
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and biological evaluation of 3-nitro-4-chromanone derivatives as potential antiproliferative agents for castration-resistant prostate cancer†
Huiqing Chen‡
a,
Yajing Xing‡b,
Jia Xieb,
Jiuqing Xieb,
Dong Xing a,
Jie Tanga,
Fan Yanga,
Zhengfang Yi*b and
Wen-Wei Qiu*a
a,
Jie Tanga,
Fan Yanga,
Zhengfang Yi*b and
Wen-Wei Qiu*a
aShanghai Engineering Research Center of Molecular Therapeutics and New Drug Development, School of Chemistry and Molecular Engineering, East China Normal University, Shanghai, 200062, China. E-mail: wwqiu@chem.ecnu.edu.cn
bShanghai Key Laboratory of Regulatory Biology, Institute of Biomedical Sciences and School of Life Sciences, East China Normal University, Shanghai, 200241, China. E-mail: zfyi@bio.ecnu.edu.cn
First published on 21st October 2019
Abstract
A series of novel 3-nitro-4-chromanones were synthesized and their in vitro cytotoxicity was evaluated on castration-resistant prostate cancer cell (CRPC) lines using the sulforhodamine B (SRB) assay. The amide derivatives showed more potent antitumor activity than their corresponding ester derivatives. Most of the tested compounds showed less toxicity towards human fibroblasts (HAF) compared with the tumor cell lines. The optimal compound 36 possessed much more potent antiproliferative activity than the positive compound cisplatin. The colony formation, cell cycle distribution, apoptosis, transwell migration and wound healing assays of 36 were performed on CRPC cell lines.
Introduction
Prostate cancer (PC) is a type of malignancy that arises in the prostate gland and it tends to develop in older men. Globally, prostate cancer is the second most common cancer among men. The International Agency for Research on Cancer (IARC) estimated 1.27 million new PC cases and 359![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths in 2018 worldwide.1 Although various anti-cancer agents are used solely or in combination with radiotherapy to treat advanced diseases, none of the conventional therapies have been proven to be highly successful for PC.2 It often finally develops into fatal castration-resistant prostate cancer (CRPC) with the ability to grow in the absence of androgens.3 CRPC is not responsive to hormonal therapy, readily re-emerges and is highly metastatic, resulting in most of the deaths in PC patients.4 The presence of androgen receptor (AR)-negative cell populations in CRPC has been identified and new therapeutic strategies targeting AR-negative PC cells would provide a potential approach for treatment of CRPC.5 The AR-negative metastatic DU145 and PC3 cell lines6 are often studied as in vitro models for CRPC.7 Studies disclosed that 3-nitro-4-chromanones possessed anticancer activity against murine leukemia L1210 (Fig. 1, A and B),8,9 human colon HT-29 (Fig. 1, B)9 and human acute myeloid leukemia U937 cell line (Fig. 1, C).10 Our previous research discovered that 3-nitro-4-chromanone derivatives (Fig. 1, D), especially compound 1 (Table 1) exhibited as potent antiproliferative activity as cisplatin against CRPC-like DU145 and PC3 cell lines.11 Herein, we have evaluated the antiproliferative activity of these newly synthesized 3-nitro-4-chromanones in DU145, PC3 and its more metastatic derivative PC3M cell lines12 and disclosed the preliminary structure–activity relationships (SAR) of these compounds. Further investigations including colony formation, cell cycle distribution, apoptosis and migration have also been performed.
000 deaths in 2018 worldwide.1 Although various anti-cancer agents are used solely or in combination with radiotherapy to treat advanced diseases, none of the conventional therapies have been proven to be highly successful for PC.2 It often finally develops into fatal castration-resistant prostate cancer (CRPC) with the ability to grow in the absence of androgens.3 CRPC is not responsive to hormonal therapy, readily re-emerges and is highly metastatic, resulting in most of the deaths in PC patients.4 The presence of androgen receptor (AR)-negative cell populations in CRPC has been identified and new therapeutic strategies targeting AR-negative PC cells would provide a potential approach for treatment of CRPC.5 The AR-negative metastatic DU145 and PC3 cell lines6 are often studied as in vitro models for CRPC.7 Studies disclosed that 3-nitro-4-chromanones possessed anticancer activity against murine leukemia L1210 (Fig. 1, A and B),8,9 human colon HT-29 (Fig. 1, B)9 and human acute myeloid leukemia U937 cell line (Fig. 1, C).10 Our previous research discovered that 3-nitro-4-chromanone derivatives (Fig. 1, D), especially compound 1 (Table 1) exhibited as potent antiproliferative activity as cisplatin against CRPC-like DU145 and PC3 cell lines.11 Herein, we have evaluated the antiproliferative activity of these newly synthesized 3-nitro-4-chromanones in DU145, PC3 and its more metastatic derivative PC3M cell lines12 and disclosed the preliminary structure–activity relationships (SAR) of these compounds. Further investigations including colony formation, cell cycle distribution, apoptosis and migration have also been performed.
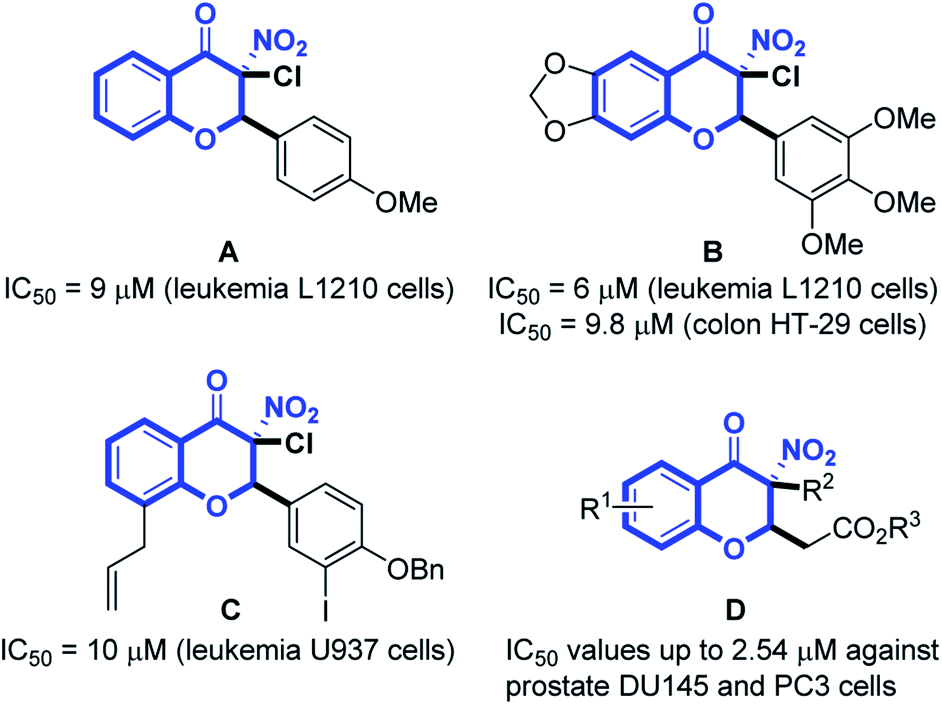 | ||
| Fig. 1 Structures of reported antitumor 3-nitro-4-chromanones. | ||
|
|||||
|---|---|---|---|---|---|
| No. | R2 | R2 | IC50b (μM) | ||
| DU145 | PC3 | HAF | |||
| a From SRB assay after 96 h of treatment.b IC50 data are an average of at least 3 independent experiments.c These compounds were reported previously.11 | |||||
| 1c | 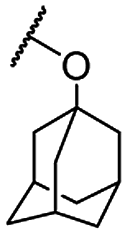 |
Me | 2.54 ± 0.27 | 10.60 ± 0.68 | >100 |
| 3a | OH | Me | >50 | >50 | >100 |
| 4c |  |
Me | >50 | >50 | >100 |
| 5b |  |
Me | >50 | >50 | >100 |
| 6 | 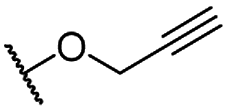 |
Me | >50 | 26.21 ± 1.42 | >100 |
| 7c | 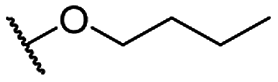 |
Me | 21.08 ± 1.26 | >50 | >100 |
| 8c | 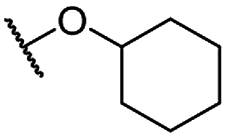 |
Me | 20.91 ± 1.36 | 32.93 ± 2.96 | >100 |
| 9c | 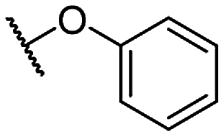 |
Me | >50 | >50 | >100 |
| 10 | 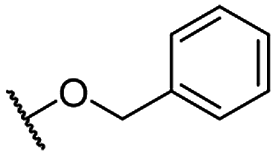 |
Me | 32.26 ± 1.51 | 20.13 ± 1.30 | >100 |
| 11 | NH2 | Me | 46.23 ± 1.67 | 23.24 ± 1.37 | >100 |
| 12 | 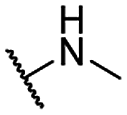 |
Me | 24.66 ± 1.39 | >50 | >100 |
| 13 | 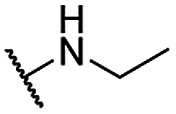 |
Me | 18.69 ± 1.27 | 9.35 ± 0.97 | >100 |
| 14 | 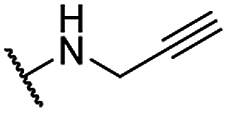 |
Me | 38.17 ± 1.58 | 12.97 ± 1.11 | >100 |
| 15 | 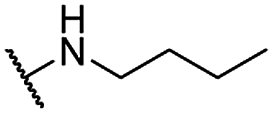 |
Me | 15.67 ± 1.20 | 9.97 ± 1.00 | >100 |
| 16 | 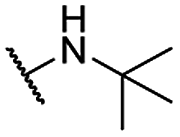 |
Me | 5.01 ± 0.70 | 10.03 ± 1.00 | >100 |
| 17 | 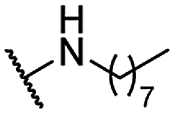 |
Me | 6.81 ± 0.83 | 4.83 ± 0.68 | >100 |
| 18 | 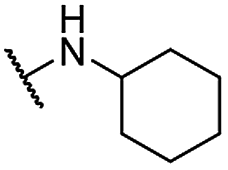 |
Me | 5.09 ± 0.71 | 3.35 ± 0.52 | >100 |
| 19 | 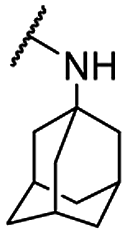 |
Me | 1.73 ± 0.24 | 3.09 ± 0.79 | >100 |
| 20 | 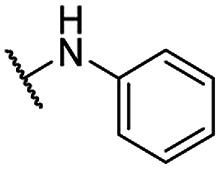 |
Me | 20.32 ± 1.31 | 6.44 ± 0.81 | >100 |
| 21 | 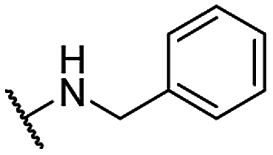 |
Me | 19.52 ± 1.29 | 9.91 ± 1.00 | >100 |
| 22 | 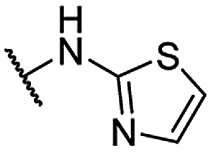 |
Me | 49.24 ± 3.04 | 36.90 ± 0.98 | >100 |
| 23 | 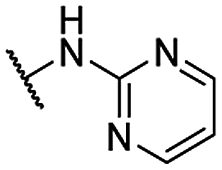 |
Me | >50 | >50 | >100 |
| 24 | 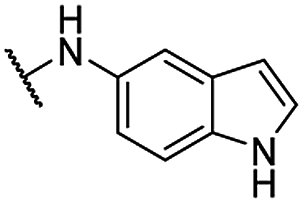 |
Me | 23.94 ± 1.89 | 15.82 ± 0.33 | >100 |
| 25 | 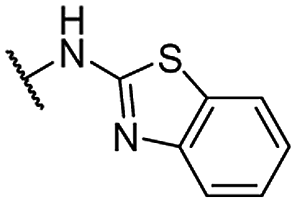 |
Me | >50 | >50 | >100 |
| 26 | 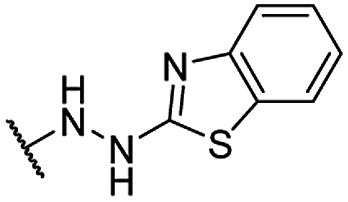 |
Me | 29.7 ± 2.21 | >50 | >100 |
| 27 | 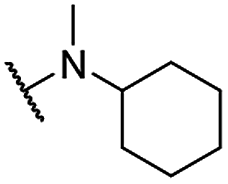 |
Me | 10.58 ± 1.02 | >50 | >100 |
| 28 | 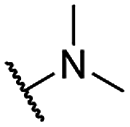 |
Me | >50 | >50 | >100 |
| 29 | 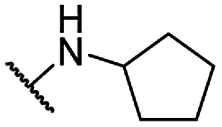 |
Me | 8.00 ± 0.90 | >50 | >100 |
| 30 | 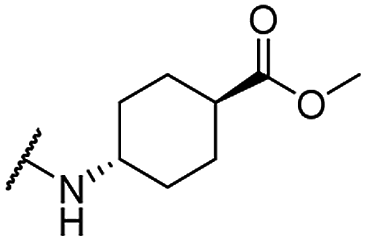 |
Me | 14.36 ± 1.16 | >50 | >100 |
| 31 | 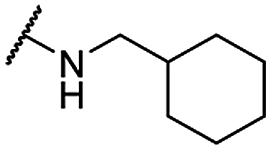 |
Me | 4.22 ± 0.63 | 43.90 ± 1.64 | >100 |
| 32 | 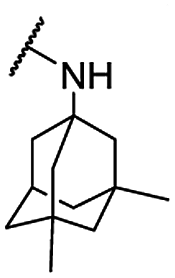 |
Me | 5.41 ± 0.73 | 11.43 ± 1.06 | >100 |
| 33 | 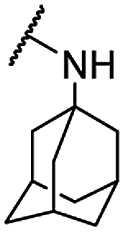 |
Et | 5.16 ± 0.71 | 6.81 ± 0.83 | >100 |
| 34 | 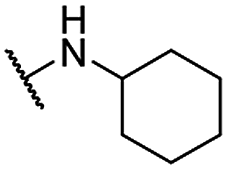 |
Et | 11.08 ± 1.04 | >50 | >100 |
| Cisplatin | 2.80 ± 0.45 | 18.20 ± 1.26 | 32.63 ± 1.51 | ||
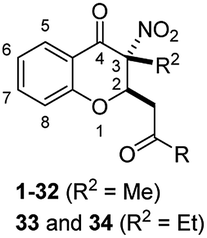
Results and discussion
Chemistry
The general strategies developed for compounds synthesis is described as follows (Scheme 1). The esters 2, previously synthesized in our group, were used as starting compounds. Hydrolysis of the esters 2 with 6 N HCl in H2O/1,4-dioxane to provide the corresponding acids 3. Afterwards, ester compounds 6 and 10 were obtained by condensation of 3 with corresponding alcohols under thionyl chloride. Amide compounds 11–43 were obtained by condensation of 3 with corresponding amines under 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDCI), 1-hydroxybenzotriazole (HOBt) and N,N-diisopropylethylamine (DIPEA) in CH2Cl2. Compound 44 was obtained by reduction of the nitro group of compound 36. Structures were well characterized with 1H NMR, 13C NMR and high-resolution mass spectrum.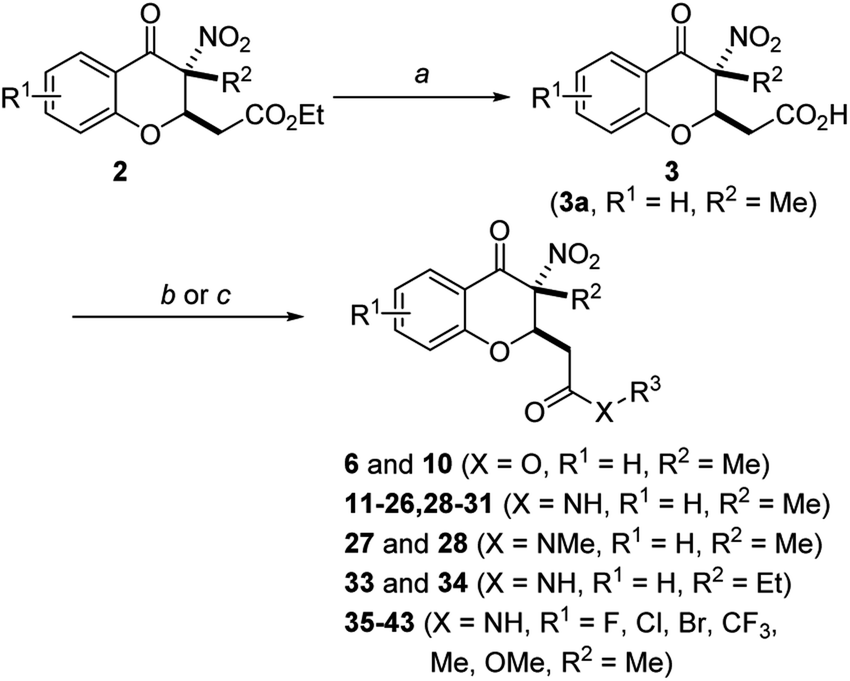 | ||
| Scheme 1 General synthetic route of compound 3–43. Reagent and conditions: (a) 6 N HCl/1,4-dioxane, 100 °C, 95% for 3a; (b) alcohols, SOCl2, 1,4-dioxane, 70 °C, 68–81%; (c) amines, HOBt, EDCI, DIPEA, DCM, rt, 64–88%. | ||
Biological activities
For the carboxylic acid (3a) and its ester compounds (4–10), the results showed that most compounds exhibiting moderate to weak antitumor activity. The carboxylic acid and ester compounds bearing small substituents (4 and 5) possessed almost no activity. The antiproliferative activity was increased to moderate as the small ester group was replaced by relatively large ester substituents (8 and 10), except phenyl ester group (9).
For C-2 amide compounds, they showed obviously improved IC50 values than their corresponding ester compounds (19, 12, 13, 14, 15, 18, 20 and 21 vs. 1, 4, 5, 6, 7, 8, 9 and 10). For alkyl amides, the antiproliferative activity was improved as the substituents increased from small amino (11, IC50 = 46.23 and 23.24 μM) and aminomethyl (12, IC50 = 24.66 and >50 μM) to large n-octylamine (17, IC50 = 6.81 and 4.83 μM) and especially cyclohexane (18, IC50 = 5.09 and 3.35 μM) and adamantanamine (19, IC50 = 1.73 and 3.09 μM) groups. For aryl amides, the phenyl amide (20) and benzyl amide (21) showed moderate antitumor activity, and most aromatic heterocyclic amides (22–26) showed moderate to weak or even no activity. Therefore, the aryl amide substituents were unfavorable substituted groups for improving antitumor activity. The cyclohexyl amide (18) and adamantane amide (19) were key structures for maintaining the potent antitumor activity. So, we further synthesized the derivatives (27, 29–34) based on the compounds 18 and 19. For the derivatives 27, 29–31 and 34 (the C-3 methyl group was replaced by ethyl group), their antitumor activity, especially in PC3 cells, decreased significantly compared with 18. The trialkylamines 27 and 28 showed the decreased activity, which means the acidic proton for amide derivative can be essential for the activity. For the derivatives 32 and 33 (the C-3 methyl group was replaced by ethyl group), the antitumor activity also decreased obviously compared with 19. In Table 1, the most potent compound 19, which bearing an adamantane amide substituent, possessed 1.6-fold (in DU145 cells) and 5.9-fold (in PC3 cells) more potent antiproliferative activity than the positive compound cisplatin. These results illustrated that the adamantane amide group showed more potent activity than other amide groups.
The second-round synthetic compounds 35–44 were obtained by modification of the benzene ring of compound 19 and their antiproliferative activity was screened using the SRB assay in DU145, PC3 and PC3M cell lines (Table 2). Results discovered that most compounds possessed moderate to potent antitumor activity, except 44. Most compounds displayed almost no toxicity on human fibroblasts (HAF), except 38. We introduced groups –CF3, –F, –Cl, –Br, –Me and –OMe into the C-6 position of the benzene ring of 19 firstly. The electron withdrawing –CF3 (35), –F (36), –Cl (37) and –Br (38) groups were favorable substituted groups for improving antitumor activity, except the compound 35 on PC3M cells. The electron donating –Me (39) and –OMe (40) groups decreased the antitumor activity obviously compared with 19. Compound 38 possessed the most potent antiproliferative activity (IC50 values were about 0.5 μM) in DU145, PC3 and PC3M cells, while it also displayed potent toxicity on HAF cells (IC50 = 3.67 μM). The second-best active compound 36 (bearing 6-F group), which IC50 on HAF was more than 100 μM. So, 36 and its –F group was regarded as the optimal compound and substituent. The –F group was also introduced into the C-5 (41), C-7 (42) and C-8 (43) positions of the benzene ring of 19 and their antitumor activity was decreased slightly compared with 36. If the nitro group was reduction to amino group at C-3 position of 36, the antitumor activity was significantly decreased (IC50 > 50 μM). We have also tested the activity of compound 36 on AR-positive prostate cancer cell lines LNCaP (IC50 = 4.4 μM) and 22RV1 (IC50 = 7.43 μM), which were much weaker than on AR-negative prostate cancer cell lines DU145 (IC50 = 1.21 μM) and PC3 (IC50 = 0.94 μM).
|
|||||
|---|---|---|---|---|---|
| No. | R1 | IC50b (μM) | |||
| DU145 | PC3 | PC3M | HAF | ||
| a From SRB assay after 96 h of treatment.b IC50 data are an average of at least 3 independent experiments.c Replacement of nitro group with amine group at 3-position. | |||||
| 19 | H | 1.73 ± 0.24 | 3.09 ± 0.79 | 7.31 ± 1.17 | >100 |
| 35 | 6-CF3 | 1.63 ± 0.21 | 1.69 ± 0.53 | 12.88 ± 1.11 | >100 |
| 36 | 6-F | 1.21 ± 0.08 | 0.94 ± 0.03 | 5.66 ± 1.05 | >100 |
| 37 | 6-Cl | 1.34 ± 0.13 | 3.07 ± 0.79 | 6.34 ± 0.80 | >100 |
| 38 | 6-Br | 0.47 ± 0.02 | 0.51 ± 0.03 | 0.45 ± 0.24 | 3.67 ± 0.56 |
| 39 | 6-Me | 1.75 ± 0.54 | 6.39 ± 0.81 | 19.20 ± 1.58 | >100 |
| 40 | 6-MeO | 2.62 ± 0.42 | 6.77 ± 0.83 | 13.06 ± 1.42 | >100 |
| 41 | 5-F | 2.27 ± 0.66 | 2.91 ± 0.70 | 9.75 ± 0.99 | >100 |
| 42 | 7-F | 2.24 ± 0.65 | 3.03 ± 0.78 | 12.46 ± 1.40 | >100 |
| 43 | 8-F | 2.35 ± 0.67 | 3.02 ± 0.78 | 9.16 ± 1.26 | >100 |
| 44c | 6-F | >50 | >50 | >50 | >100 |
| Docetaxel | 0.0076 ± 0.0005 | 0.013 ± 0.003 | 0.043 ± 0.005 | 0.157 ± 0.031 | |
| Cisplatin | 2.80 ± 0.45 | 18.20 ± 1.26 | 12.86 ± 1.11 | 32.63 ± 1.51 | |
Accordingly, although the antitumor activity of compound 36 was much weaker than the positive compound docetaxel, it was 2.3–19.4 times more potent than the positive compound cisplatin and displayed almost no toxicity on normal cells, thus it was selected for further evaluation.
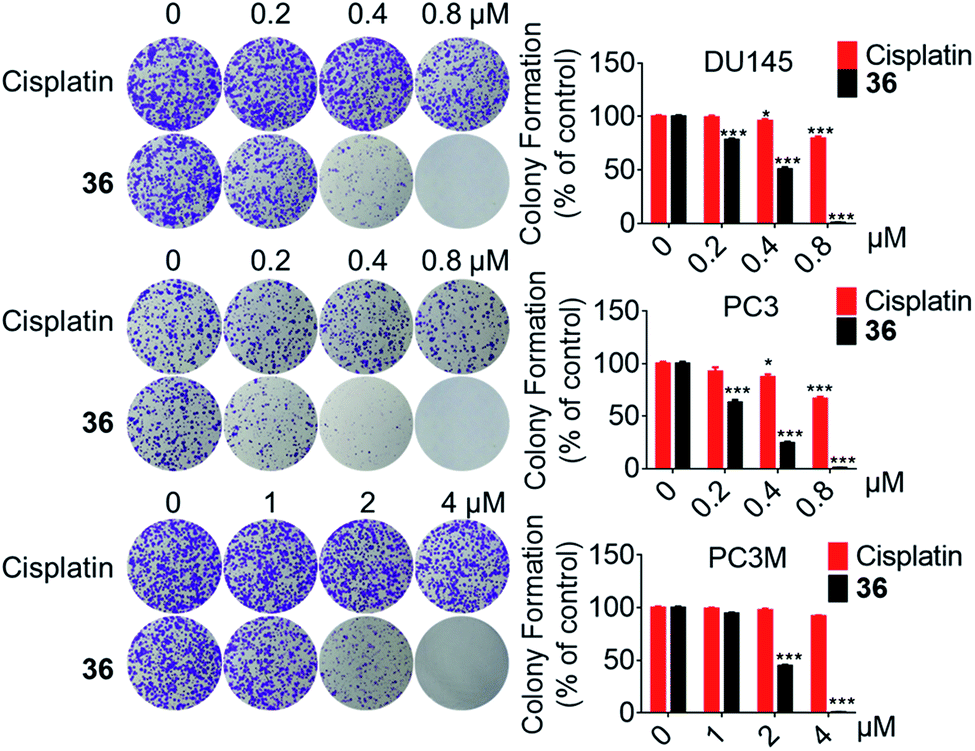 | ||
| Fig. 2 The colony formation ability of DU145, PC3 and PC3M cells was inhibited by 36. *p < 0.05, ***p < 0.001. | ||
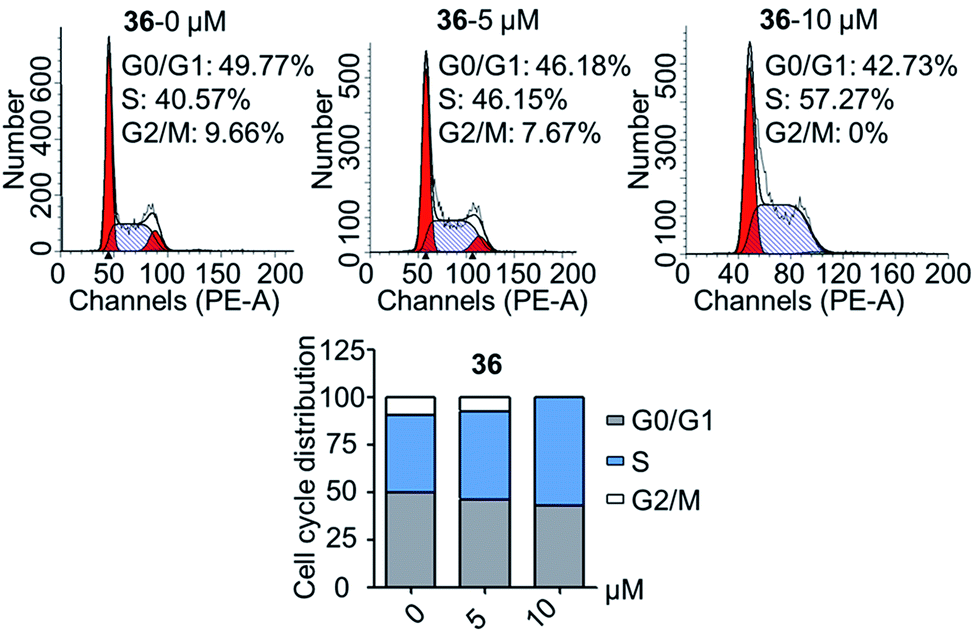 | ||
| Fig. 3 Compound 36 disrupted the cell cycle distribution of DU145 cells. | ||
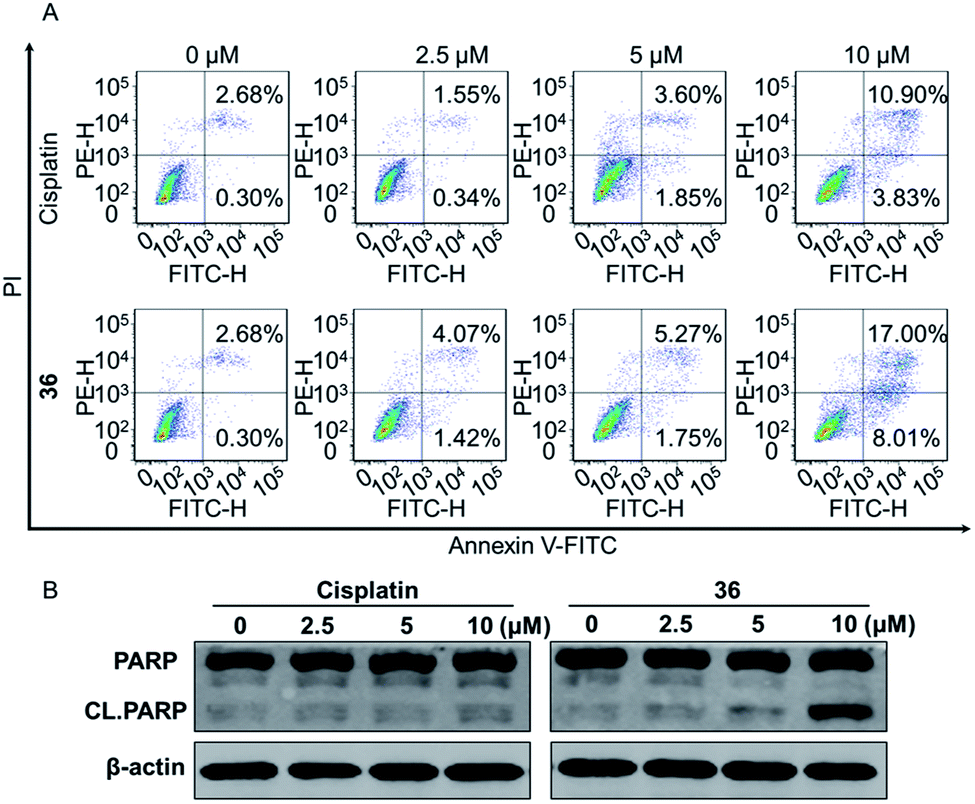 | ||
| Fig. 4 (A) Compound 36 induced cell apoptosis significantly. (B) The expression of PARP and CL.PARP upon 36 treatment. | ||
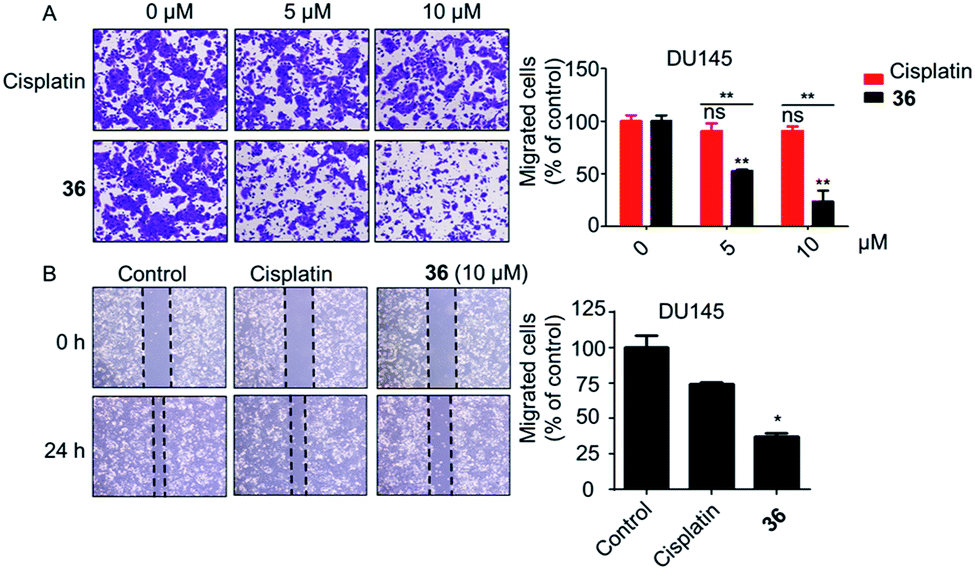 | ||
| Fig. 5 The cell migration ability of DU145 cells was notably inhibited by 36. (A) Transwell migration assay. (B) Wound healing migration assay (10 μM of 36). *p < 0.05, **p < 0.01. | ||
In conclusion, a series of 3-nitro-4-chromanone derivatives were synthesized by a facile and convenient method. Their antiproliferative activity in CRPC cell lines were assessed in vitro. The amide derivatives showed more potent antitumor activity than their corresponding ester derivatives. Most of these compounds possessed more selectivity towards tumor cells than human fibroblasts. The C-2 adamantane amide and 6-F were favorable substituents for improving antitumor activity. The optimal compound 36, which possessed 2.3–19.4 times more potent than the positive cisplatin in antitumor activity. 36 also displayed more potent than that of cisplatin in colony formation, apoptosis, transwell migration and wound healing assays. The primary mechanism studies disclosed that 36 led to S phase cell cycle arrest and promoted the PARP to CL. PARP in tumor cells. Collectively, we reported 3-nitro-4-chromanone derivatives as a series of new chemical entities for the first time. Especially 36, which displayed potent antitumor activity in CRPC cell lines in vitro, could be used as a promising lead for further development.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was partially supported by Shanghai Science and Technology Council (Grant 18ZR1411200), National Natural Science Foundation of China (21772043) and the Fundamental Research Funds for the Central Universities.References
- (a) F. Bray, J. Ferlay, I. Soerjomataram, R. L. Siegel, L. A. Torre and A. Jemal, Ca-Cancer J. Clin., 2018, 68, 394 CrossRef PubMed; (b) C.-C. Weng, P.-Y. Ding, Y.-H. Liu, J. R. Hawse, M. Subramaniam, C.-C. Wu, Y.-C. Lin, C.-Y. Chen, W.-C. Hung and K.-H. Cheng, Oncogene, 2019, 38, 2005 CrossRef CAS; (c) J. Ferlay, M. Colombet, I. Soerjomataram, C. Mathers, D. M. Parkin, M. Piñeros, A. Znaor and F. Bray, Int. J. Cancer, 2019, 144, 1941 CrossRef CAS.
- (a) G. Attard, C. Parker, R. A. Eeles, F. Schröder, S. A Tomlins, I. Tannock, C. G. Drake and J. S. de Bono, Lancet, 2016, 387, 70 CrossRef; (b) C. Parker, S. Gillessen, A. Heidenreich and A. Horwich, Ann. Oncol., 2015, 26(suppl. 5), v69 CrossRef; (c) T. Wüstemann, U. Haberkorn, J. Babich and W. Mier, Med. Res. Rev., 2019, 39, 40 CrossRef.
- (a) P. A. Watson, V. K. Arora and C. L. Sawyers, Nat. Rev. Cancer, 2015, 15, 701 CrossRef CAS; (b) Y. Yamamoto, Y. Loriot, E. Beraldi, F. Zhang, A. W. Wyatt, N. Al Nakouzi, F. Mo, T. Y. Zhou, Y. Kim, B. P. Monia, A. R. MacLeod, L. Fazli, Y. Z. Wang, C. C. Collins, A. Zoubeidi and M. Gleave, Clin. Cancer Res., 2015, 21, 1675 CrossRef CAS; (c) J. W. Russo, C. Gao, S. S. Bhasin, O. S. Voznesensky, C. Calagua, S. Arai, P. S. Nelson, B. Montgomery, E. A. Mostaghel, E. Corey, M.-E. Taplin, H. H. Ye, M. Bhasin and S. P. Balk, Cancer Res., 2018, 78, 6354 CAS.
- (a) T. A. Yap, A. D. Smith, R. Ferraldeschi, B. Al-Lazikani, P. Workman and J. S. de Bono, Nat. Rev. Drug Discovery, 2016, 5, 699 CrossRef; (b) Y. Zong and A. S. Goldstein, Nat. Rev. Urol., 2013, 10, 90 CrossRef CAS; (c) A. Davies, V. Conteduca, A. Zoubeidi and H. Beltran, European Urology Focus, 2019, 5, 147 CrossRef PubMed.
- C. Tran, S. Ouk, N. J. Clegg, Y. Chen, P. A. Watson, V. Arora, J. Wongvipat, P. M. Smith-Jones, D. Yoo, A. Kwon, T. Wasielewska, D. Welsbie, C. Degui Chen, C. S. Higano, T. M. Beer, D. T. Hung, H. I. Scher, M. E. Jung and C. L. Sawyers, Science, 2009, 324, 787 CrossRef CAS.
- A. van Bokhoven, M. Varella-Garcia, C. Korch, W. U. Johannes, E. E. Smith, H. L. Miller, S. K. Nordeen, G. J. Miller and M. S. Lucia, Prostate, 2005, 57, 205 CrossRef PubMed.
- (a) T.-C. Shih, R. W. Liu, C.-T. Wu, X. C. Li, W. W. Xiao, X. J. Deng, S. Kiss, T. Wang, X.-J. Chen, R. Carney, H.-J. Kung, Y. Duan, P. M. Ghosh and K. S. Lam, Clin. Cancer Res., 2015, 21, 1675 CrossRef; (b) M. Recagni, M. L. Greco, A. Milelli, A. Minarini, N. Zaffaroni, M. Folini and C. Sissi, Eur. J. Med. Chem., 2019, 177, 401 CrossRef CAS.
- D. Dauzonne, B. Folléas, L. Martinez and G. G. Chabot, Eur. J. Med. Chem., 1997, 32, 71 CrossRef CAS.
- A. G. de Peredo, S. Léonce, C. Monneret and D. Dauzonne, Chem. Pharm. Bull., 1998, 46, 79 CrossRef CAS PubMed.
- S. Bouchet, M. Piedfer, S. Susin, D. Dauzonne and B. Bauvois, AIMS Mol. Sci., 2016, 3, 368 CAS.
- H. Q. Chen, J. Xie, D. Xing, J. P. Wang, J. Tang, Z. F. Yi, F. Xia, W.-W. Qiu and F. Yang, Org. Biomol. Chem., 2019, 17, 1062 RSC.
- Z. Y. Xu, Y. L. Wang, Z. G. Xiao, C. Zou, X. Zhang, Z. Wang, D. L. Wu, S. Yu and F. L. Chan, Oncogene, 2018, 37, 6259 CrossRef CAS.
- (a) Y.-Y. Wang, Y. He, L.-F. Yang, S.-H. Peng, X.-L. He, J.-H. Wang, F. Lv, Y. Hao, M.-Y. Liu, Z. F. Yi and W.-W. Qiu, Eur. J. Med. Chem., 2016, 120, 13 CrossRef CAS; (b) B. Yu, H. Y. Liu, X. Y. Kong, X. L. Chen and C. L. Wu, Eur. J. Med. Chem., 2019, 163, 500 CrossRef CAS.
- Y. Ai, Y. Hu, F. H. Kang, Y. S. Lai, Y. J. Jia, Z. J. Huang, S. X. Peng, H. Ji, J. D. Tian and Y. H. Zhang, J. Med. Chem., 2015, 58, 4506 CrossRef CAS.
- A. Valentini, F. Conforti, A. Crispini, A. De Martino, R. Condello, C. Stellitano, G. Rotilio, M. Ghedini, G. Federici, S. Bernardini and D. Pucci, J. Med. Chem., 2009, 52, 484 CrossRef CAS.
- (a) L. Virág, A. Robaszkiewicz, J. M. Rodriguez-Vargas and F. J. Oliver, Mol. Asp. Med., 2013, 34, 1153 CrossRef; (b) F. J. Oliver, G. de la Rubia, V. Rolli, M. C. Ruiz-Ruiz, G. de Murcia and J. M. -de Murcia, J. Biol. Chem., 1998, 273, 33533 CrossRef CAS; (c) A. Nagarsenkar, L. Guntuku, S. D. Guggilapu, D. Bai K., S. Gannoju, V. G. M. Naidu and N. B. Bathini, Eur. J. Med. Chem., 2016, 124, 782 CrossRef CAS.
- (a) N. P. Kumar, P. Sharma, T. S. Reddy, S. Nekkanti, N. Shankaraiah, G. Lalita, S. Sujanakumari, S. K. Bhargava, V. G. M. Naidu and A. Kamal, Eur. J. Med. Chem., 2017, 127, 305 CrossRef CAS; (b) D. J. Fu, L. Zhang, J. Song, R. W. Mao, R. H. Zhao, Y. C. Liu, Y. H. Hou, J. H. Li, J. J. Yang, C. Y. Jin, P. Li, X. L. Zi, H. M. Liu, S. Y. Zhang and Y. B. Zhang, Eur. J. Med. Chem., 2017, 127, 87 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available:Information regarding materials and methods, characterization data of compounds from this study. See DOI: 10.1039/c9ra06420f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |