
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Constructing ZIF-8 derived C–ZnS/ZnMoO4@MoS2 and C–ZnS/MoS2 nanocomposites using a simple one-pot strategy to enhance photocatalytic degradation activity†
Yi-Wei Cui,
Hai-Huan Zhang and
Shi-Yong Yu *
*
Inner Mongolia Key Laboratory of Mongolian Medicine Chemistry, Inner Mongolia Key Laboratory of Coal Chemistry, School of Chemistry and Chemical Engineering, Inner Mongolia University, Hohhot 010021, Inner Mongolia, China. E-mail: syyunano@imu.edu.cn
First published on 31st October 2019
Abstract
Efficient C–ZnS/ZnMoO4@MoS2 and C–ZnS/MoS2 nanocomposite photocatalysts, using ZIF-8 derived C–ZnO as a precursor were successfully synthesized using a simple one-pot procedure. This is the first application that involves transforming ZIF-8 into C–ZnMoO4 for photocatalysis. The C–ZnS/ZnMoO4@MoS2 and C–ZnS/MoS2 heterostructures were characterized by X-ray diffraction, UV-vis, X-ray photoelectron spectroscopy, electrochemical impedance spectroscopy, photocurrent measurements, scanning electron microscopy and transmission electron microscopy. The ZM2 sample of C–ZnS/ZnMoO4@MoS2 exhibited enhanced photocatalytic activity of about 2.9 times as high as that of ZIF-8 derived C–ZnO in the reduction of tetracycline hydrochloride, and also showed obvious photocatalytic activity 1.81 and 3.33 times as high as that of a ZM3 sample of C–ZnS/MoS2 and ZIF-8 derived C–ZnO in the degradation of RhB, respectively. The improved photodegradation activity is a result of the heterogenous structure and the tighter contact between C–ZnS and C–ZnMoO4 compared with the physical contact of general heterogenous photocatalysts. The C–ZnS/ZnMoO4@MoS2 heterostructure photocatalyst is expected to be a new type of nanomaterial for the degradation of pollutants from wastewater.
1. Introduction
Wastewater containing antibiotics has become a grave threat to human health and the environment.1,2 Tetracycline hydrochloride (TC-H) as the second most widely distributed antibiotic in the world has been extensively produced and used.3,4 However, due to the unique characteristic properties of TC-H, only 10–30% of TC-H can be metabolized by animals and human beings, and 70–90% is released into the water environment via discharge of wastewater effluent and animal manure after being heavily used in the pharmaceutical industry, animal husbandry and aquaculture, which causes serious water environmental problems.5,6Among the treatments of tetracycline wastewater, photocatalysis is a simple,7 dependable and environmentally-friendly technology compared to traditional techniques, such as adsorption,8 microbial degradation9 or membrane separation.10 Compared with conventional photocatalysts, such as TiO2, C3N4 or ZnO,11–13 using MOFs as “precursors” or self-sacrificial frameworks to fabricate porous metal oxides as photocatalysts has been a research hotspot in recent years.14,15
Zeolite imidazolate frameworks (ZIFs) as a sub aggregation of MOFs are a kind of porous crystalline material in which imidazolate linkers are cross-linked to transition metal ions to form 3D tetrahedral frameworks.16–19 They have attracted extensive attention in various fields, such as heterogeneous catalysis,20–23 photocatalysis,24 sensing,25 gas adsorption26–28 and drug delivery due to their high surface area and high crystallinity as well as good chemical and thermal stability.29 However, as photocatalysts, most ZIFs cannot be excited by visible light because their band gaps are too wide to be excited: for example, ZIF-8 has a band gap of 5.1 eV.30 For the purposes of diminishing their band gaps and broadening their application in photocatalysis, MOFs can be used as precursors to directly prepare C-doped oxides and it has been proved that the prepared C-doped materials can expand the response to visible light and facilitate the absorption of visible light. Du et al. prepared porous C–ZnO by calcining ZIF-8 directly in air and they found that such ZnO showed improved photocatalytic activity compared to ZIF-8 or pure ZnO.31 Feng et al. prepared N–ZnO from the direct calcination of urea and a ZIF-8 mixture for RhB degradation, and found that such a material can enhance the visible-light harvesting ability and achieve a photocatalytic efficiency higher than that of ZIF-8 derived C–ZnO.32 However, reports about transforming ZIF-8 directly into heterojunctions are very scarce.
Molybdenum disulfide (MoS2) has been proven to be an excellent co-catalyst in photocatalytic activity by providing more active sites and inhibiting the recombination rate of electron–hole pairs. Thomas et al. reported active edge sites for electrochemical hydrogen production from MoS2.33 By contrast, ZnS and ZnMoO4 are both well-known photocatalysts, which have a wider band gap than MoS2. Such a combination can facilitate the separation of the electron–hole pairs by photoexcitation.34,35
Inspired by this, herein, we have adopted a one-pot route in which ZIF-8 derived C–ZnO was added to the raw materials for the synthesis of MoS2 and a three-phase heterojunction of C–ZnS/ZnMoO4@MoS2 and a two-phase heterojunction of ZnS/MoS2 could be finally obtained. It is worth mentioning that, because the C–ZnS and C–ZnMoO4 in the three-phase heterojunction were directly produced together from ZIF-8 derived C–ZnO in the same step, the contact between C–ZnS and C–ZnMoO4 will be tighter than the physical contact of general heterojunction photocatalysts, and this can facilitate the separation of the electron–hole pairs.
2. Experimental section
2.1 Materials
Zinc nitrate hexahydrate [Zn(NO3)2·6H2O ≥ 99.0%], absolute methanol (EtOH ≥ 99.7%) and ethanol (EtOH ≥ 99.7%) were purchased from Sinopharm Chemical Reagents Co., Ltd. 2-Methylimidazole [H-MeIM], thiourea (CH4N2S) and ammonium molybdate [(NH4)6Mo7O24·4H2O] were purchased from Aladdin (P. R. China). All chemicals were used without any further purification.2.2 Preparation
2.3 Characterization of the photocatalysts
The crystallographic phase of the as-synthesized samples was determined with a Panalytical X-Pert X-ray diffractometer (XRD) with a scanning rate of 2.5° per min. The morphology and elemental compositions of the prepared photocatalysts were observed by a scanning electron microscope (SEM) equipped with an energy dispersive X-ray (EDX) spectrometer and transmission electron microscopy (TEM, JEM-2010). X-ray photoelectron spectroscopy (XPS) was performed on an ESCALAB250 instrument (Thermo VG Corp.) The UV-vis spectra were carried out on a spectrometer (Shimadzu UV-3600). The photoluminescence (PL) spectra of the samples were recorded on an Edinburgh Instruments FLS 920 spectrometer at an excitation wavelength of 350 nm.2.4 Photocatalytic measurements
The photocatalytic activities of C–ZnS/ZnMoO4@MoS2 and C–ZnS/MoS2 were measured using the photodegradation of tetracycline hydrochloride and RhB in an aqueous solution under full spectrum light irradiation (320 nm < λ < 780 nm) using a 300 W xenon lamp. Typically, in the degradation process of tetracycline hydrochloride: 15 mg of catalyst was dispersed in 30 ml of tetracycline hydrochloride (15 mg l−1); in the degradation process of RhB: 25 mg of catalyst was dispersed in 50 ml of RhB (5 mg l−1). The reactor was stirred in the dark for 20 min before irradiation to ensure absorption–desorption equilibrium. During the process, the suspension was kept under stirring to retain the homogeneity of the suspension.2.5 Photoelectrochemical measurements
Photoelectrochemical analysis was carried out in a conventional three electrode cell. In the working cell, the fabricated electrode (a platinum wire) and reference electrodes Ag/AgCl (saturated KCl) were used. The photocatalyst powder was deposited on FTO (F doped tin oxide) glass by electrophoretic deposition. Electrochemical impedance spectroscopy (EIS) measurements were performed at frequencies from 0.1 Hz to 100 KHz and the applied voltage was the open circuit voltage. The electrolyte was Na2SO4 aqueous solution (0.2 M, pH = 7).3. Results and discussion
3.1 Structural and morphological analysis
The crystal phases of the samples (ZIF-8 C–ZnO ZM) were first identified by X-ray diffraction and the results are shown in Fig. 1. | ||
| Fig. 1 XRD patterns of (a) ZIF-8 and ZIF-8 derived ZnO and (b) ZM1, ZM2, ZM3 and ZM4. | ||
As shown in Fig. 1a, the transition from ZIF-8 to C–ZnO can be seen. The XRD pattern of ZIF-8 is consistent with the patterns reported in the literature.37 After thermal annealing, the diffraction peaks of ZIF-8 derived C–ZnO located at about 31.738°, 34.380°, 36.217°, 47.487°, 56.535°, 62.777°, 67.865° can be indexed to the (100), (002), (101), (102), (110), (103) and (112) diffraction planes of ZnO (JCPDS no. 01-076-0704), which reflects the successful transformation from ZIF-8 to ZnO.
The XRD patterns of the ZMs are shown in Fig. 1b. Six peaks located at about 26.915°, 28.502°, 30.530°, 47.563°, 51.778°, 56.394° correspond to diffraction from the (100), (002), (101), (110), (103) and (112) crystallographic planes of ZnS (JCPDS no. 00-036-1450) respectively in all samples. The peaks corresponding to ZnMoO4 can be observed at 24.302°, 36.620°, 48.844°, 53.715° corresponding to the (110), (120), (022) and (−221) diffraction planes of ZnMoO4 (JCPDS no. 00-016-0310) in ZM1, 36.620° and 53.715° corresponding to the (120) and (−221) diffraction planes of ZnMoO4 in ZM2, and 36.620° corresponding to the (120) diffraction planes of ZnMoO4 in ZM4, and no peaks corresponding to ZnMoO4 were observed in ZM3. The peaks located at 33.4° corresponding to MoS2 could be observed in ZM3. Unfortunately, no diffraction peaks corresponding to MoS2 could be discerned in the other XRD patterns, which may be because of the low content of this material in the three composites. Other than this, we also found that the main diffraction peaks corresponding to MoS2 at 37.6° and 57.8° were almost coincident with the diffraction peaks corresponding to ZnMoO4 at 36.620° and ZnS at 56.394°, which may also cause the inconspicuous peaks of MoS2 in the XRD patterns of these composites, except for ZM3 which did not contain the component of ZnMoO4. The presence of MoS2 and the composition of the catalysts will be further corroborated by SEM, TEM and XPS images.
The synthetic procedure for ZIF-8 derived C–ZnS/ZnMoO4@MoS2 and C–ZnS/MoS2 is shown in Scheme 1. As shown in Scheme 1, ZIF-8 derived C–ZnO was mixed with the raw materials for the synthesis of MoS2. During the hydrothermal reaction, when the MoS2 is forming from the reaction of thiourea and ammonium molybdate, ZIF-8 derived C–ZnO will also react with thiourea and ammonium molybdate to generate C–ZnS/ZnMoO4 or C–ZnS. The differences in the products is because the amount of thiourea will affect the acid–base properties of the solution and this may be the requirement for determining whether or not ZnMoO4 can be formed. Since the raw material in the preparation of C–ZnS/MoS2 (ZM3) was used in a larger amount than in the preparation of ZnS/ZnMoO4@MoS2 (ZM1, 2 and 4), the size of MoS2 in ZM3 was much larger than that in ZM2, eventually resulting in the different morphologies of ZM2 and ZM3, as shown in Scheme 1.
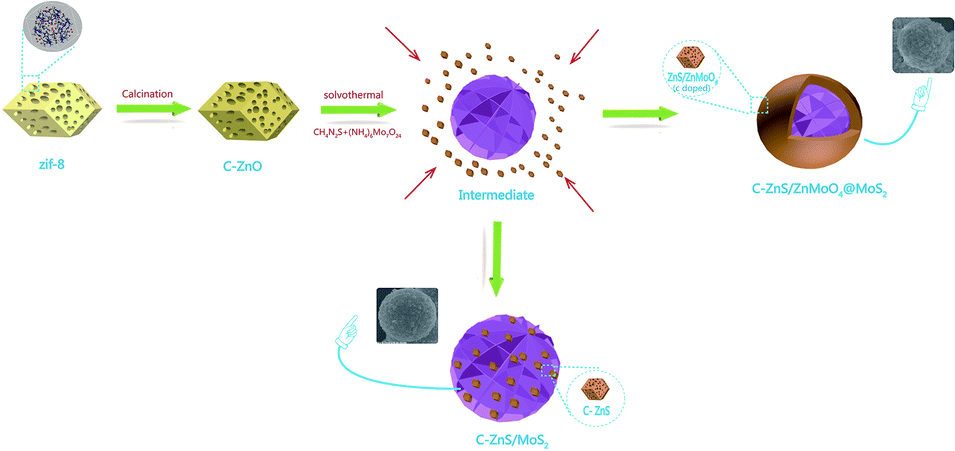 | ||
| Scheme 1 Synthetic procedure of ZIF-8 derived C–ZnS/ZnMoO4@MoS2 and C–ZnS/MoS2. | ||
The morphologies of the composites were investigated by SEM and TEM. Pure flower-like MoS2 is shown in Fig. S1.† It should be noted that pure MoS2 exhibited a smaller diameter than MoS2 in C–ZnS/MoS2 or C–ZnS/ZnMoO4@MoS2, the reason being that, in the process of preparing the catalysts, the addition of C–ZnO will increase the pH of the solution. According to research,37,38 in the process of preparing flower-like MoS2, an increase in the pH of the solution will accelerate the growth of the diameter of the obtained MoS2. The prepared ZIF-8 shown in Fig. S2† has a uniform polyhedral morphology with a diameter of 200–300 nm.
After hydrothermal treatment, the sphere-like ZM2 with a rough surface covered with 200–300 nm particles shown in Fig. 2a and b was proved by XRD (Fig. 1b) and XPS (Fig. 3) analysis to be ZIF-8 derived C–ZnS/ZnMoO4. We also found that C–ZnS/ZnMoO4 approximately retained the polyhedral morphology of ZIF-8 (inset Fig. 1b). The morphologies of MoS2 cannot clearly be observed in the SEM images because the surface of ZM2 is covered with nanoparticles of C–ZnS/ZnMoO4. However, because MoS2 is a flexible kind of material, its nanosheets can pass through the outer layer, which cannot be clearly observed in the SEM images. As shown in Fig. 2c–e, a large number of nanosheets can be found at the surface of ZM2 in the TEM images, and the high-resolution TEM (HRTEM) images (Fig. 2c and e) show clear fringes with a lattice spacing of 0.62 nm, which correspond to the (002) plane of MoS2. In addition, EDS analysis shows that selected areas of the ZM2 composites comprise Zn, S, Mo, C and O elements (inset Fig. 2a).
 | ||
| Fig. 2 (a) and (b) SEM images and EDS pattern (inset (a)) of ZM2; (d) TEM images of ZM2; (c) and (e) HRTEM images of ZM2; (f) and (g) SEM images and EDS pattern (inset (f)) of ZM3; (h) and (i) TEM images of ZM3; (j) HRTEM images of ZM3. | ||
 | ||
| Fig. 3 The high resolution XPS spectra of (a) survey spectrum, (b) C 1s, (c) Mo 3d (d) Zn 2p, (e) O 1s, (f) S 2p of (1) C–ZnS/ZnMoO4@MoS2(ZM2) and (2) C–ZnS/MoS2 (ZM3). | ||
It is worth mentioning that the carbon content can be observed both in the EDS pattern of the ZM2 and ZM3 samples (inset Fig. 2a and f), but no diffraction peaks of carbon were found in the XRD patterns of any of the samples, which indicates that the carbon was successfully doped into the lattice of ZnS/ZnMoO4 and ZnS.39 This conclusion was also further proved by XPS analysis (Fig. 3). Unlike the composition and morphology of ZM2, the nanoparticles of ZM3 were proved by XRD (Fig. 1b) and XPS (Fig. 3b) analysis to be ZIF-8 derived C–ZnS, and the particles retained the polyhedral morphology of ZIF-8 (inset Fig. 2g) loaded onto the flower-like MoS2, as shown in Fig. 2f and g. The nanosheets on the surface of ZM3 can be clearly observed in Fig. 2j and i. The HRTEM (Fig. 2j) images also show fringes with a lattice spacing of 0.62 nm corresponding to the (002) plane of MoS2. The EDS analysis shows the ZM3 to have Zn, S, Mo, C and O elements, in which the existence of oxygen might be due to the presence of water molecules absorbed on the surface of ZM3, which was further confirmed by XPS (Fig. 3).
3.2 XPS analysis
XPS analysis was carried out to further confirm the chemical composition and surface chemical state of ZM2 and ZM3 (Fig. 3). The characteristic peaks of C, Mo, Zn, O and S elements appear over the whole XPS survey spectrum of the two samples (Fig. 3a) which is consistent with the EDS results. The peaks of the high-resolution spectrum of C 1s are located at 284.6 and 285.9 eV (Fig. 3b). The main peak at 284.6 eV can be assigned to adventitious hydrocarbons and the peak at 285.9 eV can be attributed to Zn–C bonds, which further proves that carbon was doped into the lattice of ZnS/ZnMoO4 and ZnS into the C–ZnS/ZnMoO4@MoS2 and C–ZnS/MoS2 samples, respectively.In Fig. 3c(1), the high resolution spectrum for Mo 3d of ZM2 could be divided into five peaks: the binding energy at approximately 226.0 eV can be attributed to the S 2 s of MoS2 and the binding energies of 232.53 and 235.5 eV correspond to the Mo 3d5/2 and Mo 3d3/2 of Mo6+, respectively. The Mo 3d5/2 peak reveals that Mo exists in the form of a hexavalent oxide. Another two peaks at 228.3 and 231.5 eV are characteristic of the Mo 3d5/2 and Mo 3d3/2 of Mo4+ in MoS2, which further proves the existence of MoS2 in the C–ZnS/ZnMoO4@MoS2 samples (Fig. 2c and e). In Fig. 3c(2), the high resolution spectrum of Mo 3d of ZM3 can also be divided into five peaks. Compared to ZM2 in Fig. 3c(1), the main peaks of ZM3 are located at 228.7 and 231.8 eV, characteristic of Mo 3d3/2 and Mo 3d5/2 of Mo4+, and another two weaker peaks at about 232.75 and 235.35 eV were fitted as Mo6+, which might be due to the incomplete decomposition of MoO42−, consistent with the XRD results shown in Fig. 1b. The differences between the peak positions might due to the difference in the compositions of the two samples, resulting in a slight shift in the peak position.
As displayed in Fig. 3d(1), two distinct peaks at 1021.7 and 1044.7 eV correspond to Zn 2p1/2 and Zn 2p3/2, and the slight deviation is also due to the difference in the compositions of the two samples. The characteristic binding energy of 530.1 eV of ZM2 in Fig. 3e(1) for O 1s reveals that O is present in the metal oxide ZnMoO4 in C–ZnS/ZnMoO4@MoS2, and the binding energy at 531.5 eV can be attributed to the chemisorbed or dissociated oxygen or OH species on C–ZnS/MoS2. The two peaks at 162.8 and 161.7 eV correspond to the S 2p1/2 and S 2p3/2 of S2− and the slight difference in the 2p3/2 peak of ZM3, which can be found in Fig. 3f(2), is also due to the different compositions of the two samples.
3.3 Photocatalytic activity
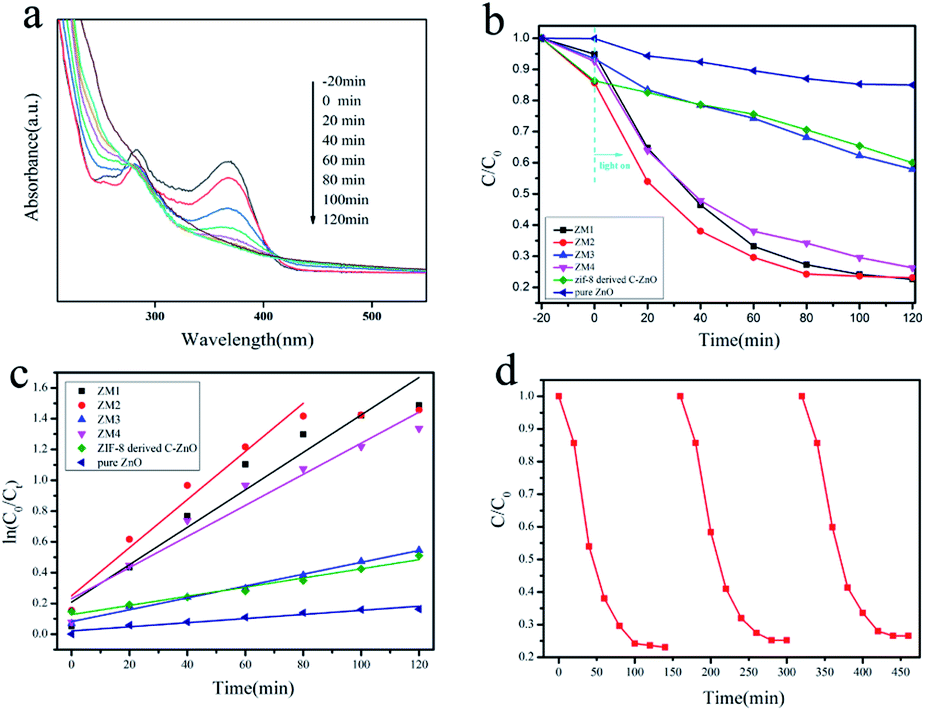 | ||
| Fig. 4 (a) Absorption spectra of TC-H solution collected during the photodegradation of ZM2; (b) the photodegradation efficiency of TC-H (C/C0) as a function of irradiation time over ZM1, ZM2, ZM3, ZM4, ZIF-8 derived C–ZnO and pure ZnO; (c) kinetic linear simulation curves of TC-H degradation with different photocatalysts under full spectrum light irradiation; (d) photocatalytic activities of ZM2 for TC-H degradation with three times of cycling use. | ||
The reaction kinetics of the TC-H degradation were further studied according to the pseudo-first-order kinetic model shown eqn (1), and the results are shown in Fig. 4c
| ln(C0/Ct) = kt | (1) |
It should be noted that TC-H was completely degraded by ZM2 in 80 minutes (Fig. 4a), so only the first five points were taken to obtain the slope (eqn (1)). As shown in Fig. 4c, the rates of TC-H photodegradation were 0.001, 0.003, 0.012, 0.016, 0.004, and 0.010 min−1 for pure ZnO, ZIF-8 derived C–ZnO, ZM1, ZM2, ZM3 and ZM4, respectively. Thus, the ZM2 samples showed excellent performance in the degradation of TC-H. In Fig. 4d, the recycling tests reveal that C–ZnS/ZnMoO4@MoS2 (ZM2) can be used repeatedly at least three times and the photocatalytic activity remains sufficiently stable without any significant deactivation.
 | ||
| Fig. 5 (a) Photodegradation efficiency of RhB (C/C0) as a function of irradiation time over ZM1, ZM2, ZM3, ZM4, ZIF-8 derived C–ZnO and pure ZnO; (b) kinetic linear simulation curves of RhB degradation with different photocatalysts under full spectrum light irradiation. | ||
In order to reveal the underlying reaction mechanism of the enhanced photocatalytic activity of ZM2, the radical and hole generation during the process of photocatalytic degradation of RhB over ZM2 was investigated by adding various scavengers. As shown in Fig. S4,† ammonium oxalate (AO) was added as an h+ scavenger, p-benzoquinone (PBQ) as an ·O2− scavenger and tert-butanol (t-BuOH) as an ·OH scavenger. It was found that there was a dramatic decrease in the photocatalytic activity when PBQ was added, indicating that ·O2− plays an important role in the photocatalytic degradation process. In addition, the photodegradation rate of RhB was also suppressed when AO and t-BuOH were added, suggesting that h+ and ·OH participate in the photodegradation process of RhB.
3.4 Optical analysis
Fig. 6 reveals the UV-vis diffuse reflection spectra of pure ZnO, ZIF-8 derived C–ZnO and ZM1-4. As shown in Fig. 6a, the main absorption intensity of pure ZnO occurs at ca. 390 nm and the edge of absorption is very low. In contrast to that, the absorption values of ZIF-8 derived C–ZnO extend into the visible light region, which confirms that it can harvest visible light effectively.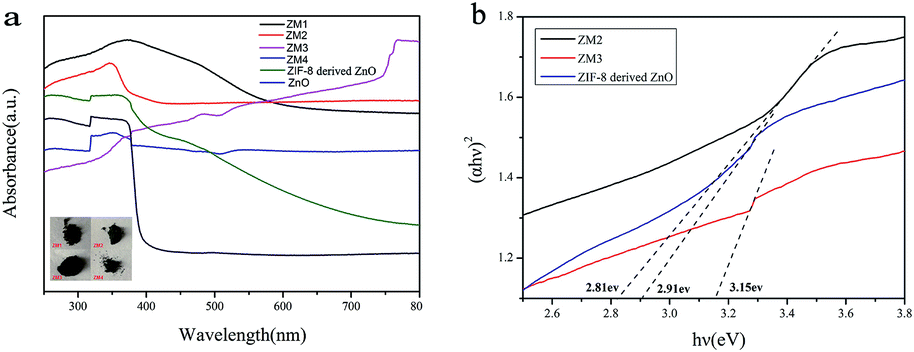 | ||
| Fig. 6 (a) UV-vis diffuse reflection spectra (inset: photographs of ZM1-4) and (b) (αhv)2 vs. hv curves of different samples. | ||
It is obvious that using ZIF-8 as a precursor to prepare catalysts has more advantages compared to using pure ZnO as a photocatalyst. After the introduction of MoS2 and transformation of ZIF-8 derived C–ZnO to C–ZnS/ZnMoO4 and C–ZnS, the absorption peaks of the ZM composite materials were red shifted and the light absorption was obviously enhanced compared to that of ZIF-8 derived C–ZnO. The enhanced absorption intensities of these materials are also consistent with the colors of the catalyst powders (inset Fig. 6a) which will be beneficial for their photodegradation activities.
On the basis of the Kubelka–Munk equation, the band gap energy of a semiconductor can be calculated using the following formula eqn (2)
| (αhv)2 = (hμ − Eg) | (2) |
3.5 Photoelectrochemical analysis
As shown in Fig. 7a, a fast and steady photocurrent response was detected for each light-on and light-off period over ZM2 and ZM3. The ZM2 samples show a significant enhancement in photocurrent density compared with that of the ZM3 samples, which further confirms the efficient transfer of photo-induced electrons of ZM2.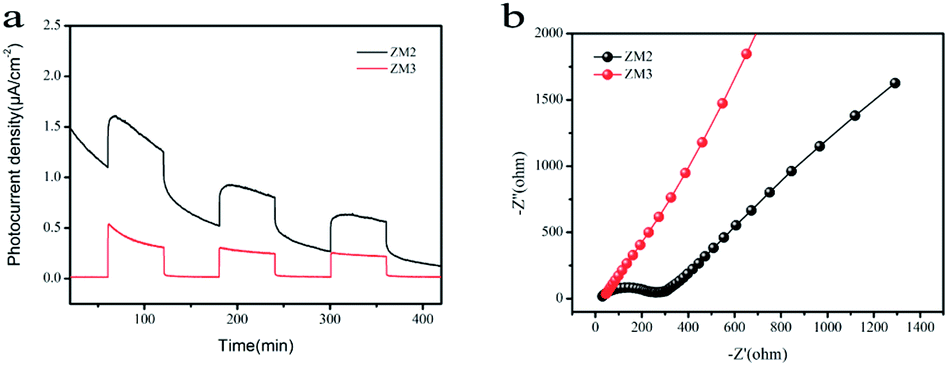 | ||
| Fig. 7 (a) Photocurrent spectra of as-synthesized samples under UV-vis light irradiation; (b) EIS Nyquist plots of the samples under dark condition. | ||
Electrochemical Impedance Spectroscopy (EIS) measurements were also carried out to figure out the interface charge transport behavior of ZM2 and ZM3. As shown in Fig. 7b, the radius of the arc on the EIS Nyquist plot reflects the reactor rate occurring at the electrode surface. The lower arc radius of ZM2 with respect to ZM3 demonstrates a faster interfacial transfer and more effective separation of photo-induced pairs occurring in the composites. These results are well consistent with the above-mentioned photocurrent tests. Thus, the photocurrent spectra and EIS Nyquist plots confirm the superior separation efficiency of the photo-induced electrons and holes and efficient charge transfer in ZM2, which are responsible for the enhanced photocatalytic activity.
The separation efficiency of the charge carriers is also shown in the photoluminescence (PL) analysis. As shown in Fig. S5,† the main emission peak is observed at around 623 nm. The ZM3 sample exhibits a lower PL intensity than ZIF-8 derived C–ZnO, and ZM2 shows the lowest emission peak among the C–ZnO, ZM3 and ZM2 samples, which proves that the construction of the heterojunction structure effectively inhibits photo-induced electron–hole pair recombination and the separation rate of the electron–hole pairs of ZM2 is better than that of ZM3, which is consistent with the results of the photoelectrochemical analysis.
3.6 Possible mechanism
On the basis of the experimental results and previous studies, a possible mechanism for the enhanced photoactivity of TC-H and RhB degradation can be proposed. As shown in Scheme 2a, under the excitation of light, C–ZnS, C–ZnMoO4 and MoS2 will all be excited at the same time, and the electrons (e−) in the valence band (VB) are easily photoexcited to the conduction band (CB), leaving holes (h+) in the VB.41 The difference between the CB and VB edge potentials of these semiconductors42,43 allows the electrons to first transfer from the CB of C–ZnMoO4 to the CB of C–ZnS; after that, all the electrons on the CB of C–ZnS will continue to transfer to the CB of MoS2. During the process, the holes leaving the VB of C–ZnS first move in the opposite direction to the electrons to the VB of C–ZnMoO4, which could result in effective electron–hole separation and the holes will finally transfer to the VB of MoS2. It is harder for photogenerated electrons and holes to recombine in the indirect band gap of the MoS2 nanosheets. Thus, the lives of photogenerated electrons and holes are prolonged, which results in enhanced photocatalytic activity. And, it should be noted that the C-doping of the structure can improve the visible-light response, which can also enhance the photocatalytic activity.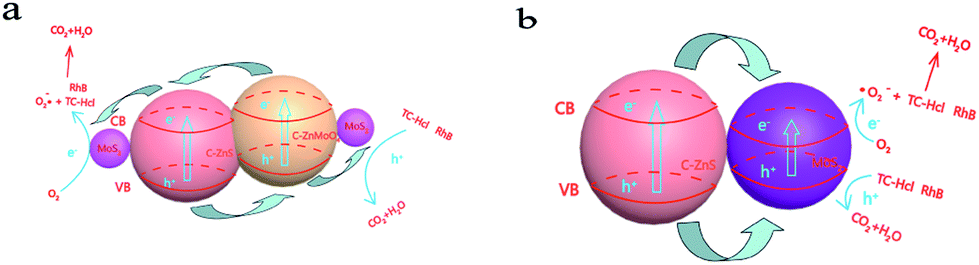 | ||
| Scheme 2 (a) Schematic illustration of the charge transfer in the C–ZnS/ZnMoO4@MoS2 composites and (b) C–ZnS/MoS2 composites under full spectrum light irradiation. | ||
C–ZnS/MoS2 is a two-phase catalyst, so the transfer of electrons and holes only occurs in two phases, which may decrease the separation of the electron–hole pairs and increase the recombination of the photo excitation pairs. Compared to C–ZnS/ZnMoO4@MoS2, C–ZnS/ZnMoO4@MoS2 is a three-phase catalyst consisting of C–ZnS, C–ZnMoO4 and MoS2, so the contact between C–ZnS and C–ZnMoO4 will be closer and intersecting compared to the physical contact of general heterojunction photocatalysts, because they were prepared from ZIF-8 derived C–ZnO at the same time using a direct one-pot route. The longer transfer steps and closer contact of the catalyst can facilitate the separation of electron–hole pairs through closer contact between the different semiconductors and enhance the photocatalytic performance of C–ZnS/ZnMoO4@MoS2.
4. Conclusion
In summary, a series of ZIF-8 derived C–ZnS/ZnMoO4@MoS2 and C–ZnS/MoS2 catalysts were successfully produced by an efficient and facile one-pot strategy. The as-prepared ZIF-8 derived C–ZnS/ZnMoO4@MoS2 and C–ZnS/MoS2 show enhanced photocatalytic activity in the photocatalytic degradation of TC-H and RhB compared to ZIF-8 derived C–ZnO or pure ZnO. C–ZnS/ZnMoO4@MoS2 also shows better photocatalytic capability over C–ZnS/MoS2. The enhanced photocatalytic capability is due to the C-doped structure and the heterojunction structure, which improve the visible-light response and facilitate the separation of photogenerated electron–hole pairs and reduce the recombination of photogenerated charge carriers. We believe that such novel ZIF-8 derived C–ZnS/ZnMoO4@MoS2 and C–ZnS/MoS2 heterojunction structures can potentially be developed into a new-type of photocatalyst.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 21661024), the Open Funds of the State Key Laboratory of Rare Earth Resource Utilization (Grant No. RERU2017009), the Natural Science Foundation of Inner Mongolia (Grant No. 2018MS02004), the Projects of Inner Mongolia Scientific research (Grant No. NJZZ14003) and the Student's Platform of Inner Mongolia University.Notes and references
- K. Kümmerer, J. Antimicrob. Chemother., 2003, 52, 5–7 CrossRef.
- M. Seifrtová, L. Nováková, C. Lino, A. Pena and P. Solich, Anal. Chim. Acta, 2009, 649, 158–179 CrossRef PubMed.
- P. Shetty, Lancet Infect. Dis., 2002, 2, 704 CrossRef.
- B. Luo, D. Xu, L. Di, G. Wu, M. Wu, W. Shi and C. Min, ACS Appl. Mater. Interfaces, 2015, 7, 17061 CrossRef CAS.
- A. K. Sarmah, M. T. Meyer and A. B. Boxall, Chemosphere, 2006, 65, 725–759 CrossRef CAS.
- R. Daghrir and P. Drogui, Environ. Chem. Lett., 2013, 11, 209–227 CrossRef CAS.
- X. Wang, Y. Zhang and S. Song, Angew. Chem., Int. Ed., 2016, 55, 4542–4546 CrossRef CAS.
- J. Rivera-Utrilla, C. V. Gómez-Pacheco, M. Sánchez-Polo, J. J. López-Peñalver and R. Ocampo-Pérez, J. Environ. Manage., 2013, 131, 16–24 CrossRef CAS.
- K. Yang, Q. Yue, J. Kong, P. Zhao, Y. Gao, K. Fu and B. Gao, Chem. Eng. J., 2016, 285, 319–330 CrossRef.
- S. F. Pan, M. P. Zhu, J. P. Chen, Z. H. Yuan, L. B. Zhong and Y. M. Zheng, Sep. Purif. Technol., 2015, 153, 76–83 CrossRef CAS.
- C. Reyes, J. Fernández, J. Freer, M. A. Mondaca, C. Zaror, S. Malato and H. D. Mansilla, J. Photochem. Photobiol., A, 2008, 184, 141–146 CrossRef.
- M. Yan, Y. Hua, F. Zhu, L. Sun, W. Gu and W. D. Shi, Appl. Catal., B, 2017, 206, 531–537 CrossRef CAS.
- S. Liu, M. Lim and R. Amal, Chem. Eng. Sci., 2014, 3, 46–52 CrossRef.
- K. Shen, L. Chen, J. Long, W. Zhong and Y. Li, ACS Catal., 2015, 5, 150730103814009 Search PubMed.
- J. Long, K. Shen, L. Chen and Y. Li, J. Mater. Chem. A, 2016, 4, 10254–10262 RSC.
- B. Li, H. M. Wen, W. Zhou and B. Chen, J. Phys. Chem. Lett., 2014, 5, 3468 CrossRef CAS PubMed.
- K. Huang, Z. Dong, Q. Li and W. Jin, Chem. Commun., 2013, 49, 10326–10328 RSC.
- K. Tan, N. Nijem, P. Canepa, Q. Gong, J. Li, T. Thonhauser and Y. J. Chabal, Chem. Mater., 2016, 24, 3153–3167 CrossRef.
- F. G. Cirujano, A. Corma and F. X. Llabrés i Xamena, Chem. Eng. Sci., 2015, 124, 52–60 CrossRef CAS.
- A. Corma, H. García and F. X. L. I. Xamena, Chem. Rev., 2010, 41, 4606–4655 CrossRef.
- D. Farrusseng, S. Aguado and C. Pinel, Chem. Inf., 2010, 48, 7502–7513 Search PubMed.
- R. Jin, Y. Yang, Y. Xing, L. Chen, S. Song and R. Jin, ACS Nano, 2014, 8(4), 3664–3670 CrossRef CAS.
- N. M. Mahmoodi, J. Abdi, M. Oveisi, M. A. Asli and M. Vossoughi, Mater. Res. Bull., 2018, 100, 357–366 CrossRef CAS.
- P. Hu, C. Chen and J. Song, Mater. Sci. Semicond. Process., 2018, 77, 40–49 CrossRef CAS.
- Z. Z. Lu, R. Zhang, Y. Z. Li, Z. J. Guo and H. G. Zheng, J. Am. Chem. Soc., 2011, 133, 4172–4174 CrossRef CAS.
- J. Yao and H. Wang, Chem. Soc. Rev., 2014, 43, 4470–4493 RSC.
- J. Yao, D. Dong, D. Li, L. He, G. Xu and H. Wang, Chem. Commun., 2011, 47, 2559–2561 RSC.
- T. G. Glover, G. W. Peterson and B. J. Schindler, Chem. Eng. Sci., 2011, 66(2), 163–170 CrossRef.
- P. Horcajada, T. Chalati, C. Serre, B. Gillet, C. Sebrie, T. Baati, J. F. Eubank, D. Heurtaux, P. Clayette and C. Kreuz, Nat. Mater., 2010, 9, 172–178 CrossRef CAS.
- G. S. Cláudia, L. Ignacio, C. Avelino and G. Hermenegildo, Chem.–Eur. J., 2010, 16, 11133–11138 CrossRef.
- Y. Du, R. Z. Chen, J. F. Yao and H. T. Wang, J. Alloys Compd., 2013, 551, 125–130 CrossRef CAS.
- Y. Feng, H. Lu, X. Gu, J. Qiu, M. Jia, C. Huang and J. Yao, J. Phys. Chem. Solids, 2017, 102, 110–114 CrossRef CAS.
- C. Feng, J. Ma and H. Li, Mater. Res. Bull., 2009, 449, 1811–1815 CrossRef.
- B. Zhu, B. Lin, Y. Zhou, P. Sun, Q. Yao, Y. Chen and B. Gao, J. Mater. Chem. A, 2013, 2, 3819–3827 RSC.
- S. Y. Zhang, H. Liu, C. C. Sun, P. F. Liu, L. C. Li, Z. H. Yang, X. Feng, F. W. Huo and X. H. Lu, J. Mater. Chem. A, 2015, 3, 5294–5298 RSC.
- J. Yao, M. He and H. Wang, CrystEngComm, 2015, 17, 4970–4976 RSC.
- M. He, J. Yao, Q. Liu, K. Wang, F. Chen and H. Wang, Microporous Mesoporous Mater., 2014, 184, 55–60 CrossRef CAS.
- B. Liu, H. Shioyama, T. Akita and Q. Xu, J. Am. Chem. Soc., 2008, 130, 5390–5391 CrossRef CAS PubMed.
- B. Lin, H. An, X. Yan, T. Zhang, J. Wei and G. Yang, Appl. Catal., B, 2017, 210, 173–183 CrossRef CAS.
- D. Lin, H. Wu, R. Zhang, W. Zhang and W. Pan, J. Am. Ceram. Soc., 2010, 93, 3384–3389 CrossRef CAS.
- S. Yang, S. Xu, D. Chen and W. Shi, Appl. Catal., B, 2017, 204, 602–610 CrossRef CAS.
- J. Zhang, X. Mao, W. Xiao and Y. Zhuang, Chin. J. Catal., 2017, 38, 2009–2020 CrossRef CAS.
- Y. Yang, Y. Zhang, Z. Fang, L. Zhang, Z. Zheng, Z. Wang, W. Feng, S. Weng, S. Zhang and P. Liu, ACS Appl. Mater. Interfaces, 2017, 9, 6950–6958 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra06591a |
| This journal is © The Royal Society of Chemistry 2019 |