
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectronic and optical properties of BxCyNz hybrid α-graphynes†
A. Freitas *a,
L. D. Machadoa,
C. G. Bezerraa,
R. M. Tromera and
S. Azevedob
*a,
L. D. Machadoa,
C. G. Bezerraa,
R. M. Tromera and
S. Azevedob
aDepartamento de Física, Universidade Federal do Rio Grande do Norte, 59072-970, Natal, RN, Brazil. E-mail: alilianefisica@yahoo.com.br
bDepartamento de Física, CCEN, Universidade Federal da Paraíba, Caixa Postal 5008, 58051-970, João Pessoa, PB, Brazil
First published on 31st October 2019
Abstract
Hybrid two-dimensional (2D) materials composed of carbon, boron, and nitrogen constitute a hot topic of research, as their flexible composition allows for tunable properties. However, while graphene-like hybrid lattices have been well characterized, systematic investigations are lacking for various 2D materials. Hence, in the present contribution, we employ first-principles calculations to investigate the structural, electronic and optical properties of what we call BxCyNz hybrid α-graphynes. We considered eleven structures with stoichiometry BC2N and varied atomic arrangements. We calculated the formation energy for each arrangement, and determined that it is low (high) when the number of boron-carbon and nitrogen-carbon bonds is low (high). We found that the formation energy of many our structures compared favorably with a previous literature proposal. Regarding the electronic properties, we found that the investigated structures are semiconducting, with band gaps ranging from 0.02 to 2.00 eV. Moreover, we determined that most of the BxCyNz hybrid α-graphynes proposed here strongly absorb infrared light, and so could potentially find applications in optoelectronic devices such as heat sensors and infrared filters.
I. Introduction
In the last decade, two-dimensional (2D) nanoscale materials have attracted considerable interest due to their innovative physical properties and potential applications in many emerging technologies. Examples of 2D materials which have already been synthesized and extensively studied include graphene1,2 and hexagonal boron nitride (h-BN).3,4 Both structures are composed of sp2-hybridized atoms arranged in a honeycomb lattice. Graphene is a zero-band-gap semiconductor, which presents high electron mobility, high thermal conductivity, high strength, and flexibility.5–8 On the other hand, h-BN is a wide-gap semiconductor (>4 eV), with mechanical properties similar to graphene, and high resistance to oxidation and degradation.9–11 In this context, it is worth pointing out that the substitution of C atoms in graphene by B and N atoms leads to the formation of BxCyNz hexagonal hybrid sheets,12–14 which have also been extensively investigated and have recently been synthesized.15,16 They present a variety of energy gaps (<2 eV) that are intermediate between those found in graphene and h-BN. Moreover, these materials can exhibit either metallic or semiconducting behavior, depending on the atomic arrangement of C, B, and N atoms.17–21 Hence, BxCyNz hexagonal sheets are promising materials for the manufacture of nanoscale electronic devices.Following the advent of two-dimensional honeycomb lattices, in recent years intense research has rapidly expanded the family of 2D materials. New structures include graphyne,22,23 graphenylene,24 phagraphene,25 T-graphene26 and others.27 These materials have been theoretically proposed and some have already been synthesized. First-principles calculations predict interesting mechanical, optical, and electronic properties for these structures, making them promising candidates for future technological applications. Among the cited examples, the group of materials called “graphynes” has attracted much attention.
Graphynes are 2D structures composed of a mixture of sp and sp2-hybridized carbon atoms. This is in contrast to graphene, where all carbon atoms are sp2-hybridized. Graphynes were initially predicted by Baughman et al.28 and it has since received considerable attention. Theoretically, these structures are formed by the combination of carbon hexagons with polyyne-like carbon linear chains (…–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–CC–…).29 Although graphyne sheets have not yet been synthesized, small graphyne flakes have already been successfully obtained.30–33 In addition, large area films of graphdiyne, a related material, were produce on a copper substrate via a cross-coupling reaction by Li et al.34 The sp and sp2 carbon atoms can be combined in various ways, and so far four graphyne types have been investigated, namely α-, β-, γ-, and 6, 6, 12-. The α-, β-, and 6, 6, 12-graphynes are metallic, and all feature Dirac cones in their electronic band structure;35,36 meanwhile, γ-graphyne is a semiconductor.37 All four of them have been proposed as suitable materials for applications in nanoelectronics. Additional applications proposed for graphynes include: energy storage,38 use as battery anodes,39 gas separation,40 and water desalination.41 Boron nitride analogues of the α-, β-, γ-, and 6, 6, 12- graphynes have also been investigated.42–44 These structures, termed BNynes, are formed by the combination of BN hexagonal rings with BN linear chains (…–BN–BN–…). First-principles calculations, performed by Zhang et al.45 and Cao et al.,46 have shown that BNynes are wide-gap semiconductors (>4 eV), with strong absorption in a wide UV photon-energy region.
C–CC–…).29 Although graphyne sheets have not yet been synthesized, small graphyne flakes have already been successfully obtained.30–33 In addition, large area films of graphdiyne, a related material, were produce on a copper substrate via a cross-coupling reaction by Li et al.34 The sp and sp2 carbon atoms can be combined in various ways, and so far four graphyne types have been investigated, namely α-, β-, γ-, and 6, 6, 12-. The α-, β-, and 6, 6, 12-graphynes are metallic, and all feature Dirac cones in their electronic band structure;35,36 meanwhile, γ-graphyne is a semiconductor.37 All four of them have been proposed as suitable materials for applications in nanoelectronics. Additional applications proposed for graphynes include: energy storage,38 use as battery anodes,39 gas separation,40 and water desalination.41 Boron nitride analogues of the α-, β-, γ-, and 6, 6, 12- graphynes have also been investigated.42–44 These structures, termed BNynes, are formed by the combination of BN hexagonal rings with BN linear chains (…–BN–BN–…). First-principles calculations, performed by Zhang et al.45 and Cao et al.,46 have shown that BNynes are wide-gap semiconductors (>4 eV), with strong absorption in a wide UV photon-energy region.
Among the investigated graphynes and BNynes, α-graphyne and α-BNyne have received considerable attention.47–50 To obtain α-graphyne, carbon linear chains (containing n atoms) are inserted in each C–C bond of graphene, leading to a honeycomb-like lattice with large hexagonal rings. Corner atoms are tri-coordinated in each hexagon, as in graphene. Similarly, α-BNyne is obtained by the insertion of BN linear chains in each B–N bond of h-BN. Ongun et al. have carried out ab initio calculations to test the structural stability and to determine the electronic and mechanical properties of α-graphyne and α-BNyne, as a function of the number of atoms n contained in the linear chains.51 The authors calculated the phonon modes and performed finite-temperature molecular dynamics, and determined that both α-graphyne and α-BNyne are stable for even values of n and unstable for odd values of n. High stability was found for n = 2. Regarding the electronic properties, they found that all α-graphynes are metallic, while all α-BNynes are wide-gap semiconductors.
An interesting possibility regarding α-graphyne and α-BNyne would be to combine the properties of these materials through the formation of BxCyNz hybrid sheets, in the same way as done for graphene and h-BN.12–14,17–21 Recently, the electronic structure and the structural stability of α-graphynes, doped with different concentrations of BN pairs, was investigated via first-principles calculations by Yun et al.52 and Deng et al.53 Both authors observed the opening of an energy-gap in the electronic spectrum of α-graphyne, for any BN pair concentration. Additionally, it was determined that the band gap was proportional to the dopant concentration. Thus, α-graphynes doped with BN pairs are semiconductors with tunable band gap. On the other hand, to our knowledge, there are presently no studies in the literature investigating island- and stripe-like atomic arrangements of B, C, and N atoms in α-graphyne sheets. Such atomic arrangements minimize formation energy and maximize stability in BxCyNz hybrid graphenes.12 With this motivation in mind, and also considering the growing interest in BxCyNz hybrid nanostructures, in this paper we perform first-principles calculations to investigate the stability, as well as the electronic and optical properties, of what we call BxCyNz hybrid α-graphynes.
II. Computational details and methods
Fig. 1 shows the BxCyNz hybrid α-graphynes investigated in this work. Similar to graphene, these structures are monolayers with hexagonal rings and three-coordinated atoms in each corner. We considered different atomic arrangements for an unit cell with 96 atoms. For comparison, unit cells for α-graphyne and α-BNyne contain only 8 atoms.51 The large unit cells allow us to investigate island-like (Fig. 1B and C) and stripe-like (Fig. 1A and D–G) atomic arrangements, as was done by Azevedo et al. for BxCyNz hybrid graphenes.12 In island-like arrangements, we have the presence of (BN)x and Cy units of different sizes and forms; meanwhile, in stripe-like arrangements, we have BN and C stripes connected along a specific direction. Thus, the BxCyNz hybrid α-graphynes have different numbers of B–N, C–C, B–C, and N–C bonds. The chosen atomic arrangements preclude the presence of energetically unfavorable B–B and N–N bonds.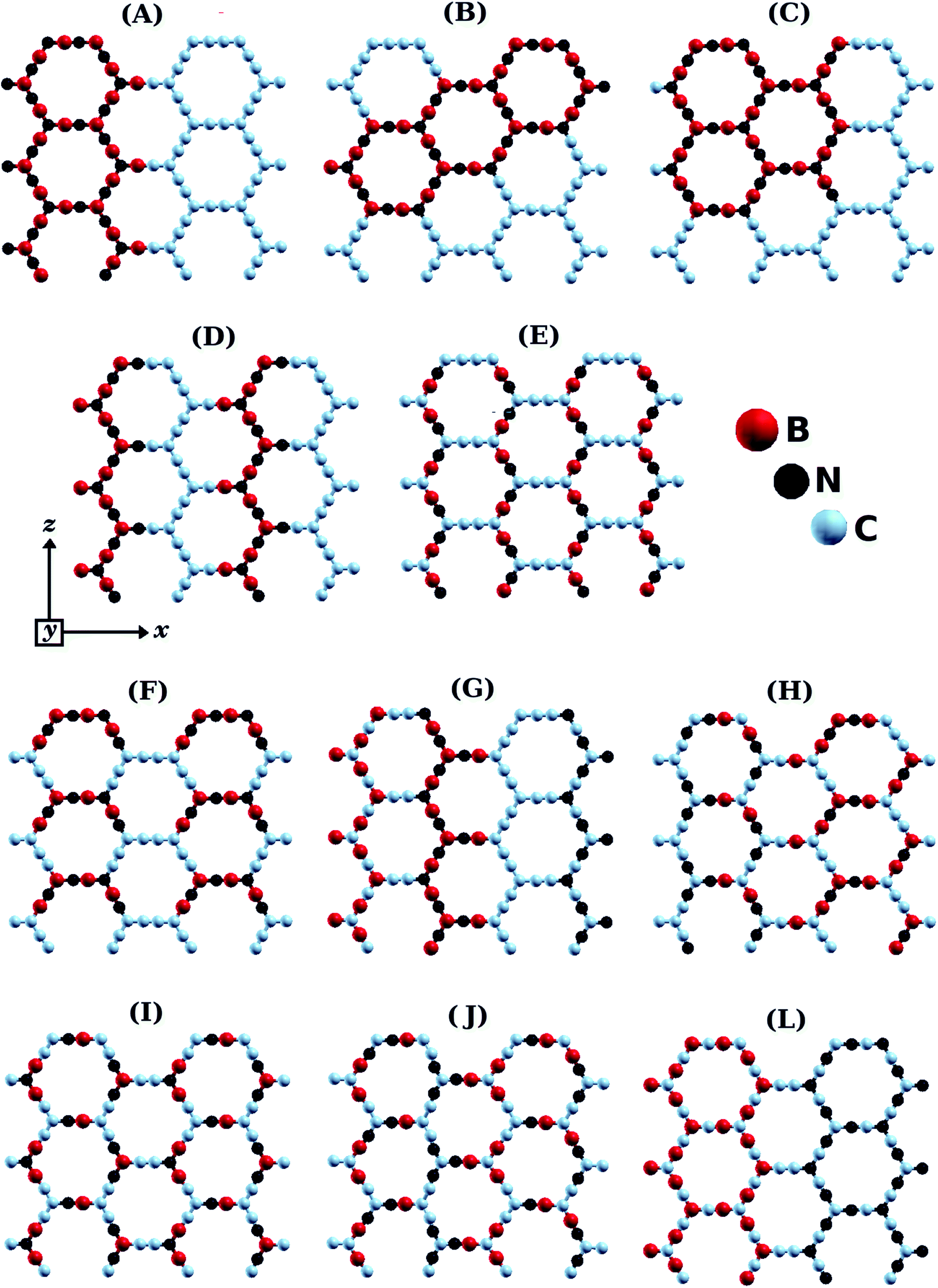 | ||
| Fig. 1 Illustration of the optimized BxCyNz hybrid α-graphynes with different atomic arrangements. | ||
The structures shown in Fig. 1 were then optimized using first-principles calculations based on density functional theory (DFT), as implemented in the SIESTA code.54,55 We used the Generalized Gradient Approximation (GGA-PBE) for the exchange-correlation term.56,57 A linear combination of numerical atomic orbitals was used to represent a double-ζ basis set with polarized functions (DZP). To modulate the strong interactions between electrons and core ions, we used the norm-conserving Troullier Martins pseudopotential58 in the Kleinman–Bylander factorized form.59 All calculations use a mesh cutoff of 150 Ry, with the Brillouin zone sampled by 3 × 2 × 2 special k-points. It was adopted a convergence criterion where the self-consistency is achieved when the maximum difference between the output and the input of each element of the density matrix, in a self-consistent field cycle, is smaller than 10−4 eV. The optimization of atomic positions was allowed to proceed until the force on each atom was less than 0.1 eV Å−1. We adopted a rectangular supercell and all calculations were performed at T = 0 K. The unit cell is repeated in the x and y directions, forming an infinite BxCyNz hybrid α-graphyne sheet. A large vacuum region of 20 Å was used along of the z direction, to avoid spurious interactions between two adjacent periodic images.
Note, however, that the DFT-LDA/GGA approach is known to underestimate band gaps,60 and this can lead to incorrect predictions regarding the optical properties of a material. Several approaches have been developed to address this issue, including the so-called hybrid functionals and the GW approximation.61–64 In particular, the hybrid functional HSE06 has been found to give a significantly improved description of the electronic properties of nanomaterials.65,66 Thus, for the BxCyNz hybrid α-graphynes investigated in this work, we also performed calculations of the electronic properties using the HSE06 functional, as implemented in the GAUSSIAN16 code.67,68 A comparison between the results obtained with the GGA and HSE06 functionals is given below.
III. Results and discussion
In order to estimate the structural stability of the BxCyNz hybrid α-graphynes analyzed in the present study, we calculate the formation energy EForm through a thermodynamic approach, which is based on the prior determination of the chemical potentials of the atomic species involved in the synthesis reaction. The details of this approach are described in ref. 13 and 14. The formation energy is defined by the following expression
 | (1) |
| μBN = μB + μN and μCC = μC + μC, | (2) |
| EForm = (ETot − nBNμBN − nCCμCC)/natom. | (3) |
The formation energies, obtained by using eqn (3), are quoted in the second column of Table 1. In this context, we observe that in each structure there is a relationship between EForm and the number of “regular” chemical bonds, C–C and B–N, and “wrong” chemical bonds, C–B and C–N. Thus, in order to take into account this fact, we define the ratio Rb/Wb, where Rb and Wb denote the number of regular and wrong chemical bonds, respectively. This relationship has also been observed in other types of hybrid nanostructures with BxCyNz stoichiometry.17,18,69 The value of the ratio Rb/Wb, for each structure, is displayed in the fifth column of Table 1, where we notice that the formation energy increases as the number of regular bonds decreases, i.e., when the ratio Rb/Wb decreases.
| Structure | Eform (eV nT−1) | Rb | Wb | Rb/Wb | Eg (GGA) (eV) | Eg (HSE06) (eV) |
|---|---|---|---|---|---|---|
| (A) |  |
102 | 6 | 17 | 0.80 | 1.20 |
| (B) | 0.08 | 98 | 10 | 9.8 | 0.21 | 0.28 |
| (C) | 0.08 | 98 | 10 | 9.8 | 0.26 | 0.47 |
| (D) | 0.10 | 96 | 12 | 8.0 | 1.43 | 2.00 |
| (E) | 0.13 | 84 | 24 | 3.5 | 1.30 | 1.80 |
| (F) | 0.14 | 60 | 48 | 1.3 | 1.10 | 1.93 |
| (G) | 0.19 | 60 | 48 | 1.3 | 0.00 | 0.02 |
| (H) | 0.22 | 54 | 54 | 1.0 | 0.00 | 0.09 |
| (I) | 0.25 | 54 | 54 | 1.0 | 0.29 | 0.34 |
| (J) | 0.32 | 42 | 66 | 0.6 | 0.10 | 0.21 |
| (L) | 0.42 | 6 | 102 | 0.1 | 0.11 | 0.05 |
The lengths of the B–N, C–C, B–C, and N–C bonds in the relaxed BxCyNz hybrid α-graphynes are shown in Fig. 2. Since the hybridization state for a given B, C, or N atom can be either sp or sp2, atoms of a given element are no longer chemically equivalent, as they are in the cases of graphene1 and h-BN.11 Depending on the atomic arrangement of the sheet, we can have up to three different types of bonds: simple (Xsp–Ysp2), double (Xsp2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Ysp2), and triple (XspYsp), where X and Y are variables that represent an B, C, or N atom. Structure A is the only that exhibits a lattice with the three types of bonds mentioned above. In this case, we observe a mixture of the following atomic chains: –C–CC–C–, –B–NB–N–, and –CCBN– (see Fig. 2). The bonds in these chains are similar to those found in polyyne- or cumulene-type carbon linear chains. On the other hand, in all other structures, B, C, and N atoms are linked together through simple and triple bonds; that is, all atomic chains are of the type –X–YZ–W– (X, Y, Z, and W are variables that represent B, C, and N atoms). Moreover, we find B–N and C–C bond lengths which are slightly smaller than those found for α-graphyne and α-BNyne in ref. 29 and 51. These subtle differences may be caused by different software performance or parameter setting.
Ysp2), and triple (XspYsp), where X and Y are variables that represent an B, C, or N atom. Structure A is the only that exhibits a lattice with the three types of bonds mentioned above. In this case, we observe a mixture of the following atomic chains: –C–CC–C–, –B–NB–N–, and –CCBN– (see Fig. 2). The bonds in these chains are similar to those found in polyyne- or cumulene-type carbon linear chains. On the other hand, in all other structures, B, C, and N atoms are linked together through simple and triple bonds; that is, all atomic chains are of the type –X–YZ–W– (X, Y, Z, and W are variables that represent B, C, and N atoms). Moreover, we find B–N and C–C bond lengths which are slightly smaller than those found for α-graphyne and α-BNyne in ref. 29 and 51. These subtle differences may be caused by different software performance or parameter setting.
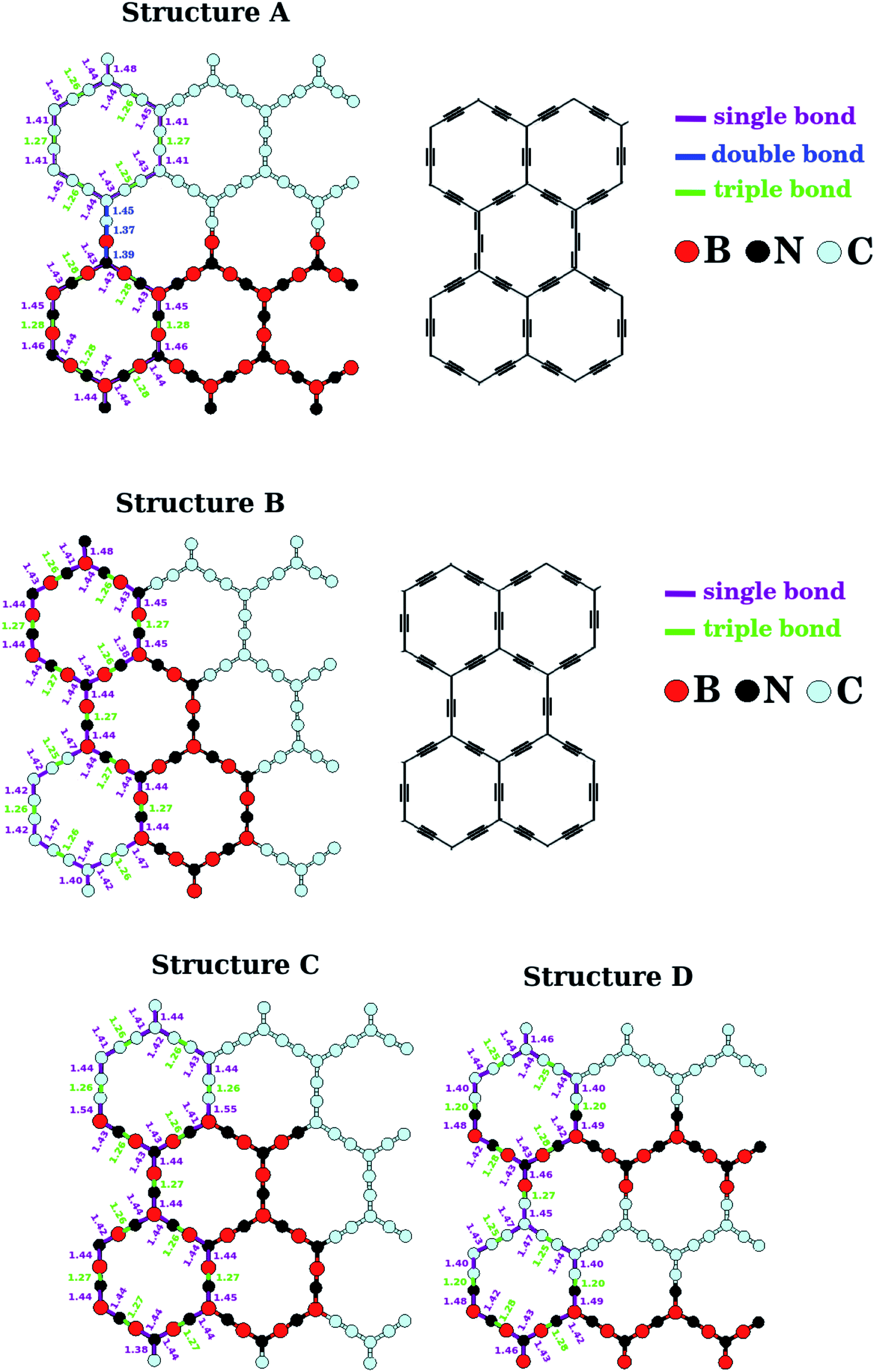 | ||
| Fig. 2 Calculated bond length for the monolayers A, B, C and D shown in Fig. 1. The bond lengths illustrated repeat themselves periodically along the zigzag direction. | ||
From the data in Table 1, we can see that structure A, with the highest Rb/Wb ratio, presents the lowest value of EForm, and so is the most stable structure. Following this line of reasoning, structures B and C, which exhibit the second lowest Rb/Wb ratio, are also the second most stable structures (the energy difference in comparison to structure A is only 0.02 eV/nat). Structure A is stable due to its atomic arrangement, with stripes of BN and C hexagons connected in a way that minimizes the number of atoms in the interface between the two materials; this arrangement also introduces single, double, and triple bonds into the lattice (see Fig. 2). On the other hand, structures J and L, which have the highest values of EForm (above 0.30 eV/nat), are the most unstable. The common feature for these structures is the low Rb/Wb ratio, which, according to our results, is not a favorable trend for BxCyNz hybrid α-graphynes. We have also calculated the formation energy for a structure based on an α-graphyne motif that was investigated by Yun et al.52 In particular, we examined the 2BN-αG monolayer,52 which has B, N, and C concentrations equivalent to those found in our structures. By using eqn (3), we found a formation energy of 0.14 eV/nat for 2BN-αG. By contrast, structure A has a formation energy of just 0.06 eV/nat. This result is not surprising, as the Rb/Wb ratio for 2BN-αG is equal to 2, a rather low value.
We also investigated the dynamical stability of the five lowest energy structures (A, B, C, D, and E) through Ab Initio Molecular Dynamics (AIMD) simulations, using the SIESTA code. This is a procedure that has been previously used to predict the thermal stability of materials.70,71 In our calculations, we evolved each structure in the NPT ensemble for 2 ps, with a time step of 1 fs. Temperature and in-plane pressure components were controlled using a Nosé–Hoover thermostat and a Parrinello–Rahman barostat. In Fig. 3(a), we present snapshots of the structures after completion of the AIMD simulations at 300 K. Careful inspection of the snapshots reveals that the thermal motion caused slight structural deformation. More importantly, inspection also reveals that the underlying atomic lattice remained intact, as no bonds were broken during the dynamics. Furthermore, we also carried out calculations at higher temperatures for structure A. Our results, presented in Fig. 3(b), indicate that this particular structure should remain stable even at temperatures as high as 2000 K.
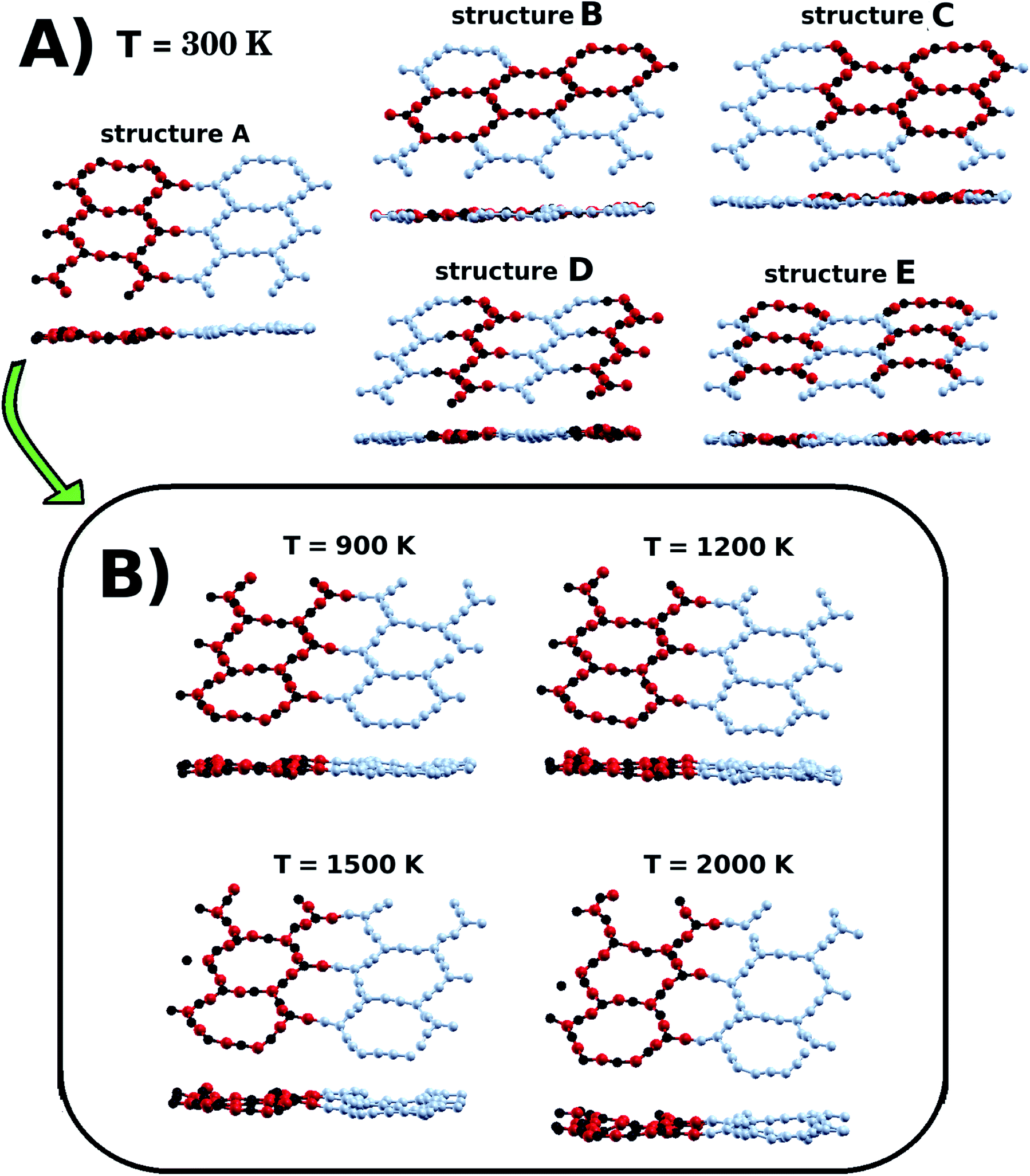 | ||
| Fig. 3 In (A) we have snapshots from molecular dynamics simulations of the BxCyNz hybrid α-graphynes, after thermalization at T = 300 K. In (B) we have the snapshots of structure A after thermalization at various higher temperatures. | ||
Let us now discuss the electronic properties of the eleven BxCyNz hybrid α-graphynes investigated in this work. We performed the calculations using the PBE and HSE06 exchange-correlation functionals using the SIESTA and GAUSSIAN16 packages, respectively. The energy gap values Eg are shown in the sixth and seventh columns of Table 1 – we found Eg values ranging from 0.0 to 1.43 eV for the PBE approach and 0.02 to 2.00 eV for the HSE06 one. We notice that, in general, our results are as expected, since the PBE approach underestimates Eg values. Only for structure L, we obtained a higher value with the PBE approach. However, in this particular case, the difference between the gap values is so small (0.05 eV) that we can consider both approaches predict the same electronic behavior. At the same time, we verified that, in the PBE approach, only structures G and H are metallic. All the other structures behave as semiconductors, in both approaches, irrespective of the atomic arrangement in the unit cell. This behavior is due to the electronic configuration of the lattice, with a mixture of sp2 and sp orbitals. In Fig. 4 we present their calculated electronic band structure, with 100 k-points along the Γ–X–Z direction. Moreover, it is important to note that the energy gap values determined for the BxCyNz hybrid α-graphynes are intermediate to those found in α-graphyne and α-BNyne.51
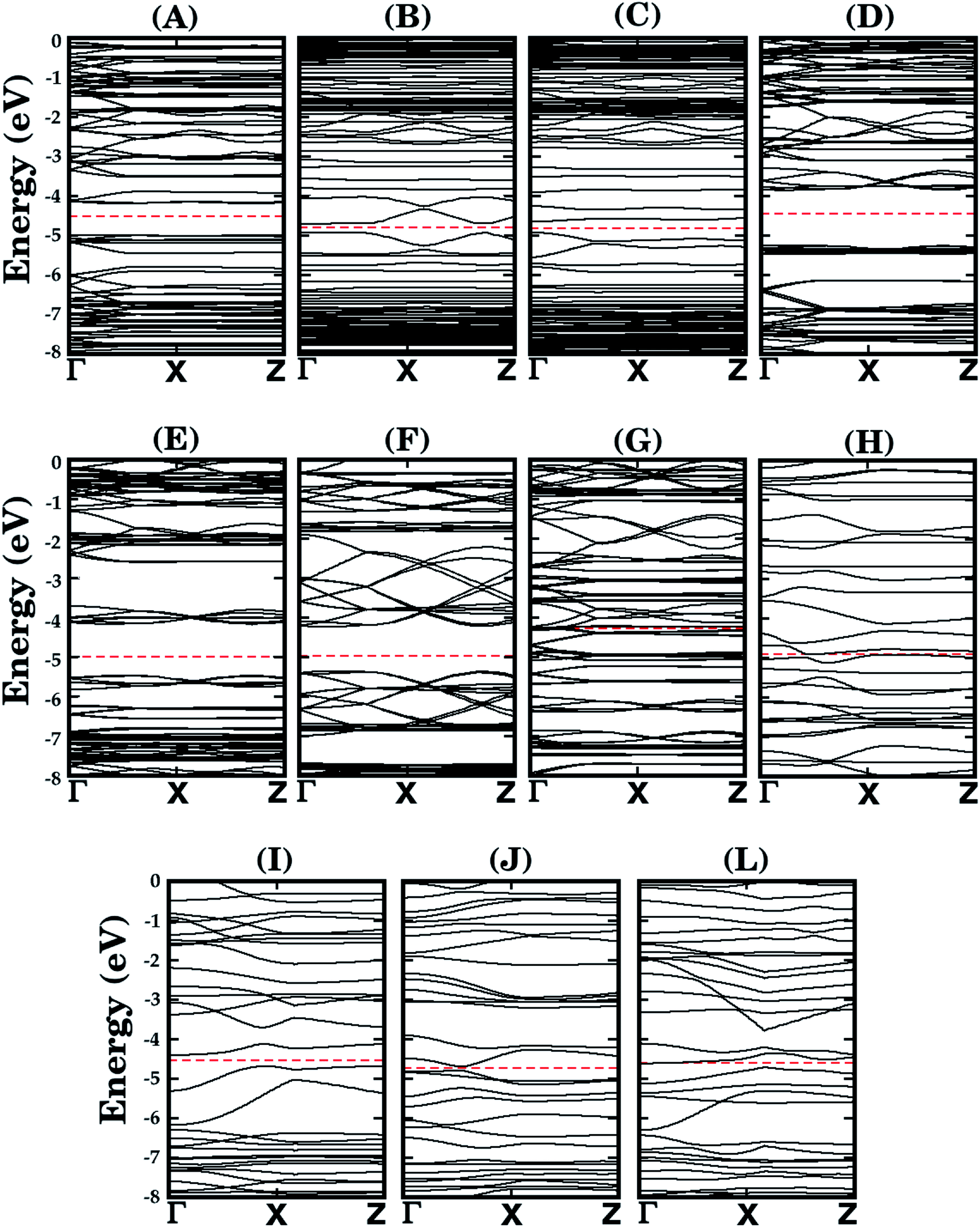 | ||
| Fig. 4 Calculated band structures for the monolayers shown in Fig. 1. The dashed line represents the Fermi level. | ||
Regarding the electronic states near the Fermi energy (Ef), projected density of states (PDOS) calculations reveal that, in general, carbon atoms are the major contributors to these states, see Fig. 5. Complementary local density of states (LDOS) calculations are also displayed in Fig. 5, and they reveal that the bottom of the conduction band and the top of the valence band are associated with pz orbitals of C atoms, in full agreement with PDOS results. Finally, no relationship was observed between energy gap values and Rb/Wb bond ratios, which indicates that the energy gap of a BxCyNz hybrid α-graphyne depends strongly on the particulars of its atomic arrangement. As a side note, in Fig. 6 we show the calculated spin polarization for selected structures. Calculations reveal a spin value of zero for all BxCyNz hybrid α-graphynes; this means their valence bands are completely filled, without unpaired electrons.
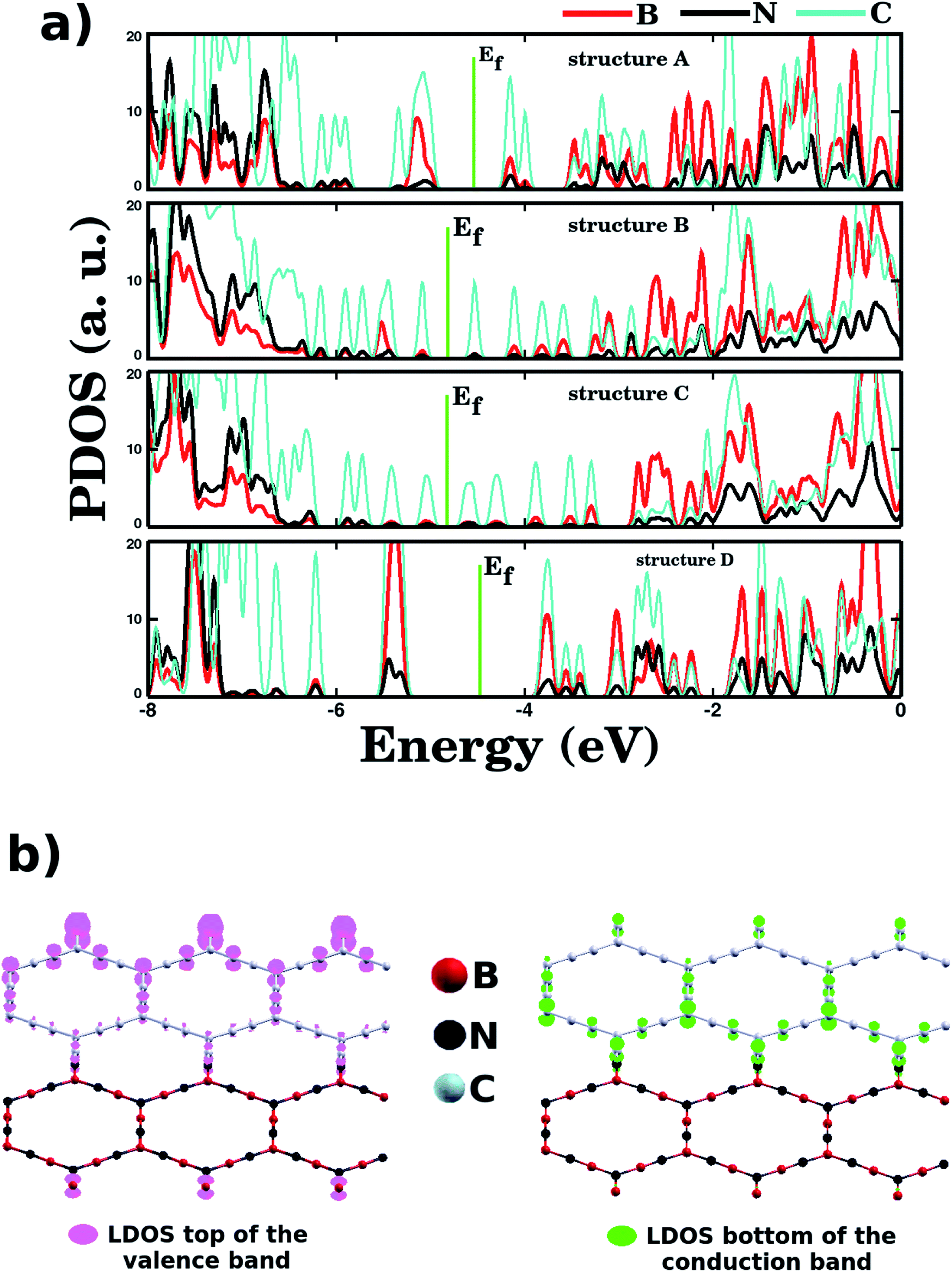 | ||
| Fig. 5 (a) The calculated projected density of states (PDOS) of selected BxCyNz hybrid α-graphynes. The Fermi energy Ef is indicated by the dotted vertical line. (b) The local density of states (LDOS) associated with the bottom of the conduction band and the top of the valence band for the α-graphyne (A) structure. | ||
 | ||
| Fig. 6 Densities of states (spin up and down) for selected BxCyNz hybrid α-graphynes. The total spin is zero for all investigated structures. | ||
In order to analyze the optical properties of the BxCyNz hybrid α-graphynes we have calculated the optical absorption spectrum as function of the photon energy. The analysis of this spectrum is an important starting point for predicting if the investigated structures are promising for future applications in optoelectronic devices. Firstly, the frequency-dependence of the complex dielectric function ε(ω) = ε1(ω) − iε2(ω) is calculated. The imaginary part is directly proportional to the optical absorption spectrum, which is calculated from the time dependent perturbation theory in the simple dipole approximation, which is given by
 | (4) |
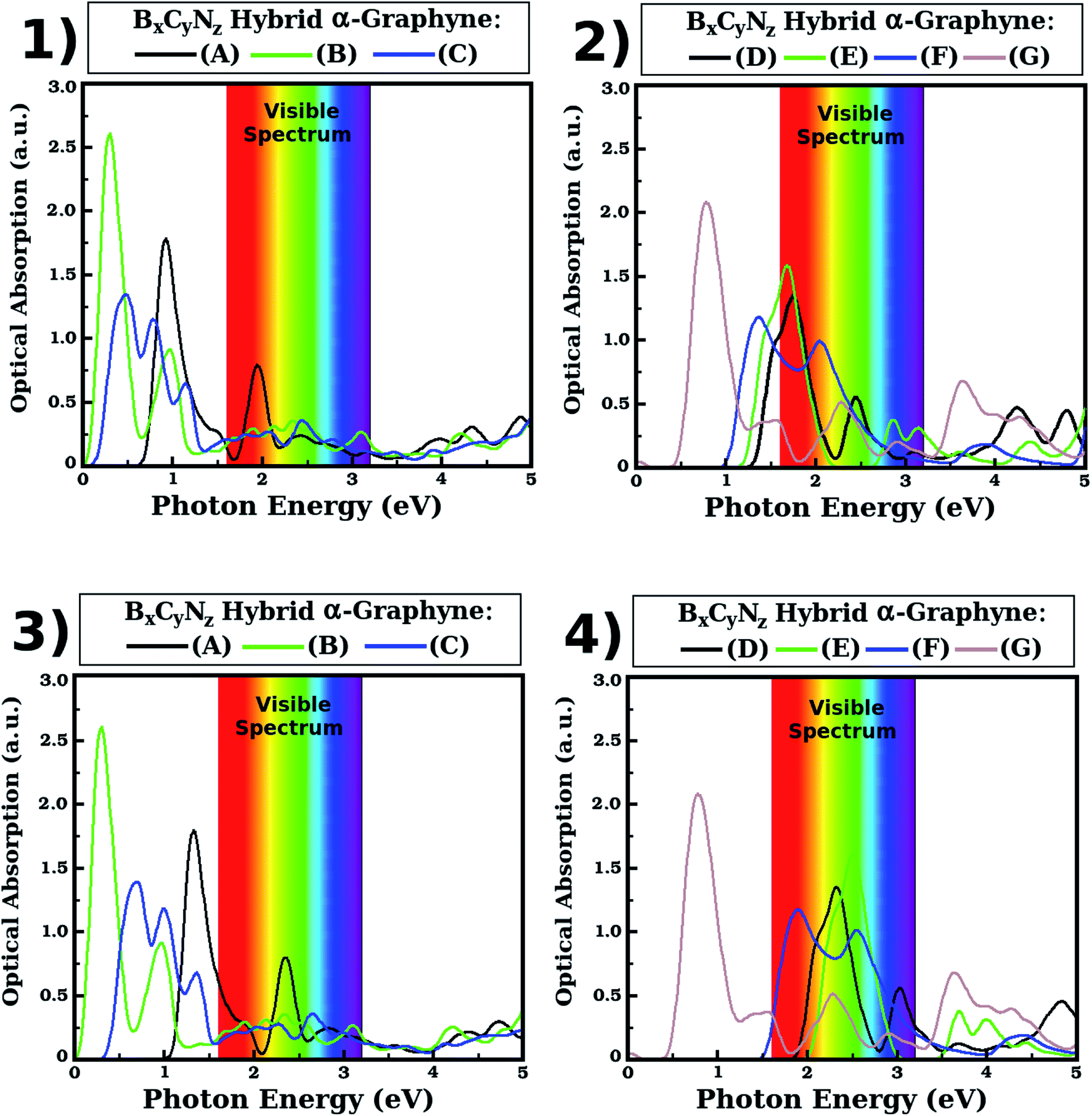 | ||
| Fig. 7 Optical absorption spectrum as function of the photon energy for selected BxCyNz hybrid α-graphynes, with the light polarization perpendicular to the α-graphynes's plane. At (1) and (2), the absorption spectrum was obtained from calculations with the PBE functional. At (3) and (4), the absorption spectrum was corrected using the scissor operator available in the SIESTA code. This correction method uses the difference between the energy gap values obtained with the HSE06 and PBE functionals. | ||
We found that the optical absorption of the BxCyNz hybrid α-graphynes is in the infrared region for the three lowest energy structures (A, B and C) and for structure G, while for some of the other structures absorption occurs in the visible region. Indeed, as can be seen in Fig. 7, for structures A, B, C and G the first absorption peak occurs in the infrared at about 1.34, 0.30, 0.70 and 0.78 eV. For structures D, E and F, the first absorption peak occurs in the visible region at about 2.32 (green), 2.51 (green) and 1.90 (red) eV. These peaks correspond to a first optical transition between the top of the valence band and the bottom of the conduction band in carbon atoms, as we can see in the projected density of states (Fig. 5). Given that the maximum optical absorption occurs in the infrared for the three lowest energy hybrid α-graphynes, we note that these materials are likely good candidates for use in nanoscale optical devices that operate in this region of the electromagnetic spectrum, such as heat sensors. They could also be potentially used in infrared radiation filters, which are typically used to absorb heat.
Regarding the other absorption peaks, we note that some structures still present peaks in the infrared region. Specifically, the second peak for structure B is found approximately at 0.98 eV, while the second and third peaks for structure C occur approximately at 1.00 and 1.36 eV. Meanwhile, the second absorption peak for structures A, D and F occurs in the visible region, at about 2.36 (green), 3.04 (violet) and 2.54 (green) eV. For structures E and G, the second absorption peak is in the ultraviolet region (at about 3.70 and 3.64 eV, respectively). Additionally, with the exception of structure C, we found that the third absorption peak occurs either in the visible (green) or in the ultraviolet region.
Additionally, we carried out first-principles calculations to investigate the electronic properties of a particular B–C–N hybrid α-graphyne nanoribbon, which is illustrated in Fig. 8. Such structure was constructed based on the atomic arrangement of monolayer A (see Fig. 1), which corresponds to the most stable structure among all investigated BxCyNz hybrid α-graphynes. The proposed nanoribbon is composed of BN and C stripes, which are perpendicular to the periodic direction, with armchair symmetry. Hydrogen atoms were also included at the nanoribbon edges. After the optimization process, notice that C–C and B–N bonds undergo deformation, modifying the shape of the local hexagons. Band structure calculations reveal that this nanoribbon is a semiconductor material, with an energy gap of 0.83 eV, which is very close to that of monolayer A (a difference of 0.03 eV). Finally, observe in the PDOS results (also displayed in Fig. 8) that electronic states near the Fermi level are associated with carbon and boron atoms.
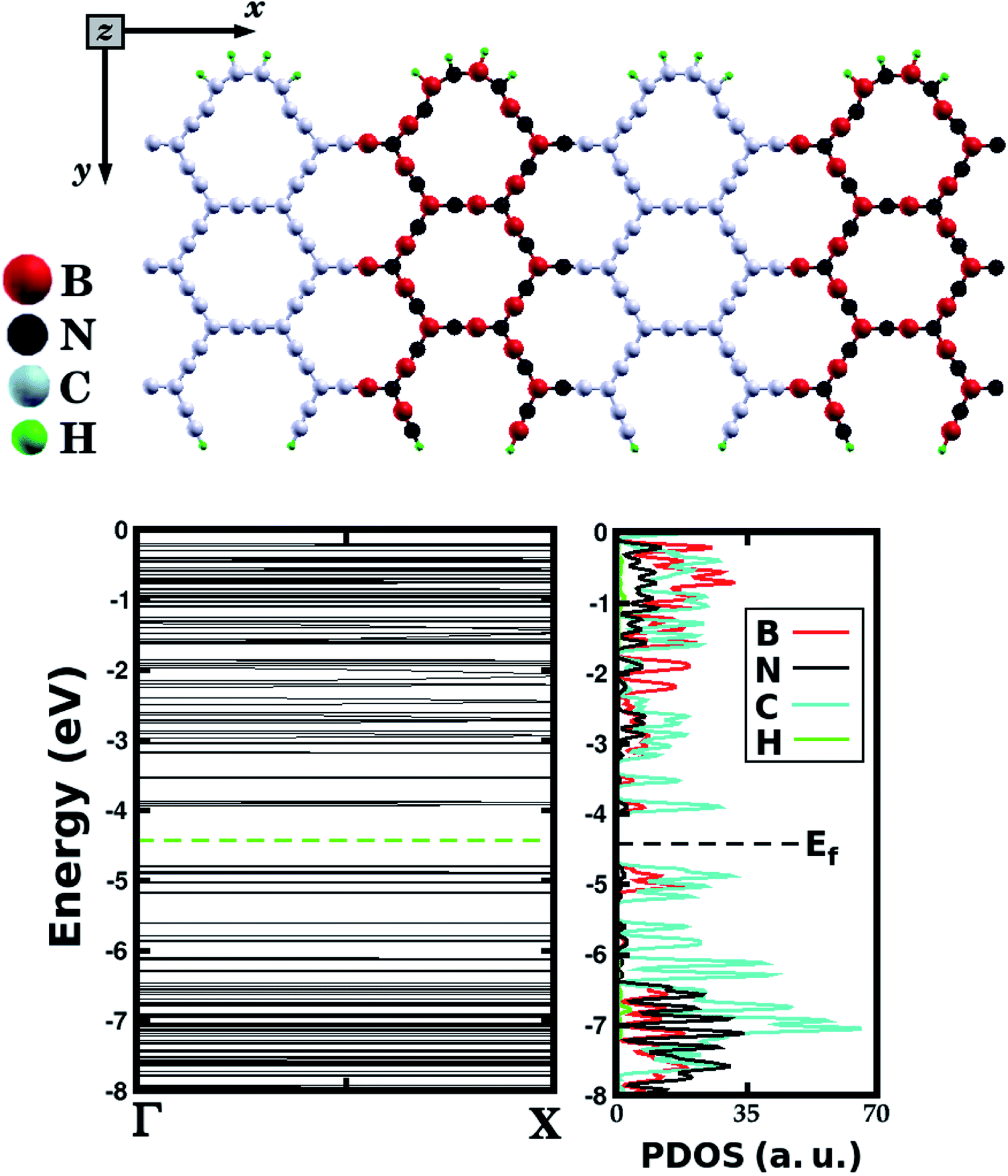 | ||
| Fig. 8 Optimized B–C–N hybrid α-graphyne nanoribbon with armchair edge. The calculated band structure and the projected density of states (PDOS) are also shown. The Fermi energy Ef is indicated by the horizontal dotted line. | ||
IV. Conclusions
In summary, we employed DFT calculations to investigate the stability, as well as the electronic and optical properties, of eleven two-dimensional structures composed of boron, nitrogen, and carbon. In the proposed monolayers, atoms were positioned following an α-graphyne pattern, and hence were termed BxCyNz hybrid graphynes. Unlike previous investigations into such hybrid materials, we also considered island- and stripe-like atomic arrangements of B, C, and N. Overall, we found that the stability of the proposed structures increased when the number of carbon-boron and carbon-nitrogen bonds decreased and, consequently, that island- and stripe-like arrangements were more stable than mixed arrangements of B, C, and N. On the other hand, in our calculations we found no relationship between the number of carbon–boron and carbon–nitrogen bonds and the electronic properties. We determined that BxCyNz hybrid α-graphynes are semiconducting, with band gap values ranging from 0.02 to 2.00 eV. We also performed PDOS and LDOS calculations, to investigate the electronic states near the Fermi level (Ef). Our results reveal that both states at the top of the valence band and at the bottom of the conduction band are associated with carbon atoms. Finally, we determined the optical absorption spectra of the proposed structures. We found that eight of the proposed monolayers present absorption peaks in various regions of the infrared, with the remaining three presenting absorption peaks in the visible spectrum. Our calculations indicate that the BxCyNz hybrid α-graphynes are viable candidates for optoelectronic devices that operate in the infrared region of the electromagnetic spectrum, such as heat sensors and infrared filters.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the Brazilian Research Agencies CNPq, PNPD/CAPES, and INCT – Nanomateriais de Carbono. We are also grateful to the CLIMA cluster at UFRN, and to the Laboratory of Computational Physics at UFPB. LDM would also like to thank the support of the High Performance Computing Center at UFRN (NPAD/UFRN).References
- A. H. C. Neto, F. Guinea, N. M. R. Peres, K. S. Novoselov and A. K. Geim, Rev. Mod. Phys., 2009, 81, 109–162 CrossRef.
- A. K. Geim, Science, 2009, 324, 1530–1534 CrossRef CAS.
- D. Pacile, J. C. Meyer, C. O. Girit and A. Zettl, Appl. Phys. Lett., 2008, 92, 133107 CrossRef.
- C. H. Jin, F. Lin, K. Suenaga and S. Iijima, Phys. Rev. Lett., 2009, 102, 195505 CrossRef.
- S. D. Sarma, A. Shaffique, E. H. Hwang and R. Enrico, Rev. Mod. Phys., 2011, 83, 407–470 CrossRef.
- E. Gibney, Nature, 2015, 522, 274 CrossRef CAS.
- C. Lee, X. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385–388 CrossRef CAS.
- X. Ma and H. Zhang, Nanoscale Res. Lett., 2013, 8, 440 CrossRef.
- W. Han, L. Wu, Y. Zhu, K. Watanabe and T. Taniguchi, Appl. Phys. Lett., 2008, 93, 223103 CrossRef.
- Y. Miyamoto, M. L. Cohen and S. G. Louie, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 52, 14971–14975 CrossRef CAS.
- S. Azevedo, J. Kaschny, M. Caio and F. de Brito Mota, Eur. Phys. J. B, 2009, 67, 507–512 CrossRef CAS.
- S. Azevedo and R. De Paiva, Europhys. Lett., 2006, 75, 126–132 CrossRef CAS.
- S. Azevedo, M. Mazzoni, R. Nunes and H. Chacham, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 205412 CrossRef.
- S. Azevedo, J. R. Kaschny, C. Castilho and F. B. Mota, Nanotechnology, 2007, 18, 495707 CrossRef PubMed.
- L. Ci, L. Song, C. Jin, D. Jariwala, D. Wu, Y. Li, A. Srivastava, Z. F. Wang, K. Storr, L. Balicas, F. Liu and P. M. Ajayan, Nat. Mater., 2010, 9, 430–435 CrossRef CAS.
- C. Huang, C. Chen, M. Zhang, L. Lin, X. Ye, S. Lin, M. Antonietti and X. Wang, Nat. Commun., 2015, 6, 7698 CrossRef.
- M. S. Mazzoni, R. W. Nunes, S. Azevedo and H. Chacham, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 073108 CrossRef.
- Y. Tateyama, T. Ogitsu, K. Kusakabe, S. Tsuneyuki and S. Itoh, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, R10161–R10164 CrossRef CAS.
- A. K. Manna and S. K. Pati, J. Phys. Chem. C, 2011, 115, 10842–10850 CrossRef CAS.
- A. Freitas, S. Azevedo, M. Machado and J. R. Kaschny, Appl. Phys. A, 2012, 108, 185–193 CrossRef CAS.
- A. Du, Z. Zhu, G. Lu and S. C. Smith, J. Am. Chem. Soc., 2009, 131, 1682–1683 CrossRef CAS.
- M. Becton, L. Zhang and X. Wang, Phys. Chem. Chem. Phys., 2014, 16, 18233 RSC.
- W. S. Cranford, B. D. Brommer and J. M. Buehler, Nanoscale, 2012, 4, 7797–7809 RSC.
- A. Freitas, L. D. Machado, C. G. Bezerra, R. M. Tromer, L. F. C. Pereira and S. Azevedo, RSC Adv., 2018, 8, 24847 RSC.
- Z. Wang, X. Zhou, X. Zhang, Q. Zhu, H. Dong, M. Zhao and A. R. Oganov, Nano Lett., 2015, 15, 6182–6186 CrossRef CAS.
- Y. Liu, G. Wang, Q. Huang, L. Guo and X. Chen, Phys. Rev. Lett., 2012, 108, 225505 CrossRef.
- G. R. Bhimanapati, Z. Lin, V. Meunier, Y. Jung, J. Cha, S. Das, D. Xiao, Y. Son, M. S. Strano, V. R. Cooper, L. Liang, S. G. Louie, E. Ringe, W. Zhou, S. S. Kim, R. R. Naik, B. G. Sumpter, H. Terrones, F. Xia, Y. Wang, J. Zhu, D. Akinwande, N. Alem, J. A. Schuller, R. E. Schaak, M. Terrones and J. A. Robinson, ACS Nano, 2015, 9, 11509–11539 CrossRef CAS PubMed.
- R. H. Baughman, H. Eckhardt and M. J. Kertesz, J. Chem. Phys., 1987, 87, 6687–6698 CrossRef CAS.
- D. Sundholm, L. N. Wirz and P. Schwerdtfeger, Nanoscale, 2015, 7, 15886–15894 RSC.
- J. M. Kehoe, J. H. Kiley, J. J. English, C. A. Johnson, R. C. Petersen and M. M. Haley, Org. Lett., 2000, 2, 969–972 CrossRef CAS.
- F. Diederich, Nature, 1994, 369, 199–207 CrossRef CAS.
- M. M. Haley, S. C. Brand and J. J. Pak, Angew. Chem., Int. Ed., 1997, 36, 836–838 CrossRef CAS.
- K. Tahara, Y. Yamamoto, D. E. Gross, H. Kozuma, Y. Arikuma, K. Ohta, Y. Koizumi, Y. Gao, Y. Shimizu, S. Seki, K. Kamada, J. S. Moore and Y. Tobe, Chem.–Eur. J., 2013, 19, 11251–11260 CrossRef CAS.
- G. Li, Y. Li, H. Liu, Y. Guo, Y. Li and D. Zhu, Chem. Commun., 2010, 46, 3256–3258 RSC.
- W. Z. Wu, W. L. Guo and X. C. Zeng, Nanoscale, 2013, 5, 9264–9276 RSC.
- D. Malko, C. Neiss, F. Vines and A. Gorling, Phys. Rev. Lett., 2012, 108, 086804 CrossRef.
- V. R. Coluci, S. F. Braga, S. B. Legoas, D. S. Galvao and R. H. Baughman, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 035430 CrossRef.
- H. J. Hwang, Y. Kwon and H. Lee, J. Phys. Chem. C, 2012, 116, 20220–20224 CrossRef CAS.
- H. J. Hwang, J. Koo, M. Park, N. Park, Y. Kwon and H. Lee, J. Phys. Chem. C, 2013, 117, 6919–6923 CrossRef CAS.
- H. Y. Zhang, X. J. He, M. W. Zhao, M. Zhang, L. X. Zhao, X. J. Feng and Y. H. Luo, J. Phys. Chem. C, 2012, 116, 16634–16638 CrossRef CAS.
- J. Kou, X. Zhou, H. Lu, F. Wu and J. Fan, Nanoscale, 2014, 6, 1865–1870 RSC.
- N. Narita, S. Nagai, S. Suzuki and K. Nakao, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 11009–11014 CrossRef CAS.
- J. Zhou, K. Lv, Q. Wang, X. S. Chen, Q. Sun and P. J. Jena, Chem. Phys., 2011, 134, 174701 Search PubMed.
- L. D. Pan, L. Z. Zhang, B. Q. Song, S. X. Du and H. J. Gao, Appl. Phys. Lett., 2011, 98, 173102 CrossRef.
- Y. Zhang, J. Yun, K. Wang, X. Chen, Z. Yang, Z. Zhang, J. Yan and W. Zhao, Comput. Mater. Sci., 2017, 136, 12–19 CrossRef CAS.
- X. Cao, Y. Li, X. Cheng and Y. Zhang, Chem. Phys. Lett., 2011, 502, 217–221 CrossRef CAS.
- S. Behzad, Phys. E, 2016, 83, 211–214 CrossRef CAS.
- J. Yun, Y. Zhang, K. Wang and Z. Zhang, Comput. Mater. Sci., 2016, 123, 79–84 CrossRef CAS.
- A. N. Enyashina, A. A. Sofronova, Y. N. Makurina and A. L. Ivanovskii, J. Mol. Struct., 2004, 684, 29–33 CrossRef.
- C. Sun, Y. Liu, J. Xu, B. Chi, C. Bai, S. Li, X. Zhao and X. Li, Phys. E, 2015, 70, 190–197 CrossRef CAS.
- V. O. Ongun and S. Ciraci, J. Phys. Chem. C, 2013, 117, 2175–2182 CrossRef.
- J. Yun, Y. Zhang, M. Xu, J. Yan, W. Zhao and Z. Zhang, J. Mater. Sci., 2017, 52, 10294–10307 CrossRef CAS.
- X. Deng, M. Si and J. Dai, J. Chem. Phys., 2012, 137, 201101 CrossRef.
- D. Sánchez-Portal, P. Ordejón, E. Artacho and J. M. Soler, Int. J. Quantum Chem., 1997, 65, 453–461 CrossRef.
- J. M. Soler, E. Artacho, J. D. Gale, A. García, J. Junquera, P. Ordejón and D. Sánchez-Portal, J. Phys.: Condens. Matter, 2002, 14, 2745 CrossRef CAS.
- W. Kohn and L. J. Sham, Phys. Rev. Lett., 1965, 140, A1133–A1138 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- N. Troullier and J. L. Martins, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 43, 1993–2006 CrossRef CAS.
- L. Kleinman and D. M. Bylander, Phys. Rev. Lett., 1982, 48, 1425–1428 CrossRef CAS.
- M. A. Marques, J. Vidal, M. J. Oliveira, L. Reining and S. Botti, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 035119 CrossRef.
- T. O. Wehling, E. Sasioglu, C. Friedrich, A. I. Lichtenstein, M. I. Katsnelson and S. Blugel, Phys. Rev. Lett., 2011, 106, 236805 CrossRef CAS.
- S. Lee, S. Kim and K. Kim, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 155436 CrossRef.
- B. Dong, H. Guo, Z. Liu, T. Yang, P. Tao, S. Tang, R. Saito and Z. Zhanga, Carbon, 2018, 131, 223 CrossRef CAS.
- I. Alcon, F. Vines, I. de P. R. Moreira and S. T. Bromley, Nat. Commun., 2017, 8, 1957 CrossRef.
- J. Heyd and G. E. Scuseria, J. Chem. Phys., 2004, 121, 1187 CrossRef CAS.
- P. Deak, B. Aradi, T. Frauenheim, E. Janzen and A. Gali, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 153203 CrossRef.
- M. J. Frisch, et al., Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- A. V. Krukau, O. A. Vydrov, A. F. Izmaylov and G. E. Scuseria, J. Chem. Phys., 2006, 125, 224106 CrossRef.
- A. C. M. Carvalho, C. G. Bezerra, J. A. Lawlor and M. S. Ferreira, J. Phys.: Condens. Matter, 2014, 26, 015303 CrossRef CAS.
- G. Zhu, Q. Sun, Y. Kawazoe and P. Jena, Int. J. Hydrogen Energy, 2015, 40, 3689 CrossRef CAS.
- E. Montes and U. Schwingenschlogl, Phys. Rev. B, 2016, 94, 035412 CrossRef.
- P. Nayebi, M. Emami-Razavi and E. Zaminpayma, J. Phys. Chem. C, 2016, 120, 4589 CrossRef CAS.
- M. P. Ljungberg, O. Vanska, P. Koval, S. W. Koch, M. Kira and D. Sanchez-Portal, New J. Phys., 2017, 19, 033019 CrossRef.
- M. Kolos and F. Karlicky, Phys. Chem. Chem. Phys., 2019, 21, 3999 RSC.
Footnote |
| † PACS numbers: 73.20.At, 73.20.Hb, 71.15.Mb. |
| This journal is © The Royal Society of Chemistry 2019 |