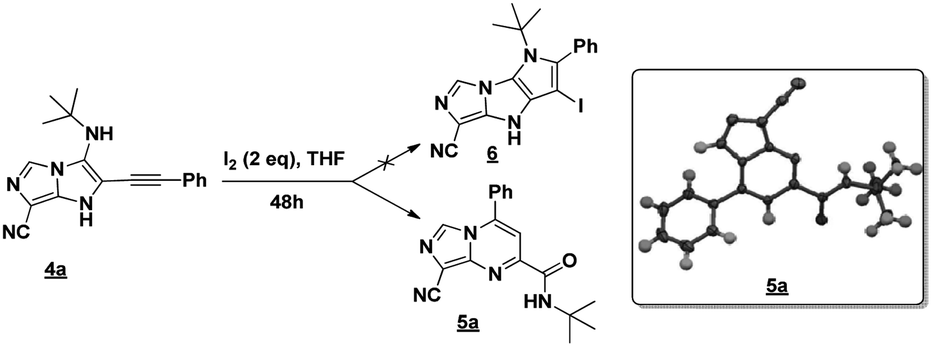 | ||
| Scheme 1 Preparation of imidazo[1,5-a]pyrimidine 5a by rearrangement of 4a. | ||
Encouraged by this preliminary result, other amounts of I2 were also investigated. Adding 4 equiv. of I2 instead of 2 equiv. increased the yield of 5a to 54% with 31% of starting material recovered (entry 3, Table 2). To our delight, when using 6 equiv. of I2, a complete conversion of the starting material into the rearranged product was achieved and 5a was isolated in 71% yield (entry 4, Table 2). To further improve the reaction yield, 8 equiv. of I2 were also examined, unfortunately, in this case only the degradation was observed (entry 5, Table 2). To gain more insight into the plausible mechanism of this rearrangement, oxygen free conditions were tested by trying the reaction under argon. However, only traces of product were isolated (entry 6, Table 2). Adding 6 equiv. of water into the reaction was associated by a complete degradation of the starting material (entry 7, Table 2). Next, we tried to extend our trials by changing the source of iodine. With iodine monochloride, unfortunately, no reaction occurred, only the starting material was recovered (entry 8, Table 2). Even with N-iodosuccinimide or hydriodic acid, the desired product was isolated in traces and in low yields, respectively (entries 9 and 10, Table 2).
In an endeavor to expand the scope of this method, the reactivity of various 3-(tert-butylamino)-1H-imidazo[1,5-a]imidazole-7-carbonitriles 4b–4j was investigated (Table 3).
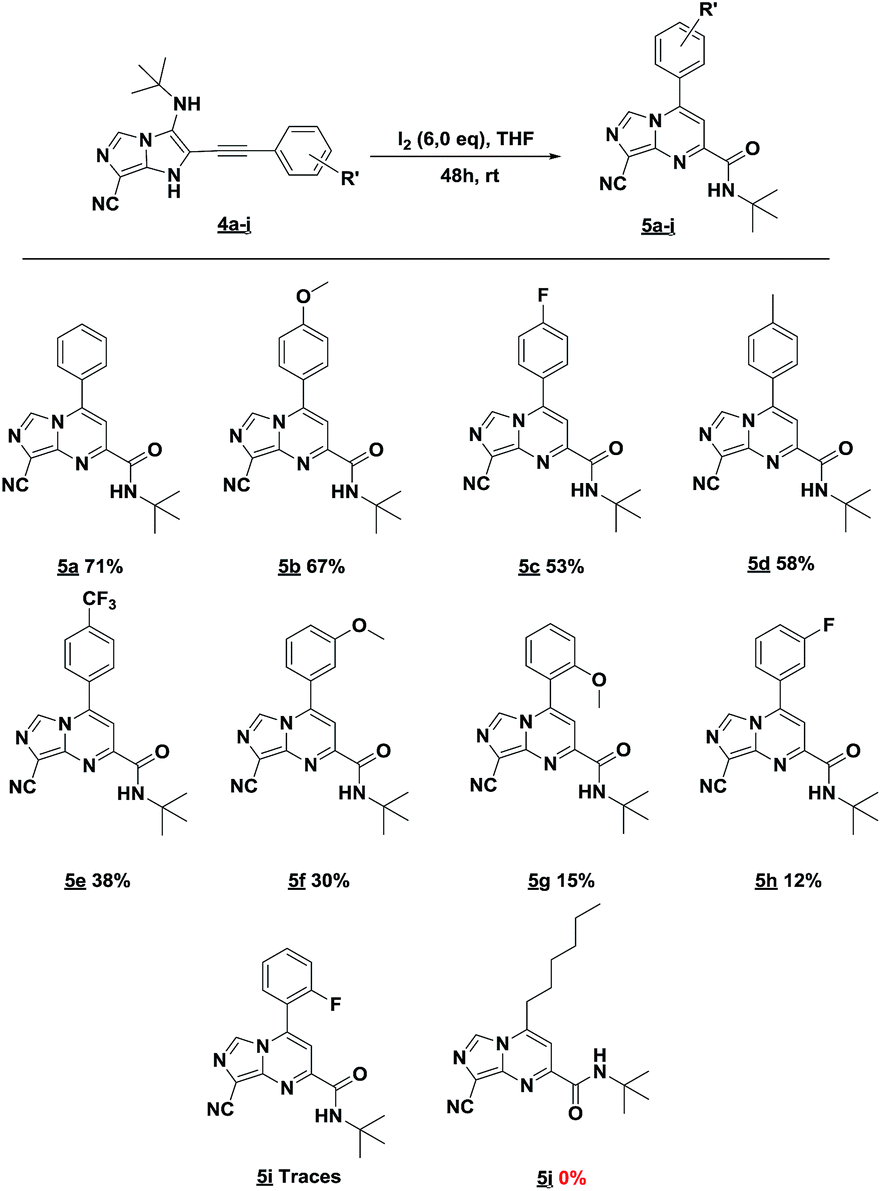 |
The reaction proceeded with electron-donating and electron withdrawing groups. Moderate yields were obtained with para-substituted substrates on the aromatic ring (Table 3, products 5b–e) while lower yields were observed with ortho and -meta-substituted analogues (Table 3, products 5f–h). Unluckily, for substrate containing fluorine group at ortho position, only traces of desired compound 5i were obtained. Also, we investigated the rearrangement with an aliphatic aldehyde, but unfortunately, no reaction was observed.
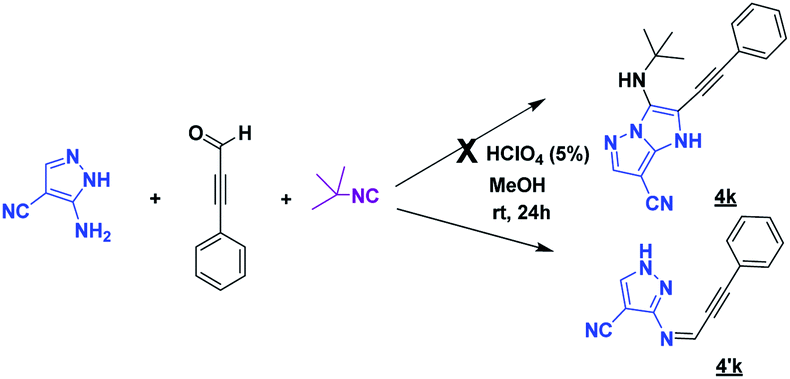
After having successfully developed a chemical library of functionalized imidazo[1,5-a]pyrimidines in only two steps, we extended the methodology to prepare various pyrazolo[1,5-a]pyrimidines in which the pyrazole core is fused with a pyrimidine. For this aim, the 5-aminoimidazole-4-carbonitrile was replaced by 3-aminopyrazole-4-carbonitrile by using the reaction conditions of MCR (Scheme 2). This latter has been serving as a basic scaffold of some synthetic compounds identified for the treatment of several diseases such as inflammation,14,15 tuberculosis16 and cancer.17
 | ||
| Scheme 2 MCR of 3-aminopyrazole-4-carbonitrile. | ||
Unfortunately, after 24 h of reaction, only the imine intermediate 4′k was obtained. To prepare the desired compound 4k, the optimization of the reaction conditions was needed. Indeed, we examined the following different conditions reported in the literature18,19 (Table 4).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | T (°C) | t (h) | 4k (%) | 4′k (%) |
| 1 | HClO4 (5%) | MeOH | r.t. | 24 | 0 | 98 |
| 2 | TFA (20%) | EtOH | r.t. | 24 | 36 | 40 |
| 3 | TFA (20%) | EtOH | 50 | 24 | 35 | 42 |
| 4 | ZrCl4 (10%) | MeOH | r.t. | 24 | 40 | 46 |
| 5 | ZrCl4 (10%) | MeOH | 50 | 16 | 43 | 41 |
| 6 | ZrCl4 (10%) | PEG-400 | 50 | 2 | 54 | 0 |
| 7 | ZrCl4 (10%) | PEG-400 | r.t. | 1 | 76 | 0 |
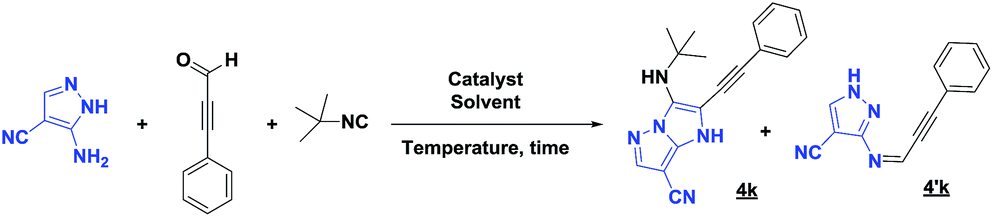
Based on the best reaction conditions obtained with 10% of zirconium tetrachloride and PEG-400, we extend this reaction with two different substituted aminopyrazoles (Table 5). Afterwards, the obtained imidazo[1,2-b]pyrazoles 4k–m were involved in the rearrangement reaction to give the corresponding pyrazolo[1,5-a]pyrimidines 5k–m as shown in Table 5.
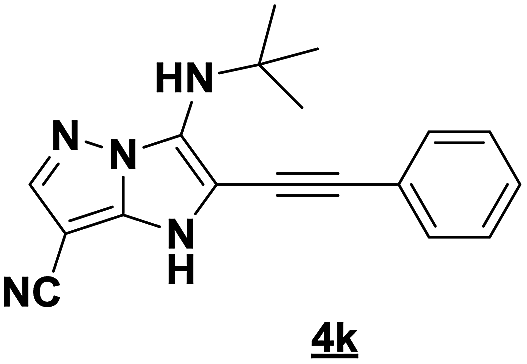
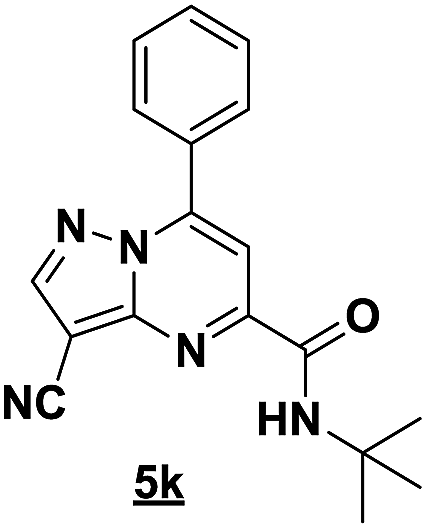
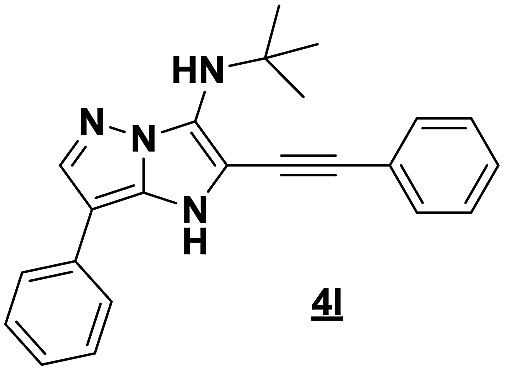
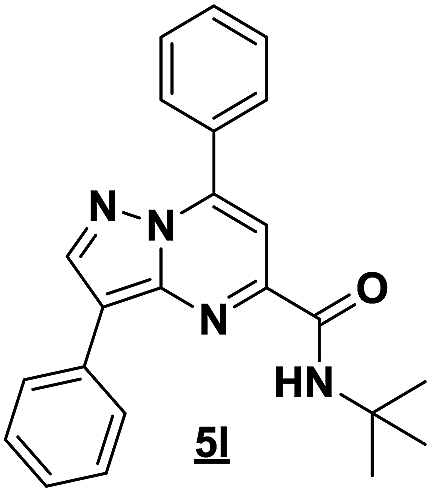
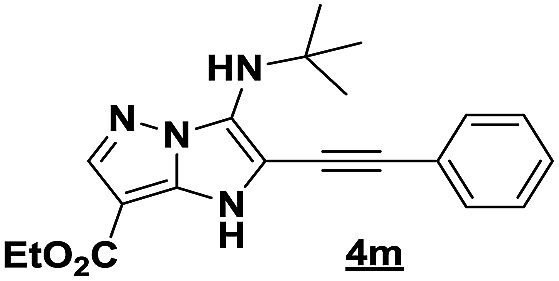
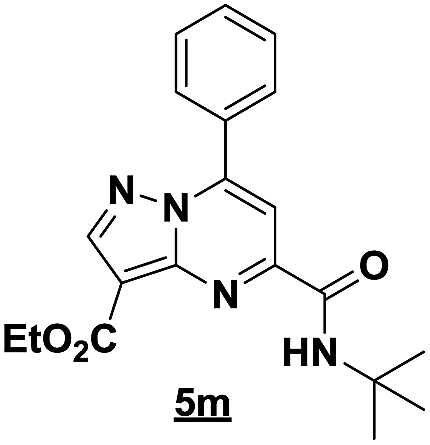
With the nitrile group, the rearranged compound 5k was obtained in an average yield of 35% while the starting materials containing phenyl or ester groups on the pyrazole ring gave the products 5l and 5m in good yields of 56 and 67%, respectively.
For a plausible mechanism, we think that, during the cyclization/aromatization, I2 in the presence of O2 (air) led to the formation of H2O2 (ref. 20) which may open the imidazo[1,5-a]imidazole 4a and then by cyclisation resulting from the nucleophilic attack of the N1 of the imidazole on the benzylic position led to imidazo[1,5-a]pyrimidine 5a (Scheme 3).
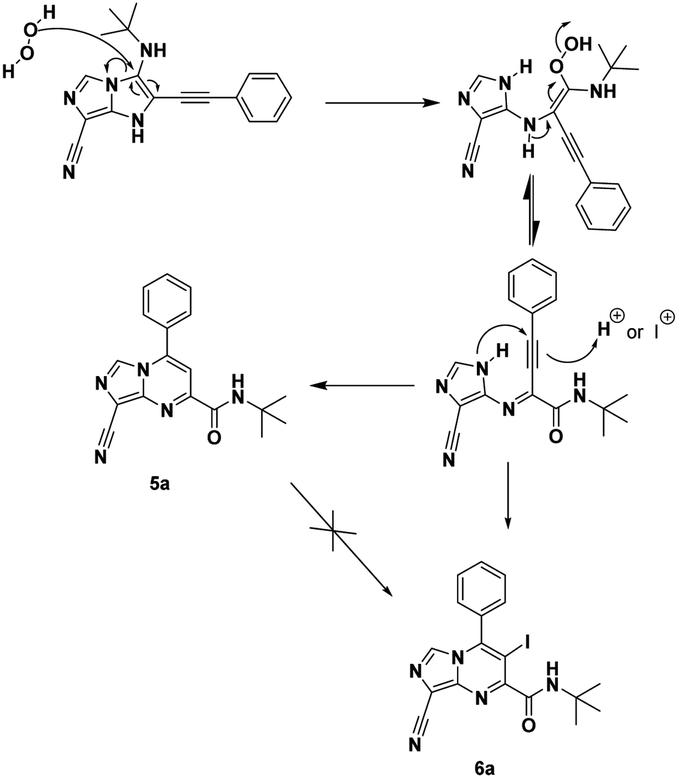 | ||
| Scheme 3 Plausible mechanism and synthesis of 5a. | ||
To support our hypothesis, we first carried out the reaction under the same conditions using the radical scavenger 1,4-cyclohexadiene (1,4-CHD), the results revealed the formation of the product which explained the non-radical reaction pathway. Next, we performed the reaction in the presence of hydrogen peroxide (3 equiv.) under argon, the result showed the formation of the iodinated product 6a in 31% during 24 h (Scheme 3). Whereas, when the reaction was conducted without an oxidant under argon no reaction occurred (see Table 2, entry 6). This shows the involvement of H2O2 in the reaction mechanism.
The formation of the product 6a can be explained by an electrophilic addition of the iodinium cation I+ during the cyclisation. To ensure this, no reaction took place when 5a was treated under the same conditions (Scheme 3), which means that the product 6a is not formed by iodination of 5a. The iodination agent I+ was produced in situ from the combination of I2 and H2O2.21
The structure of this compound was elucidated by 1D and 2D NMR. In addition, the 1H–13C HMBC experiment indicated that the iodination occurs on the pyrimidine moiety rather than the imidazole moiety.
However, when another oxidant was added to the reaction mixture (K2S2O8, 1.1 equiv.) under air, the reaction yield was improved to 79% (Scheme 4).
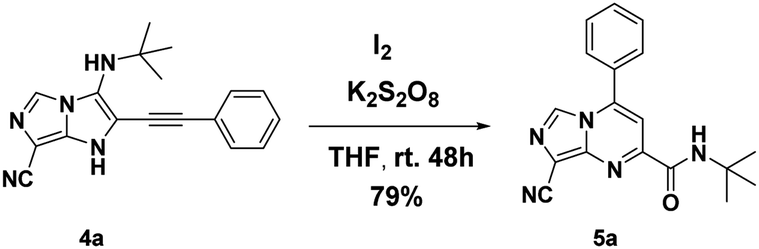 | ||
| Scheme 4 Synthesis of 5a using I2/K2S2O8. | ||
Conclusions
In summary, we have reported an exceptional rearrangement reaction starting from 3-(tert-butylamino)-2-(arylethynyl)-1H-imidazo[1,5-a]imidazoles or 3-(tert-butylamino)-2-(arylethynyl)-1H-imidazo[1,2-b]pyrazoles which were readily prepared by MCR reaction. This rearrangement reaction is unique and gives access rapidly and easily in two steps to a large diversity of functionalized imidazo- and pyrazolo-[1,5-a]pyrimidines, using mild reaction conditions in the presence of 6 equivalents of I2 from available starting materials.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by CNRST of Morocco, University of Orleans, University Hassan II of Casablanca, and the Euromed University of Fes.References
- T. B. Nguyen, M. Corbin, P. Retailleau, L. Ermolenko and A. Al-Mourabit, Org. Lett., 2015, 17, 4956–4959 CrossRef CAS PubMed.
- B. Pujala, A. K. Agarwal, S. Middya, M. Banerjee, A. Surya, A. K. Nayak, A. Gupta, S. Khare, R. Guguloth, N. A. Randive, B. U. Shinde, A. Thakur, D. I. Patel, M. Raja, M. J. Green, J. Alfaro, P. Avila, F. Perez de Arce, R. G. Almirez, S. Kanno, S. Bernales, D. T. Hung, S. Chakravarty, E. McCullagh, K. P. Quinn, R. Rai and S. M. Pham, ACS Med. Chem. Lett., 2016, 7, 1161–1166 CrossRef CAS PubMed.
- J. Liu and X. Wang, ACS Comb. Sci., 2011, 13, 414–420 CrossRef CAS PubMed.
- A. Twarda-Clapa, S. Krzanik, K. Kubica, K. Guzik, B. Labuzek, C. G. Neochoritis, K. Khoury, K. Kowalska, M. Czub, G. Dubin, A. Dömling, L. Skalniak and T. A. Holak, J. Med. Chem., 2017, 60, 4234–4244 CrossRef CAS PubMed.
- J. Liu and T. W. Lee, Annu. Rep. Med. Chem., 2011, 46, 103–115 CAS.
- R. Alajarin, J. J. Vaquero, J. Alvarez-Builla, M. Fau de Casa-Juana, C. Sunkel, J. G. Priego, P. Gomez-Salcp and R. Torres, Bioorg. Med. Chem., 1994, 2, 323–329 CrossRef CAS PubMed.
- A. El-Mekabaty and A. A. Fadda, J. Heterocycl. Chem., 2018, 2303–2308 CrossRef CAS.
- A. S. Hassana, M. F. Madyb, H. M. Awadd and T. S. Hafez, Chin. Chem. Lett., 2017, 28, 388–393 CrossRef.
- M. Baenziger, E. Durantie and C. Mathes, Synthesis, 2017, 49, 2266–2274 CrossRef CAS.
- M. Baenziger, E. Durantie and C. Mathes, Synthesis, 2017, 49, 2266–2274 CrossRef CAS.
- (a) M. Driowya, H. Allouchi, J. M. Gally, P. Bonnet and G. Guillaumet, New J. Chem., 2018, 42, 5728–5741 RSC; (b) M. Loubidi, J. Jouha, Z. Tber, M. Khouili, F. Suzenet, M. Akssira, M. A. Erdogan, F. A. Kose, T. Dagcı, G. Armagan, L. Saso and G. Guillaumet, Eur. J. Med. Chem., 2018, 145, 113–123 CrossRef CAS PubMed; (c) S. Faarasse, S. El Kazzouli, F. Suzenet and G. Guillaumet, J. Org. Chem., 2018, 83, 12847–12854 CrossRef CAS PubMed; (d) S. Faarasse, S. El Kazzouli, M. Naas, J. Jouha, F. Suzenet and G. Guillaumet, J. Org. Chem., 2017, 82, 12300–12306 CrossRef CAS PubMed; (e) M. Loubidi, C. Pillard, A. El Hakmaoui, P. Bernard, M. Akssira and G. Guillaumet, RSC Adv., 2016, 6, 7229–7238 RSC; (f) A. El Bouakher, S. Massip, C. Jarry, Y. Troin, I. Abrunhosa-Thomas and G. Guillaumet, Eur. J. Org. Chem., 2015, 556–569 CrossRef CAS; (g) M. Naas, S. El Kazzouli, E. M. Essassi, M. Bousmina and G. Guillaumet, Org. Lett., 2015, 17, 4320–4323 CrossRef CAS PubMed; (h) R. Belaroussi, A. El Bouakher, M. Marchivie, S. Massip, C. Jarry, A. El Hakmaoui, G. Guillaumet, S. Routier and M. Akssira, Synthesis, 2013, 45, 2557–2566 CrossRef CAS.
- (a) K. Groebke, L. Weber and F. Mehlin, Synlett, 1998, 661–663 CrossRef CAS; (b) C. Blackburn, B. Guan, P. Fleming, K. Shiosaki and S. Tsai, Tetrahedron Lett., 1998, 39, 3635–3638 CrossRef CAS; (c) H. Bienayme and K. Bouzid, Angew. Chem., Int. Ed., 1998, 37, 2234–2237 CrossRef CAS.
- Z. Tber, M.-A. Hiebel, A. El Hakmaoui, M. Akssira, G. Guillaumet and S. Berteina-Raboin, J. Org. Chem., 2015, 80, 6564–6573 CrossRef CAS PubMed.
- L. Nagarapu, J. Mateti, K. Gaikwad, R. Bantu, M. Sheeba Rani and J. Subhashini, Bioorg. Med. Chem. Lett., 2011, 21, 4138–4140 CrossRef CAS PubMed.
- A. Bekhit, A. Ashour, S. Abdel Ghany, A. Bekhit and A. Baraka, Eur. J. Med. Chem., 2008, 43, 456–463 CrossRef CAS PubMed.
- V. Pathak, H. K. Maurya, S. Sharma, K. K. Srivastava and A. Gupta, Bioorg. Med. Chem. Lett., 2014, 24, 2892–2896 CrossRef CAS PubMed.
- S. C. Pirol, B. Çalıskan, I. Durmaz, R. Atalay and E. Banoglu, Eur. J. Med. Chem., 2014, 87, 140–149 CrossRef PubMed.
- S. K. Guchhait and C. Madaan, Synlett, 2009, 628–632 CrossRef CAS.
- A. Demjén, M. Gyuris, J. Wölfling, L. G. Puskás and I. Kanizsai, Beilstein J. Org. Chem., 2014, 10, 2338–2344 CrossRef PubMed.
- (a) B. Deka, P. K. Baruah and M. L. Deb, Org. Biomol. Chem., 2018, 16, 7806–7810 RSC; (b) M. Asikainen, O. Jauhiainen, O. Aaltonen and A. Harlin, Green Chem., 2013, 15, 3230–3235 RSC; (c) M. C. Milenković and D. R. Stanisavljev, J. Phys. Chem. A, 2012, 116, 5541–5548 CrossRef PubMed; (d) G. Schmitz, Phys. Chem., 2010, 12, 6605–6615 CAS.
- A. R. Pourali and M. Ghanej, Chin. J. Chem., 2006, 24, 1077–1079 CrossRef CAS and references cited therein.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1900146. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra04609g |
| This journal is © The Royal Society of Chemistry 2019 |