
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePd nanoparticles on green support as dip-catalyst: a facile transfer hydrogenation of olefins and N-heteroarenes in water†
M. Nasiruzzaman Shaikh
Center of Research Excellence in Nanotechnology (CENT), King Fahd University of Petroleum and Minerals (KFUPM), Dhahran, Saudi Arabia. E-mail: mnshaikh@kfupm.edu.sa
First published on 9th September 2019
Abstract
Chemo- and regioselective hydrogenation methods using highly green sources, particularly from metal nanoparticles on plant stem as support and water, is an intensive research area, which is highly relevant to the development of green chemistry and technology in the 21st century. Here, the synthesis and activity of a heterogeneous catalytic system (called “dip-catalyst”) for the transfer hydrogenation of a series of styrenyl, unfunctionalized olefins, quinoline and other N-heteroarenes, are presented. It consists of Pd nanoparticles (15–20 nm) anchored on bio-processed jute plant (Corchorus genus) stem as the support. Pd nanoparticles were decorated on the green support (GS) jute stem by the in situ reduction of K2PdCl4 in aqueous medium at 70 °C, using formic acid as the reductant. The Pd@GS was characterized using SEM, EDS, XRD, FTIR, XPS and TEM. Elemental mapping revealed uniform distribution of Pd on the cellulose matrix of the jute stem. The catalyst was successfully applied to the chemoselective transfer hydrogenation of numerous styrenyl, unfunctionalized olefins. Its high functional group tolerance was investigated during the olefins hydrogenation in water. Furthermore, Pd@GS was capable of quantitative hydrogenation to selectively produce 1,2,3,4-tetrahydroquinoline (THQ) in water using stoichiometric amounts of tetrahydroxydiboron (THDB) at 60 °C with turn over frequency (TOF) 4938 h−1. This system is stable in water and displays excellent recyclability; it could be used for 32 consecutive cycles, without losing its original crystallinity or requiring replenishment.
Introduction
Transition metal-mediated hydrogenation1,2 using Fe,3 Co,4 Ni,5 Pd,6,7 Pt,8 Rh,9,10 Ir,11 Ru12 or Mo13 is a well-established procedure in research and industrial applications. However, this approach usually requires high hydrogen pressure, resulting in over reduction or functional group intolerance of the substrate. To overcome this shortcoming, transfer hydrogenation14,15 is an alternative. In this context, considerable efforts have been devoted to design Pd-supported transfer hydrogenation homogeneous catalysts.16–18 Since these catalysts are well dispersed at the molecular level, every individual catalytic entity can act as an active and accessible mechanistic site to which the reactant is exposed. This leads to an overall high catalytic efficiency. Unfortunately, they suffer from some serious drawbacks such as cumbersome separation, usage of high volume of toxic solvents and continuous loss of precious metal.19 Furthermore, their restricted reusability impede them from wide commercialization. Nevertheless, the current environmental related governmental legislation imposes severe restriction to eliminate toxic chemical processing and move towards the safer and greener technological endeavours.To overcome these challenges, immobilization of the catalyst is one of the obvious choices. It is well known that the activity of heterogeneous catalytic systems rely heavily on the nature of support.20 Hence, a prudent choice of support is vital. Silica,21,22 polymers,23,24 zeolites,25,26 alumina,27,28 magnetic nanoparticles,29,30 and carbon31 have emerged as excellent support systems. They reportedly achieve almost quantitative conversion and selectivity. Furthermore, their selection is an artifact of particle size, shape and porosity.32 However, due to small size and high surface energy, the active metal nanoparticles tend to aggregate on the support, thereby lowering the number of accessible sites for the reactant; this results in low catalytic activity and selectivity. Moreover, facile leaching of nanoparticles from the surface of the support, due to weak binding, restricts the prospect of reusability.33
Recently, a great deal of attention has been devoted to develop solid supports from biomass, such as activated carbon, carbon nanofibers and hydrophilic carbons, as they are treated as sustainable support materials in heterogeneous catalysis.34 Among these, activated carbon supports were widely used as metal nanoparticle anchors for catalytic applications; however, they yielded relatively discouraging performance.35 The major reasons of failure could be (1) poor pore connectivity, resulting in limited mass-transfer and longer diffusion times, (2) lack of proper functional group on the support, hence hindering the metal nanoparticles' interaction and, (3) hydrophobic surface.
In this context, a new concept of “dip-catalyst” has emerged in order to achieve better efficiency, high turnover frequency (TOF), ease of fabrication, higher reusability and greener synthesis routes.36,37 The term “dip-catalyst” refers to the convenient deployment, high recyclability, easy insertion/removal which can turn the reaction on/off almost instantaneously. The area of “dip-catalysis” has remained largely unexplored. Recently, a few reports related to the fabrication of “dip-catalyst” have appeared in the literature.38 Hariprasad et al. reported Pd-PVA based thin films with high catalytic activity and reusability.39 Zheng et al. reported a paper-based composite material consisting of metal ions on filter paper, and achieved excellent activity and recyclability.40 Rosa et al. dispersed Pd nanoparticles on cellulose acetate and successfully applied it for the C–C bond formation reaction.41
Encouraged by such distinctive features of having highly improved activity and excellent recyclability over the conventional support-based catalysts, we continue to contribute to the development of highly recyclable heterogeneous catalysts.29,42–44 Here, we report a “dip-catalyst” comprising Pd nanoparticles supported on bio-processed jute stem (Pd@GS), for the transfer hydrogenation of olefins, quinoline and other N-heteroarenes in water (Fig. 1). The excellent conversion and selectivity over sensitive organic functional group and its reusability for tens of consecutive cycles was studied systematically.
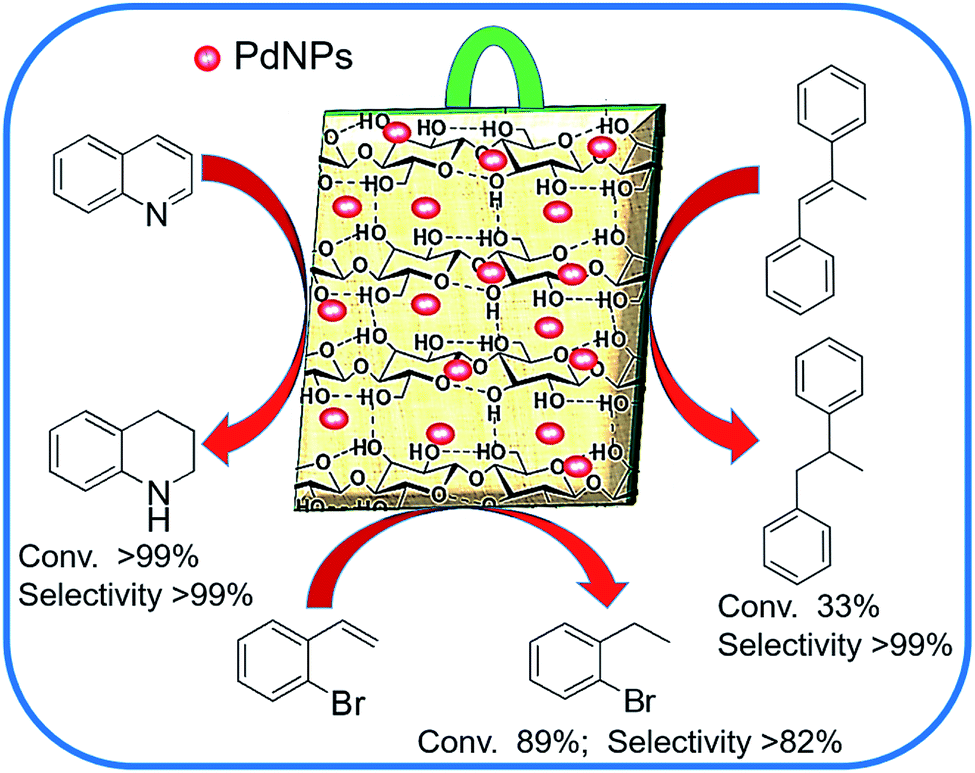 | ||
| Fig. 1 Graphical presentation of versatility of Pd@GS as “dip catalyst”. | ||
Experimental
Materials and methods
All chemicals were purchased from Sigma-Aldrich and were used as-received unless otherwise stated. Standard procedures were followed for the dry and deoxygenated solvents. Schlenk line techniques were used to carry out reactions under inert atmosphere wherever needed. Deionized (DI) water (specific conductivity: 18.2 MΩ) was used in all the experiments. Fourier-Transform Infrared (FTIR) spectroscopic data were recorded on a Nicolet 720 in the wave number range of 500 to 4000 cm−1, using ATR. The TEM samples were prepared by dropping an ethanolic suspension on a copper grid and drying at room temperature. The amount of Pd in the catalyst was determined by inductively coupled plasma optical emission spectrometry (ICP-OES; PlasmaQuant PO 9000 – Analytik Jena). The samples were first digested in a dilute mixture of HNO3 and HCl. Calibration curves were prepared for Pd using standard solutions (ICP Element Standard Solutions, Merck). Samples for SEM (Lyra 3, Tescan) imaging were prepared from a chunk of decorated jute stem on alumina stubs and coated with gold in an automatic gold coater (Quorum, Q150T E). For elemental analysis and mapping, the energy dispersive X-ray spectra (EDS) were collected on a Lyra 3 attachment, using an Oxford Instruments. Catalytic products were identified by a Shimadzu 2010 Plus gas chromatograph attached with a mass spectrometer (GC-MS, Japan). The disappearance of the reactant and sequential appearance of the product was recorded in real time, identifying the species in terms of their molecular ion (M+) by comparing and matching them with the available Wiley library of the mass spectrum database, in addition to the identification of mass fragments.Synthesis of Pd@GS
Usually, the jute plants are harvested after they bloom. The fully grown stalks are cut, trimmed, bundled and kept immersed in ponds or stream to rot (known as retting). This frees the fibers from the stem, which are removed manually. The stems are dried for 7 days in sunlight, cut into thin slices (∼2 cm × 0.5 cm × 0.1 cm) and dried in an oven at 100 °C for 5 h. Pd nanoparticles in the range of 15–20 nm on jute stem were prepared as described below and its schematic illustration is shown in Fig. 2. The oven dried jute-stem thin slices of the above dimensions were immersed in K2PdCl4 solution in DI water (32 mg/20 mL) and soaked for 2 h. The vial containing the suspension was heated at 70 °C for 1 h. 100 μL formic acid was added to reduce the Pd precursors while the mixture was stirred for 15 min and the color changed to black. Heating was continued for another hour to complete the reduction and lodging of the nanoparticles onto the substrate. The contents were allowed to settle down. The thin jute slices completely covered with black Pd nanoparticles were pulled out (Fig. 3a). They were dried in open air for 24 h and samples were prepared for characterization.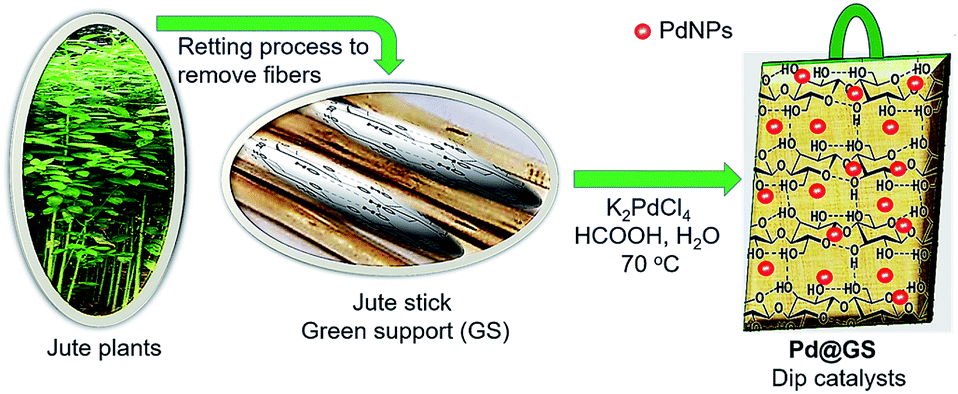 | ||
| Fig. 2 Schematic illustration for the synthesis of Pd@GS. | ||
 | ||
| Fig. 3 Images of (a) the pure GS and prepared Pd@GS catalyst and SEM images of (b) pure green support (c) Pd@GS; inset (d) is a magnified view showing uniform distribution of Pd nanoparticles. | ||
Olefin hydrogenation
The catalytic hydrogenation was performed in a 20 mL screw-capped vial under the conditions stated below. To the suspension of tetrahydroxydiboron (THDB, 3.1 mmol) in 5 mL water, the substrate (1 mmol) was added and stirred. The vial was capped and introduced into a pre-heated (70 °C) oil bath. After thermal equilibrium, the Pd@GS strip was added to the vial, which was stirred magnetically. The progress of the reaction was monitored by TLC (thin layer chromatography) and GC by periodically withdrawing small aliquots from the vial. The product was extracted in dichloromethane (DCM), dried with magnesium sulfate and passed through short silica gel column. The conversion and selectivity were determined by GC and identified and quantified by GC-MS.Quinoline hydrogenations
The transfer hydrogenation of quinoline was conducted in a 10 mL glass reaction tube reactor fitted with magnetic stirrer and a Teflon screw cap. THDB (4.1 mmol), quinoline (1 mmol) and DI water (5 mL) were added and stirred @ 550 rpm. A strip of Pd@GS catalyst was inserted and stirred at 60 °C at the same speed. The progress of the reaction was monitored by TLC and GC as in olefin hydrogenation. After cooling, the reaction mixture was extracted with DCM, dried with magnesium sulfate and the solvent removed under vacuum. The residue was re-dissolved in minimum amount of DCM and passed through a short silica gel column using a mixture of hexane and ethyl acetate (8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) as the eluent. GC was used to measure the conversion and selectivity and the products were identified by GC-MS.
:2) as the eluent. GC was used to measure the conversion and selectivity and the products were identified by GC-MS.
Results and discussion
Synthesis and characterization
The fully grown jute was cut, trimmed. Stems were extracted through the retting process. Since, the jute stick is endowed with highly fibrous, porous, lightweight, voluminous and woody structure, it has extremely high surface area. About 3 million tons of jute sticks are produced annually in India; it is economically cheap, affordable and a biodegradable green precursor. The jute stem is a lignocellulosic raw material, composed of α-cellulose (40.8%), hemicellullose (22.10%), lignin (23.5%), and others.45 Anchoring of Pd nanoparticles was achieved by immersing the jute strips, which contain large number of primary and secondary hydroxyl groups, into potassium tetrachloropalladate (K2PdCl4) solution in DI water and allowing it to be sufficiently soaked in the metal precursor solution for 2 h. The yellow palladium precursor was reduced in situ to black Pd nanoparticles by using formic acid as reductant at 70 °C.The morphological and nanostructural features of pure jute stem and Pd@GS are revealed in SEM images shown in Fig. 3. Pure jute strips are highly fibrous – a characteristic that remained intact after the decoration with Pd nanoparticles via in situ reduction in the acidic media. Formation of spherical Pd nanoparticles with uniform distribution all over the strips is also evident. This was further corroborated by confined area elemental mapping and EDS quantification, as shown in Fig. 4.
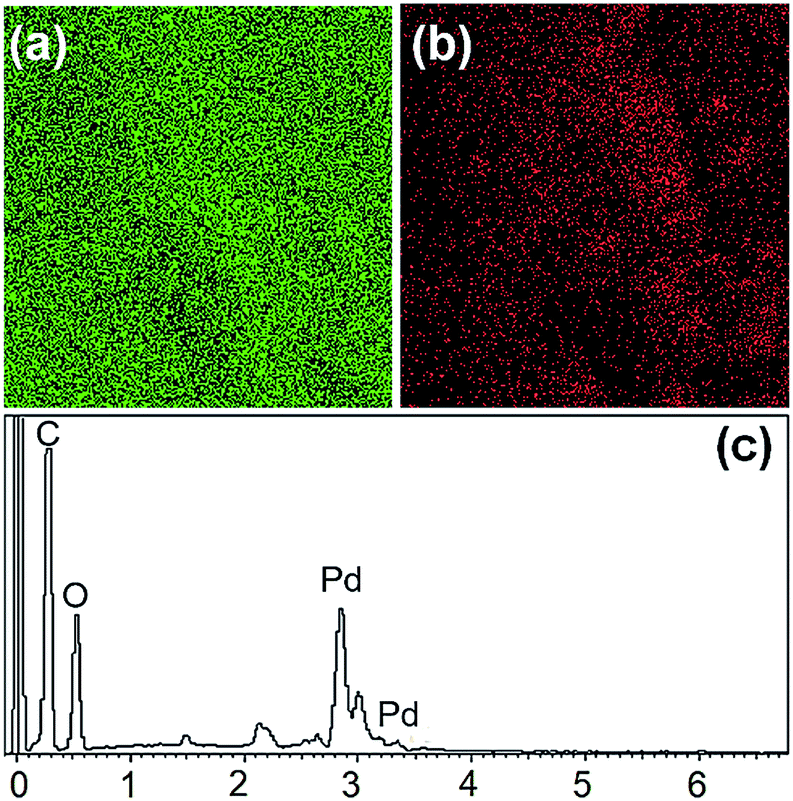 | ||
| Fig. 4 Elemental mapping of (a) carbon and (b) Pd. The EDS signature of the Pd@GS catalyst is shown in (c). | ||
TEM images show uniform distribution of Pd with average diameter of 15–20 nm (Fig. 5a and b), which was further confirmed by measuring the particles size using dynamic light scattering technique. The selected area electron diffraction (SAED) image is shown in Fig. 5c. The calculated d-value could be assigned to the 〈111〉 reflection of the fcc structured Pd nanoparticles.46
 | ||
| Fig. 5 (a) TEM image of Pd nanoparticles anchored on the surface (b) HRTEM image and (c) SAED of Pd@GS. | ||
FTIR spectroscopic technique was used to evaluate the decoration. The abundant number of hydroxyl group on the pure jute stem were detected by a characteristic peak at around 3345 cm−1; there was no significant change in the signature after Pd decoration (see ESI†).
The XPS analysis was performed on the as-synthesized Pd@GS to identify the electronic states of Pd. The overall composition is depicted in the global XPS survey (Fig. 6a) which confirmed the presence of C, O and Pd. Formation of Pd0 on the surface of jute stem is confirmed by the deconvolution in the Pd doublet peaks (Fig. 6b) with binding energies of 335.3 eV (3d5/2) and 340.5 eV (3d3/2) associated with Pd0;47 peaks at 337.2 eV and 342.4 eV are associated with Pd2+ due to the formation of PdO, probably resulting from partial oxidation of the Pd nanoparticles at the ambient.48
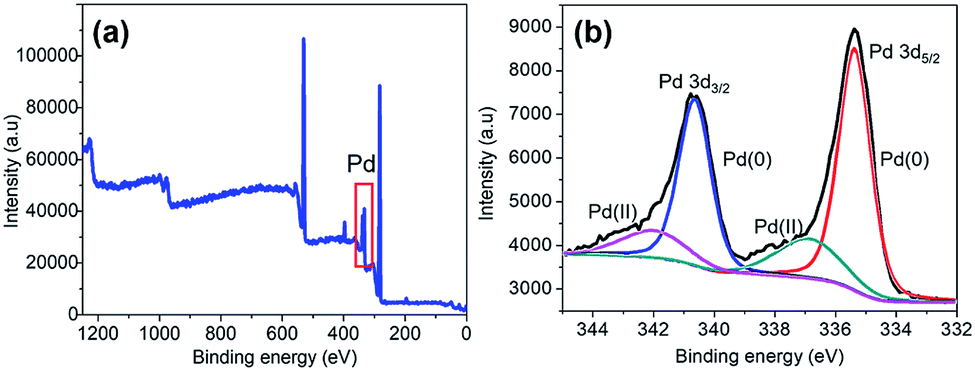 | ||
| Fig. 6 XPS signatures of Pd@GS: (a) the survey data and (b) oxidation states of Pd decorating the support surface. | ||
The Pd content of each strip was quantified by the ICP-OES technique. The optimum amount of palladium nanoparticles was measured by varying the concentration of the K2PdCl4 solutions. The Pd content was found to increase with higher concentration of pre-reduced Pd and then remained constant with an average Pd content determined to be 0.0288 ± 0.008 mg per strip. Hence, the reproducibility of the Pd nanoparticles loading imply the natural limit to its highly hydroxylated surface. This higher loading could be attributed to the combination of strong van der Waals forces and hydrophobic interaction between metal nanoparticles and the jute-stick surface, leading to the nanoparticles immobilization.49,50
Catalysis
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Subst.a (R) | Solv. | THDB (equiv.) | Temp. (°C) | Time (h) | Conv.b (%) | Sel.c (%) | TOF (h−1) |
| a 1 mmol of substrate with 3.1 mmol THDB with Pd@GS as catalyst.b Measured by GC.c Identified by GC-MS.d Without Pd@GS but with 3 equivalent of THDB; nd-not determined.e Conducted reaction using only GS. | ||||||||
| 1 | H | H2O | 3.1 | 70 | 1 | 77 | >99 | 2851 |
| 2 | H2O | 3.1 | 50 | 2 | 48 | 99 | 889 | |
| 3 | MeOH | 3.1 | 70 | 1 | 30 | >99 | 1111 | |
| 4d | H2O | 3.1 | 70 | 1 | <8 | nd | nd | |
| 5e | H2O | 3.1 | 70 | 10 | nd | nd | nd | |
| 6 | p-CH3 | H2O | 3.1 | 70 | 1 | >99 | >99 | 3704 |
| 7 | p-OCH3 | H2O | 3.1 | 70 | 1 | 98 | >99 | 3630 |
| 8 | p-Cl | H2O | 3.1 | 70 | 1 | >99 | >99 | 3704 |
| 9 | m-NO2 | H2O | 3.1 | 45 | 20 | >99 | <2 | 185 |
| 10 | THF | 1.1 | 45 | 24 | 48 | 81 | 74 | |
| 11 | THF | 2.1 | 45 | 24 | 88 | 72 | 136 | |
| 12 | DCM | 2.1 | 45 | 24 | 82 | 60 | 126 | |
| 13 | o-Br | H2O | 2.1 | 40 | 24 | >99 | <5 | 154 |
| 14 | DCM | 1.1 | 40 | 48 | 40 | 75 | 31 | |
| 15 | THF | 1.1 | 40 | 48 | 89 | 82 | 69 | |
| 16 | THF | 1.1 | 50 | 48 | 93 | 67 | 77 | |
Wide applicability of the catalyst was examined by employing styrene with various functional groups – both electron donating and electron withdrawing groups. No significant effect of the electron donating or electron withdrawing groups was seen in the case of styrenyl olefins. For example, p-methyl styrene and p-vinyl anisole were quantitatively hydrogenated with maximum selectivity (entries# 6 and 7). A similar trend was observed with choloro-substituted styrene (entry# 8); no dehalogenation was observed. The reactivity of Pd@GS towards the reduction of m-nitrostyrene (where under similar reaction conditions, both olefin and nitro functional groups are prone to simultaneous reduction) was also evaluated. At 45 °C, the conversion was complete but non-selective, where both functional groups were completely and simultaneously reduced in 20 h, in water.
Selectivity against two or more competitive functional groups toward hydrogenation process is highly challenging. Reaction conditions, including choice of solvent, need critical optimization. In DCM, 82% of the m-nitrostyrene was converted and the selectivity towards m-nitroethylbenzene reached up to 60% while for the remaining 40% both nitro and olefin functional groups were completely reduced to form m-aminoethylbenzene, at 45 °C with the use of 2.1 equivalent of THDB as reductant.
Under similar reaction conditions with THF the overall conversion was 88% with significant enhancement in selectivity (72%) towards m-nitroethylbenzene (entry# 11). The effect of temperature and variation of mole equivalent of THDB in selectivity were also evaluated in THF. The results are shown in Fig. 7a. On increasing the mole equivalent of THDB from 1.1 to 4.1, the conversion increased from 48% to >99%. However, the selectivity drastically reduced from ∼81% to ∼47% for the 4.1 mole equivalent of THDB. These results are instructive in that reaction parameters can be subtly tweaked to reduce the alkenyl group in styrene in the presence of an easily reducible nitro functional group with very high selectivity.
 | ||
| Fig. 7 (a) Variation in the conversion and selectivity of m-nitrostyrene at 45 °C with THDB mole equivalent in THF (b) conversion and selectivity of o-bromostyrene with 1.1 equivalent of THDB in different solvents. EB: ethylbenzene. | ||
Another interesting example is the reduction of o-bromostyrene to form the corresponding olefin hydrogenated product (cf. Fig. 7b), o-bormoethylbenzene at 40 °C. In most cases, hydrogenation of bromostyrene occurs via either hydrogenolysis, where dehalogenation is obviated, or through polymerization, or both take place simultaneously. Yet, it is to our delight that our Pd@GS catalyst demonstrated excellent reactivity and selectivity without hydrogenolysis nor polymerization. In water as solvent, at 40 °C, o-bromostyrene yielded only ethylbenzene with complete debromination in 24 h (entry# 13). But when the reaction was performed at 40 °C in DCM with 1.1 equivalent of THDB the conversion achieved was about 40% in 48 h with very high (75%) selectivity towards the formation of o-bromoethylbenzene and significant amount of (22%) ethylbenzene with 3% of styrene was also formed (entry# 14) as side product. By changing the solvent to THF, the conversion was significantly improved to 89% and the highest selectivity (82%) towards o-bromoethylbenzene was achieved (entry# 15). To the best of our understanding, it is highest selectivity available in literature under such very mild reaction conditions. On increasing the temperature at 50 °C, the conversion was slightly improved to 93% with the sacrifice of selectivity, which reached up to 67% o-bromoethylbenzene and 33% de-brominated product (entry# 16).
Nonetheless, this reaction could be tuned to synthesize bromoalkyl benzene from bromostyrene and at the same time it is also worthy to mention that debrominated alkylbenzene, which bears significant value in literature,51 can also be prepared from the bromostyrene by simply changing the reaction condition using this catalytic system. This dual nature of the catalyst may mark it as a practical catalyst.
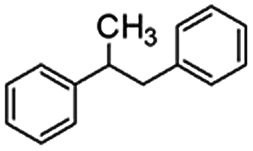
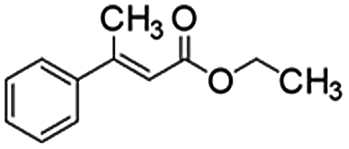
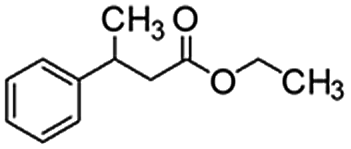
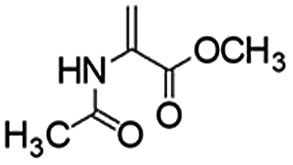
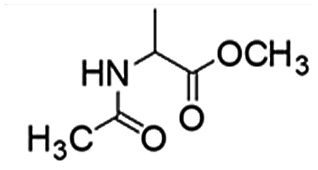
Following that report, we employed various olefins to test the wide applicability of Pd@GS. The results are shown in Table 2 which summarizes the comparison of reactivity of Pd@GS towards differently substituted linear and cyclic alkenes. For instance, cyclic alkene (entry# 1) was hydrogenated in THF at 60 °C in 20 h with 49% conversion. Hydrogenation was also conducted with the tri-substituted alkenes such as trans-α-methylstilbene (entry# 2) and ethyl trans-β-methylcinnamate (entry# 3) in THF at 70 °C. 33% conversion was achieved with trans-α-methylstilbene with maximum selectivity. Clear contrast was observed when the functionalized olefins, for instance methyl-2-acetamido propionate (entry# 4), were employed for hydrogenation.

| Entry | Catalyst | Substratea | Product | Solv. | Time (min) | Conv.b (%) | Sel.c (%) | TOF (h−1) |
|---|---|---|---|---|---|---|---|---|
| a Substrate used 1 mmol and THDB 3.1 mmol at 60 °C.b Conversions were measured by GC.c Identified by GC-MS; nd-not determined.d Reaction performed at 40 °C. | ||||||||
| 1 | Pd@GS | 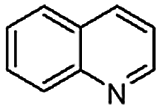 |
 |
H2O | 120 | 85d | >99 | 1574 |
| 2 | H2O | 40 | >99 | >99 | 4938 | |||
| 3 | DCM | 60 | nd | nd | nd | |||
| 4 | THF | 60 | nd | nd | nd | |||
| 5 | MeOH | 60 | 89 | nd | nd | |||
| 6 | Pd@GS | 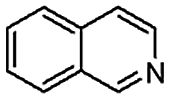 |
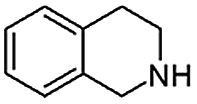 |
H2O | 300 | >99 | >99 | 741 |
| 7 | Pd@GS | 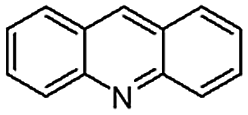 |
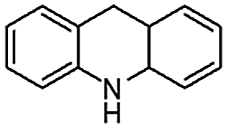 |
H2O | 90 | 99 | >99 | 2640 |
| 8 | Pd@GS | 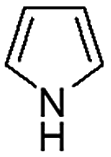 |
 |
H2O | 120 | >99 | >99 | 1652 |
| 9 | Pd@GS | 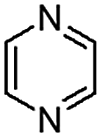 |
 |
H2O | 120 | 98 | >99 | 1815 |
| 10 | GS |  |
 |
H2O | 60 | nd | nd | nd |
| 11 | PdNPs | H2O | 60 | >99 | >99 | 3667 | ||
| 12 | Pd/C | H2O | 60 | 98 | >99 | 3593 | ||
| 13 | Pd@PVP | H2O | 60 | 95 | >99 | 3518 | ||
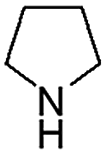
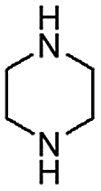
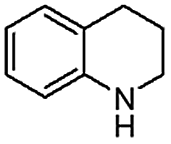
When temperature was increased to 60 °C, the reaction proceeded smoothly with >99% conversion and >99% selectivity in 40 min. It can be observed that turn over frequency (TOF) was significantly higher (TOF 4938 h−1) compared to the TOF (1574 h−1) at lower temperature (entries# 1 and 2). Changing solvent to DCM and THF affected the yield adversely; no hydrogenation occurred (entries# 3 and 4). With methanol, 89% conversion was achieved (entry# 5). This could be ascribed to the participation of lone e-pair of the oxygen atom in MeOH solvent in the formation of boron adduct in the transition state. Conversion was improved with increasing amount of THDB and best results were obtained with 4.1 equivalent of THDB. It would seem that large amount of gaseous hydrogen escapes reaction medium without interacting with the metal to form metal-hydride intermediate, which is responsible for the hydrogenation of heterocyclic compounds. Under the above optimized reaction conditions, other N-heterocycles were subjected to transfer hydrogenation reaction in water and smoothly converted to hydrogenated product contain nitrogen ring with maximum selectivity. But surprisingly, though isoquinoline was quantitatively hydrogenated with >99% selectivity (entry# 6), it took much longer time (5 h) than the quinoline. In case of polycyclic aromatic heterocycles, such as acridine, the reactions proceeded smoothly with excellent yield and selectivity (entry# 7). Five and six membered ring compounds, pyrrole and pyrazine, were also employed as substrate and the catalyst Pd@GS, was found be highly reactive to convert the substrate into the hydrogenated product in water with TOF 1652 h−1 and 1815 h−1 respectively (entries# 8 and 9). There was no conversion was observed only in presence of GS but in absence of PdNPs and THDB (entry# 10). To compare the catalytic activity of Pd@GS with standard catalyst (entries# 11–13), it was observed that our “dip-catalyst”, Pd@GS, performance was having an edge in terms of reactivity and gain superiority in terms of re-usability over the standard catalyst used.
These examples demonstrates the superior effect Pd nanoparticles immobilized onto the green support. The high reactivity aspect makes one believe that Pd nanoparticles on the cellulosic support effectively suppress the agglomeration of the bear surface nanoparticle, assisting in achieving rather high catalytic activity in comparison to the similar Pd-supported systems.54,55 The exclusive selectivity towards py-THQ depends on the interaction of the oraganonitrogen with the metal center in the strip.56 Availability of the large number of aliphatic 1° and 2° hydroxyl group on the cellulosic jute stick also affect the instantaneous microenvironment around the metal centers benignly.57 The possibility of hydrogen bonding interaction mediating the heterocyclic ring hydrogenation faster than that of the carbocyclic ring of the substrate, resulting exclusive selectivity towards py-THQ, cannot be ruled out.
Recyclability studies
One of the extraordinary feature of “dip-catalyst” is the ease of deployment in the system and its effortless post-reaction removal. Hence, reusability of spent catalyst in subsequent cycles for transfer hydrogenation of quinoline under optimized reaction condition is possible. To investigate its robustness and sustainability, the catalyst were extracted from the reaction mixture, thoroughly washed with water and dichloromethane and, dried for use in next cycle with fresh batch of quinoline as the substrate and THDB as reductant in aqueous medium. This procedure was repeated multiple times and catalyst was used in 32 consecutive cycles with excellent catalytic propensity without appreciable degradation of catalytic activity (Fig. 8a). This was corroborated by ICP-OES examination; samples were extracted at certain intervals and analyzed to determine if any Pd was leached out. As can be seen from Fig. 8b, no trace of Pd was detected in the solution. The surface morphology and stability of the recycled catalyst showed no severe damage to the jute-stick surface or no agglomeration of the Pd nanoparticles (see ESI†). The XPS analysis revealed that the peak at 335.4 eV and 340.6 eV reflect the preservation of Pd0 oxidation state. A small shoulder at 336.4 eV corresponding to PdO could be ascribed to be due to long exposure to aerobic condition.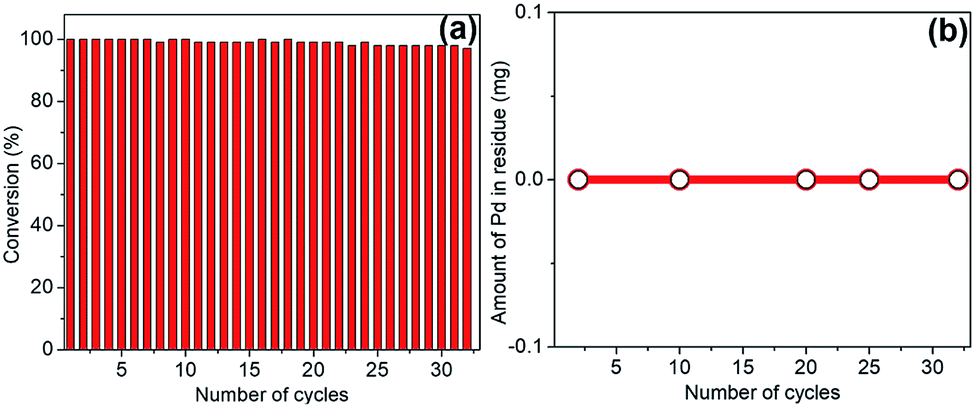 | ||
| Fig. 8 (a) Reusability potential of the catalyst for quinoline hydrogenation in water using THDB reductant. (b) ICP-OES data showing the evidence of no leaching of Pd from the dip-catalyst after a large number of cycles. | ||
From the foregoing discussion, it appears that the excellent survivability of the catalyst comes from strong and appropriate bonding with huge number of hydroxyls groups on the support.
Conclusions
Dip catalyst based on Pd nanoparticles supported on the naturally-occurring surface architecture of jute-stick was fabricated for its excellent transfer hydrogenation activity under mild conditions in water. The natural cellulosic surface provided favorable interaction for Pd nanoparticles. This dip catalyst was gainfully employed for chemoselective transfer hydrogenation of olefin with excellent conversion and exceptional organic functional group tolerance using THDB, and provided very high TOF. Quinoline and other N-heteroarenes were selectively and quantitatively hydrogenated at 60 °C within a very short span of time (cf. 40 min) under atmospheric condition. The catalyst was found to possess impressive stability and could be used over 32 consecutive cycles without adversely affecting its original reactivity or surface morphology and/or chemical composition. The unique advantage of effortless catalyst monitoring, insertion or removal between the cycles were demonstrated. The use of this naturally-occurring support, ease of handling, low cost fabrication, convenient deployment, favorable organic functional groups on support to provide enhanced stability, impressive conversion and remarkable chemoselectivity, use of water as reaction medium and exceptional reusability may lead to commercialization of this catalyst for industrial applications. The environmentally benign catalyst fabrication strategy and remarkable catalytic transfer hydrogenations activity for a series of olefins and N-heteroarenes offers a new route to develop catalysts with other metal nanoparticles for various catalytic transformations. Currently some such catalytic reactions are under study in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
MNS gratefully acknowledges the National Plan for Science, Technology and Innovation (MAARIFAH)-King Abdulaziz City for Science and Technology through the Science and Technology (KACST) Unit at King Fahd University of Petroleum and Minerals (KFUPM), the Kingdom of Saudi Arabia, award number 15-NAN4650-04. The author also gratefully acknowledges Dr Md. Abdul Aziz for providing the jute sticks.Notes and references
- Z. Zhang, N. A. Butt and W. Zhang, Chem. Rev., 2016, 116, 14769–14827 CrossRef CAS PubMed.
- P. Etayoa and A. Vidal-Ferran, Chem. Soc. Rev., 2013, 42, 728–754 RSC.
- N. M. Rezayee, D. C. Samblanet and M. S. Sanford, ACS Catal., 2016, 6, 6377–6383 CrossRef CAS.
- J. Yuwen, S. Chakraborty, W. W. Brennessel and W. D. Jones, ACS Catal., 2017, 7, 3735–3740 CrossRef.
- M. Shevlin, M. R. Friedfeld, H. Sheng, N. A. Pierson, J. M. Hoyt, L.-C. Campeau and P. J. Chirik, J. Am. Chem. Soc., 2016, 138, 3562–3569 CrossRef CAS PubMed.
- Y. Duan, L. Li, M.-W. Chen, C.-B. Yu, H.-J. Fan and Y.-G. Zhou, J. Am. Chem. Soc., 2014, 136, 7688–7700 CrossRef CAS.
- Y. T. Xia, J. Ma, X. D. Wang, L. Yanga and L. Wu, Catal. Sci. Technol., 2017, 7, 5515–5520 RSC.
- X. Zhang, L. J. Durndell, M. A. Isaacs, C. M. A. Parlett, A. F. Lee and K. Wilson, ACS Catal., 2016, 6, 7409–7417 CrossRef CAS.
- J. Wen, R. Tan, S. Liu, Q. Zhao and X. Zhang, Chem. Sci., 2016, 7, 3047–3051 RSC.
- A. Vandekerkhove, L. Claes, F. De Schouwer, C. V. Goethem, I. F. J. Vankelecom, B. Lagrain and D. E. De Vos, ACS Sustainable Chem. Eng., 2018, 6, 9218–9228 CrossRef CAS.
- X. C. Zhang, Y. H. Hu, C. F. Chen, Q. Fang, L. Y. Yang, Y. B. Lu, L. J. Xie, J. Wu, S. Li and W. Fang, Chem. Sci., 2016, 7, 4594–4599 RSC.
- H. Konnerth and M. H. G. Prechtl, Green Chem., 2017, 19, 2762–2767 RSC.
- S. Chakraborty and H. Berke, ACS Catal., 2014, 4, 2191–2194 CrossRef CAS.
- D. Wang and D. Astruc, Chem. Rev., 2015, 115, 6621–6686 CrossRef CAS PubMed.
- R. A. W. Johnstone, A. H. Wilby and I. D. Entwistle, Chem. Rev., 1985, 85, 129–170 CrossRef CAS.
- A. Vasseur, R. Membrat, D. Palpacelli, M. Giorgi, D. Nuel, L. Giordano and A. Martinez, Chem. Commun., 2018, 54, 10132–10134 RSC.
- P. Sharma and Y. Sasson, Green Chem., 2019, 21, 261–267 RSC.
- Y. Song, H. Wang, S. Liang, Y. Yu, L. Li and L. Wu, J. Catal., 2018, 361, 105–115 CrossRef CAS.
- N. Mrsaic, L. Lefort, J. A. F. Boogers, A. J. Minnaard, B. L. Feringa and J. G. de Vries, Adv. Synth. Catal., 2008, 350, 1081–1089 CrossRef.
- M. Cerro-Alarcón, A. Maroto-Valiente, I. Rodríguez-Ramos and A. Guerrero-Ruiz, Carbon, 2005, 43, 2711–2722 CrossRef.
- A. R. McDonald, C. Müller, D. Vogt, G. P. M. van Klinka and G. van Koten, Green Chem., 2008, 10, 424–432 RSC.
- Z. An, J. He, Y. Dai, C. Yu, B. Li and J. He, J. Catal., 2014, 317, 105–113 CrossRef CAS.
- X. Chena, W. Wang, H. Zhu, W. Yanga and Y. Ding, J. Mol. Catal., 2018, 456, 49–56 CrossRef.
- P. Ju, S. Wu, Q. Su, X. Li, Z. Liu, G. Lib and Q. Wu, J. Mater. Chem. A, 2019, 7, 2660–2664 RSC.
- A. W. Petrov, D. Ferri, O. Kröcher and J. A. van Bokhoven, ACS Catal., 2019, 9, 2303–2312 CrossRef CAS.
- J. Lee, Y. S. Ryou, S. Hwang, Y. Kim, S. J. Cho, H. Lee, C. H. Kim and D. H. Kim, Catal. Sci. Technol., 2019, 9, 163–168 RSC.
- T. Guo, X. Niec, J. Dub and J. Li, Chem. Eng. J., 2019, 361, 1345–1351 CrossRef CAS.
- K. Yakabi, A. Jones, A. Buchard, A. Roldan and C. Hammond, ACS Sustainable Chem. Eng., 2018, 6, 16341–16351 CrossRef CAS.
- M. N. Shaikh, M. A. Aziz, A. N. Kalanthoden, A. Helal, A. S. Hakeem and M. Bououdina, Catal. Sci. Technol., 2018, 8, 4709–4717 RSC.
- R. Abu-Reziq, H. Alper, D. Wang and M. L. Post, J. Am. Chem. Soc., 2006, 128, 5279–5282 CrossRef CAS PubMed.
- G. Sun, J. An, H. Hu, C. Li, S. Zuo and H. Xia, Catal. Sci. Technol., 2019, 9, 1238–1244 RSC.
- W. Dong, L. Zhang, C. Wang, C. Feng, N. Shang, S. Gao and C. Wang, RSC Adv., 2016, 6, 37118–37123 RSC.
- J. Li, X. Zhou, N.-Z. Shang, C. Feng, S.-T. Gao and C. Wang, New J. Chem., 2018, 42, 16823–16828 RSC.
- X. Xu, Y. Li, Y. Gong, P. Zhang, H. Li and Y. Wang, J. Am. Chem. Soc., 2012, 134, 16987–16990 CrossRef CAS PubMed.
- Y. Gong, P. Zhang, X. Xu, Y. Li, H. Li and Y. Wang, J. Catal., 2013, 297, 272–280 CrossRef CAS.
- Z. Xiang, Y. Chen, Q. Liu and F. Lu, Green Chem., 2018, 20, 1085–1094 RSC.
- V. Sharma, A. Bahuguna and V. Krishnan, Adv. Mater. Interfaces, 2017, 4, 1700604 CrossRef.
- E. Hariprasad and T. P. Radhakrishnan, Chem.–Eur. J., 2010, 16, 14378–14384 CrossRef CAS PubMed.
- E. Hariprasad and T. P. Radhakrishnan, ACS Catal., 2012, 2, 1179–1186 CrossRef CAS.
- G. C. Zheng, L. Polavarapu, L. M. Liz-Marzan, I. Pastoriza- Santos and J. Perez-Juste, Chem. Commun., 2015, 51, 4572–4575 RSC.
- V. W. Faria, D. G. Oliveira, M. H. Kurz, F. F. Gonçalves, C. W. Scheeren and G. R. Rosa, RSC Adv., 2014, 4, 13446–13451 RSC.
- M. N. Shaikh, A. Helal, A. N. Kalanthoden, B. Najjar, M. A. Aziz and H. D. Mohamed, Catal. Commun., 2019, 119, 134–138 CrossRef CAS.
- M. N. Shaikh, M. A. Aziz, A. Helal, M. Bououdina, Z. H. Yamani and T.-J. Kim, RSC Adv., 2016, 6, 41687–41695 RSC.
- M. N. Shaikh, M. Bououdina, A. A. Jimoh, M. A. Aziz, A. Helal, A. S. Hakeem, Z. H. Yamani and T.-J. Kim, New J. Chem., 2015, 39, 7293–7299 RSC.
- L. Nayak, A. Lakshmanan and D. P. Ray, J. Indian Chem. Soc., 2012, 89, 1723–1727 CAS.
- T. Teranishi and M. Miyak, Chem. Mater., 1998, 10, 594–600 CrossRef CAS.
- F. G. de Rivera, I. Angurell, M. D. Rossell, R. Erni, J. Llorca, N. J. Divins, G. Muller, M. Seco and O. Rossell, Chem.–Eur. J., 2013, 19, 11963–11974 CrossRef PubMed.
- S. Y. Xu, X. P. Yan, Q. Bu and H. A. Xia, Cellulose, 2017, 24, 2403–2413 CrossRef CAS.
- G. Zheng, K. Kaefer, S. Mourdikoudis, L. Polavarapu, B. Vaz, S. E. Cartmell, A. Bouleghlimat, N. J. Buurma, L. Yate, A. R. de Lera, L. M. Liz-Marzán, I. Pastoriza-Santos and J. Pérez-Juste, J. Phys. Chem. Lett., 2015, 6, 230–238 CrossRef CAS PubMed.
- H. Tian and J. He, Langmuir, 2016, 32, 12269–12282 CrossRef CAS PubMed.
- A. Goswami, A. K. Rathi, C. Aparicio, O. Tomanec and M. Petr, ACS Appl. Mater. Interfaces, 2017, 9, 2815–2824 CrossRef CAS PubMed.
- R. L. Halterman, in Comprehensive Asymmtric Catalysis, ed. E. N. Jacobson, A. Paltz and H. Yamamoto, Springer, New York, 1999, p. 183 Search PubMed.
- S. P. Cummings, T.-N. Le, G. E. Fernandez, L. G. Quiambao and B. J. Stokes, J. Am. Chem. Soc., 2016, 138, 6107–6110 CrossRef CAS PubMed.
- Q. Xuan and Q. Song, Org. Lett., 2016, 18, 4250–4253 CrossRef CAS PubMed.
- J. Wang, Y.-L. Qin, X. Liu and X.-B. Zhang, J. Mater. Chem., 2012, 22, 12468–12470 RSC.
- R. H. Fish, E. Baralt and S. J. Smith, Organometallics, 1991, 10, 54–56 CrossRef CAS.
- Y. Zhang, M. E. Grass, S. E. Habas, F. Tao, T. Zhang, P. Yang and G. A. Somorjai, J. Phys. Chem. C, 2007, 111, 12243–12253 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra06285h |
| This journal is © The Royal Society of Chemistry 2019 |