
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltra-small dispersed CuxO nanoparticles on graphene fibers for miniaturized electrochemical sensor applications†
Jinfeng Zenga,
Xiaoteng Dingb,
Liwei Chena,
Le Jiaoa,
Yuze Wanga,
Christopher D. Windlec,
Qing Han *a and
Liangti Qua
*a and
Liangti Qua
aKey Laboratory of Photoelectronic/Electrophotonic Conversion Materials, Key Laboratory of Cluster Science, Ministry of Education of China, School of Chemistry and Chemical Engineering, Beijing Institute of Technology, Beijing 100081, P. R. China. E-mail: qhan@bit.edu.cn
bCollege of Life Sciences, Qingdao University, Qingdao 266071, P. R. China
cSolar Energy and Advanced Materials Group, Department of Chemical Engineering, University College London, Torrington Place, London, WC1E 7JE, UK
First published on 9th September 2019
Abstract
A graphene microfiber (GF) modified with ultrafine CuxO nanoparticles (CuxONPs/GF) has been fabricated by direct annealing of electrodeposited nano-sized copper-based metal organic frameworks (HKUST-1) and used as an electrode for nonenzymatic H2O2 sensing. Benefiting from the unique microfiber architecture and synergetic effects, as well as strong coupling between components with many active sites and boosted electron transport, the CuxONPs/GF electrode shows prominent sensitivity, selectivity and long-term operational stability for the detection of H2O2. Further work successfully applied this CuxONPs/GF electrode to detection of H2O2 in real samples such as milk and human serum. These results indicate that the CuxONPs/GF is a promising mini-sized sensor in electrochemical analysis.
Introduction
The demand for electrochemical sensors with high sensitivity, low cost, excellent selectivity, and facile miniaturization has stimulated extensive research into developing versatile materials with remarkable electrocatalytic activity. Compared to conventional macro-electrodes,1 fiber-type electrodes with a one-dimensional microstructure, particularly carbon fiber microelectrodes possessing one dimensional diffusion, have been widely fabricated and used in sensors due to their low cost, small volume, portability and good biocompatibility. However, the poor electrocatalytic activity and low current response of carbon fiber microelectrodes seriously restrict their application in sensing.Graphene has potential as an electrode for electrochemical analysis owing to its large specific surface area, excellent electron mobility, high mechanical properties and good biocompatibility. Recently, we reported a graphene fiber assembled with chemically reduced graphene sheets, which exhibited promising electrochemical sensing activity, mainly owing to the inherent defects and residual oxygenated functional groups as active sites. However, the weak electrocatalytic properties of graphene restrict its wide application in electrochemical analysis. It is revealed that combining graphene fiber with catalytically active nanomaterials such as metals and metal oxides is an efficient way to enhance the electrocatalytic activity.2–12 Copper oxide nanoparticles (CuxONPs) are an attractive alternative electroactive species due to their abundance, low cost, good chemical stability and unique electronic properties.13,14 Hybridizing graphene with CuxONPs is an effective way, where the composite materials have fully exploited the structural merits of individual components and improved the analytic performance. So far, various strategies, including thermal conversion,15 ion exchange,16 wet chemistry,17 microwave,18 template growth,19 and anion-assisted approaches20 have been employed to prepare CuxONPs/graphene hybrids. Unfortunately, as-prepared CuxONPs from these approaches suffer from aggregation on graphene-based materials, leading to poor dispersion and a low number of active sites with poor electrocatalytic performance. In addition, although CuxONPs/graphene composites have been widely investigated and demonstrated with great potential for electrochemical analysis, there is still a lack of exploration for its miniaturized sensors. Thus, it is urgent to design and develop a simple and effective method for the production of CuxONPs/graphene fiber-based microsensor with a high number of active sites and highly efficient electrocatalytic activity.
Hydrogen peroxide (H2O2) is indispensable to ecosystems and is widely used in foodstuffs, the environment, medicine and industry. Excess H2O2 can lead to toxicity for humans and to the environment.21–24 Although electrochemical enzyme-based sensors have good selectivity and high performance, their applications are limited because of their poor stability and high cost. Therefore, the development of non-enzymatic sensors has drawn tremendous attention. Herein, we report a graphene microfiber electrode modified by well-dispersed and ultrafine CuxO nanoparticles (CuxONPs/GF) derived from copper-based metal organic frameworks (HKUST-1) by a direct electrodeposition self-assembly technique for nonenzymatic electrochemical H2O2 detection. The unique nanoarchitecture and synergetic effect as well as strong coupling between components endows them with a high density of active sites and enhanced electron transport leading to improved electrochemical micro-sensing. The resulting hybrid fiber electrode exhibits a very low detection limit of 0.023 μM and a rapid response time of 1 s. Our work provides a straightforward method for synthesizing highly efficient nonenzymatic micro-sensing electrodes for H2O2 detection.
Experimental
Synthesis of graphene fiber (GF)
Graphene oxide (GO) was synthesized by oxidation of graphite powder according to the modified Hummers' method.25 Then, graphene oxide fibers (GOF) were prepared by a wet spinning strategy. Typically, 3 mL GO (20 mg mL−1) was spun into a methanol solution saturated by potassium chloride (KCl) with a speed of 0.1 m s−1 by an injector (with a diameter of 0.25 mm), followed by drying. To obtain GF, the GOF was heated to 200 °C for 2 h at first and then further heated to 1000 °C for 2 h with a heating rate of 10 °C min−1 under N2/H2 (v/v = 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) atmosphere.
:1) atmosphere.
Synthesis of HKUST-1 nanoparticles decorated GF (HNPs/GF)
A two-electrode system was used for the preparation of HNPs/GF (HNPs = HKUST-1 nanoparticles). Firstly, 0.2416 g of cupric nitrate trihydrate (Cu(NO3)2·3H2O), 0.2102 g of 1,3,5-benzenetricarboxylic acid (H3BTC) and 0.7748 g of tetrabutylammonium hexafluorophosphate (NBu4PF6) were dispersed in 20 mL of N,N-dimethylformamide (DMF) solution, and the mixture was treated by ultrasound for 30 min to obtain a homogeneous solution. Next, the as-synthesized GF (2.0 mm length) and a commercial Pt plate were used as the working electrode and counter electrode, respectively. The HNPs/GF was fabricated by the electrodeposition of HNPs on the GF surface in the above mixed solution (degassed with N2 for 30 min) at −1.5 V for 60 s, the obtained sample is denoted HNPs60/GF. For comparison, a series of HNPs/GF samples with different amounts of HNPs were obtained by adjusting the electrodeposition time (30 s, 90 s, and 120 s) in the same mixed solution, which were denoted HNPs30/GF, HNPs90/GF and HNPs120/GF, respectively.Synthesis of CuxONPs/GF
The CuxONPs/GF was synthesized by direct annealing of HNPs/GF (HNPs30/GF, HNPs60/GF, HNPs90/GF and HNPs120/GF) samples from room temperature to 400 °C in N2 with a ramp rate of 10 °C min−1 and stabilized for 2 h, then cooled to room temperature naturally, and denoted CuxONPs30/GF400, CuxONPs60/GF400, CuxONPs90/GF400 and CuxONPs120/GF400, respectively.In order to study the influence of the annealing temperature on the performance of the final samples, a series of CuxONPs/GF samples were obtained by annealing the HNPs60/GF sample at 300 °C and 500 °C, respectively. The obtained samples were denoted CuxONPs60/GF300 and CuxONPs60/GF500, respectively.
Synthesis of CuxONPs/GF electrode
The CuxONPs/GF was glued with silver conductive adhesive onto stainless steel sheets (2.5 × 0.5 cm). The end of the tip was sealed with molten paraffin. The exposed CuxONPs/GF was cut down to 2.0 mm in length and used as the electrode. The exposed stainless steel sheet was then connected with the electrochemical workstation.Results and discussion
The procedure for the fabricated CuxONPs/GF is shown in Fig. 1. Firstly, GO suspension (Fig. 1a) was converted into a GO fiber (GOF, Fig. 1b) by a wet spinning strategy. After drying naturally, the GOF was thermally reduced to graphene fiber (GF, Fig. 1c). The obtained GF with a diameter of 44 μm appears as wrinkled sheet-like surface morphologies (Fig. S1†). Then, electrodeposition self-assembly of the copper MOF (HKUST-1 nanoparticles, HNPs) occurred in a mixed solution containing cupric nitrate trihydrate (Cu(NO3)2·3H2O) and 1,3,5-benzenetricarboxylic acid (H3BTC), in which HNPs grew in situ on the surface of GF after 60 s of electrodeposition (HNPs60/GF, Fig. 1d), which was confirmed by X-ray diffraction (XRD, Fig. S2†). As shown in Fig. 2a, the obtained HNPs60/GF shows fiber features with a diameter of 45 μm, larger than that of the bare GF. Fig. 2b demonstrates that the HNPs60 with an octahedral structure have a uniform size of 200 nm and are well distributed over the surface of GF. Further annealing at 400 °C led to pyrolysis of HNPs60 with simultaneous formation of CuxO nanoparticles (CuxONPs60, the size is about 10 nm) well distributed on the sheets of GF (CuxONPs60/GF400, Fig. 2c–e). High-resolution transmission electron microscopy (HR-TEM) of CuxONPs/GF (Fig. 2f) indicates clear lattice fringes with a spacing of 0.24, 0.27 and 0.30 nm, corresponding to the Cu2O(111), CuO(110) and Cu2O(110), respectively.26–30 Element mapping reveals the coexistent and uniform distribution of C, O and Cu elements for CuxONPs60/GF400 (Fig. 3a). An X-ray photoelectron spectrum (XPS) of the bare GF shows C 1s (284.6 eV) and O 1s (531.2 eV) peaks. Apart from the C 1s and O 1s peaks observed in the GF, the CuxONPs60/GF400 shows the two apparent Cu 2p3/2 and Cu 2p1/2 features at 934.2 eV and 944.4 eV with a Cu atomic percentage of 0.76% (Fig. 3b), indicating the presence of CuxONPs60, which is consistent with the energy-dispersive X-ray spectroscopy measurement (EDS, Fig. S3 and Table S1†). The ratio of C/O decreases from 11.1 (GF) to 8.4 (CuxONPs60/GF400), demonstrating an increase in the oxygen content resulting from the introduction of CuxO. In addition, the chemical states of copper in CuxONPs60/GF400 are revealed by the high-resolution XPS spectra (Fig. 3c). The signals of Cu 2p3/2 and Cu 2p1/2 can each be deconvoluted into two peaks of Cu2O (933.4 eV and 935.4 eV) and CuO (941.9 eV and 944.2 eV)31 ascribed to the oxidation products of HNPs after annealing.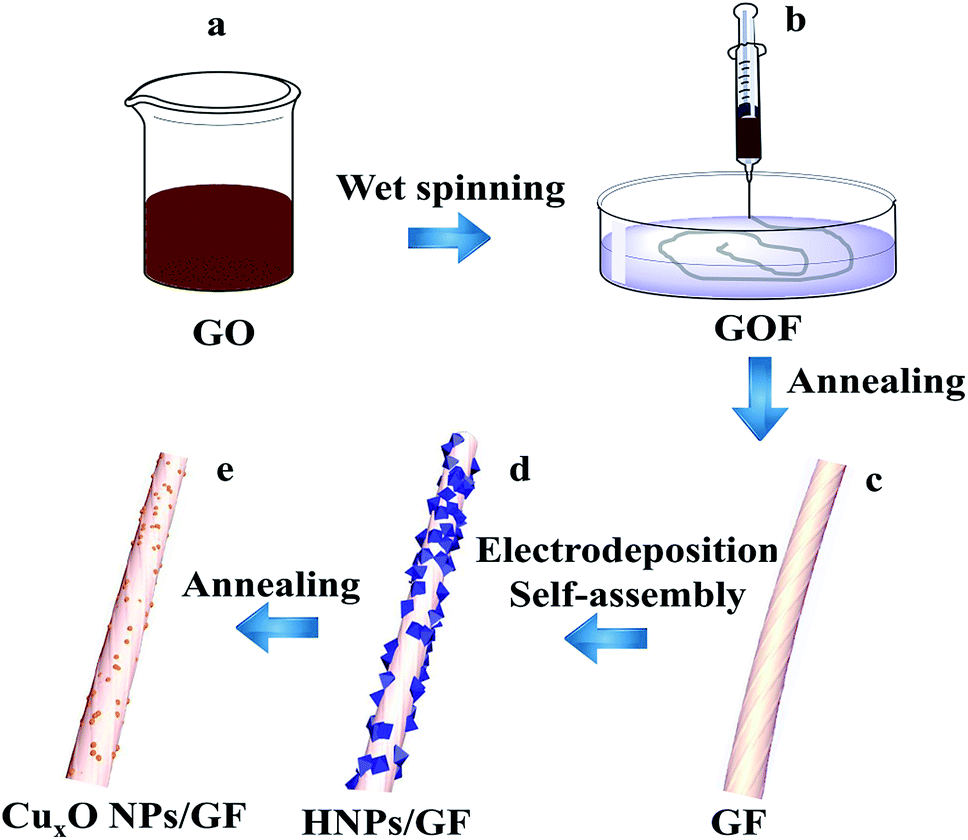 | ||
| Fig. 1 Schematic illustration of the preparation of CuxONPs/GF. (a) GO solution, (b) GOF, (c) GF, (d) HNPs/GF, (e) CuxONPs/G. | ||
 | ||
| Fig. 2 (a) Scanning electron microscopy (SEM) images of HNPs60/GF. (b) The enlarged view of the rectangle area in (a). The inset in (b) is an enlarged SEM image of a single HNPs60. (c) SEM image of CuxONPs60/GF400. (d) The enlarged view of the rectangle area in (c). (e) Transmission electron microscopy (TEM) image of sheets within the CuxONPs60/GF400. (f) The enlarged view of the rectangle area in (e). | ||
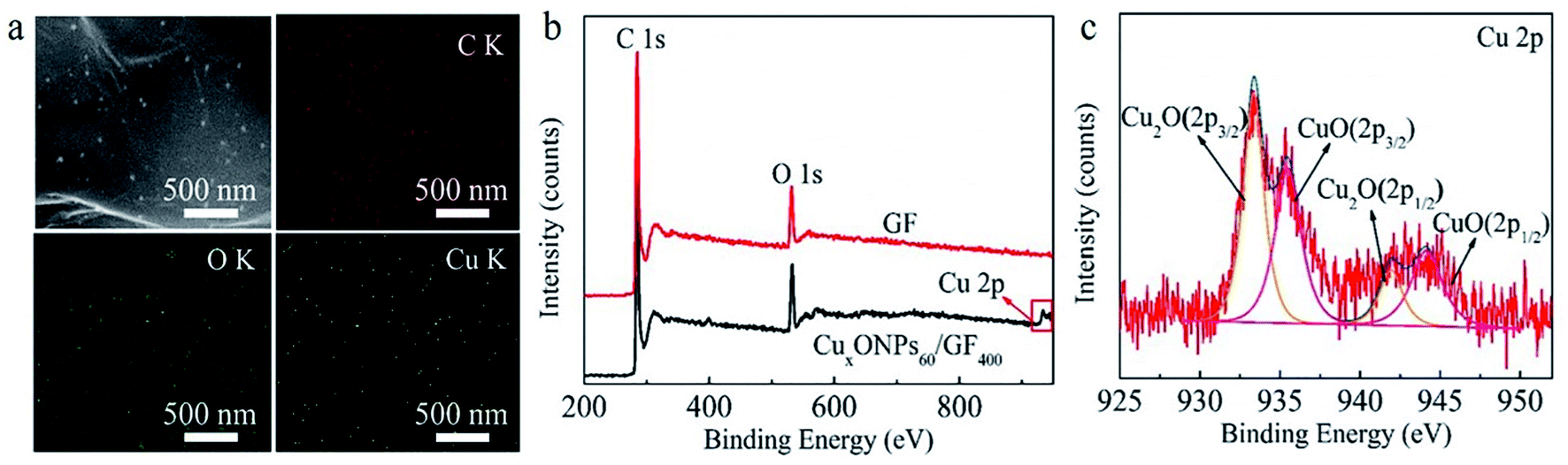 | ||
| Fig. 3 (a) Scanning transmission electron microscopy (STEM) and C-, O- and Cu-element mappings for the sheets within the CuxONPs60/GF400. (b) XPS spectra of the CuxONPs60/GF400 and the bare GF. (c) High-resolution Cu 2p XPS spectrum of CuxONPs60/GF400. | ||
H2O2-sensing properties
The electrochemical properties of the CuxONPs60/GF400 electrode are investigated in 0.1 M PBS saturated with N2 at a scan rate of 50 mV s−1. Considering the electrodeposition self-assembly time has a significant effect on the content and size of CuxONPs, as well as the catalytic performance of the CuxONPs/GF electrode, various CuxONPs/GF electrodes were obtained by adjusting the electrodeposition self-assembly time (30 s, 90 s and 120 s), which were denoted CuxONPs30/GF400, CuxONPs90/GF400, and CuxONPs120/GF400, respectively. The SEM images and EDS analysis of the CuxONPs/GF samples (Fig. S4 and Table S1†) imply that with the increasing time of electrodeposition self-assembly, the content and size of the CuxONPs increase. As a result, there is almost no reductive peak at the CuxONPs30/GF400 electrode (Fig. 4a), which can be attributed to only trace amounts of CuxO with inferior electrocatalytic activity (Fig. S4a and b and Table S1†). In comparison, an obviously reductive peak (about −0.05 V) was observed for the CuxONPs60/GF400 electrode, which was attributed to the successful electrode surface reaction process.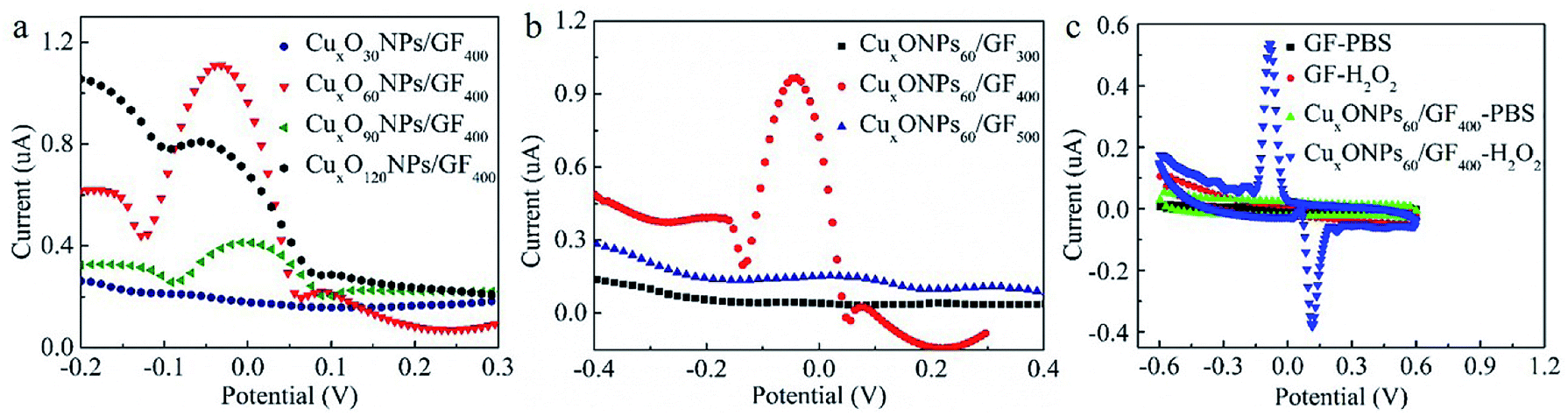 | ||
| Fig. 4 (a) Differential pulse voltammograms (DPVs) of the CuxONPs30/GF400, CuxONPs60/GF400, CuxONPs90/GF400 and CuxONPs120/GF400 in 0.1 M phosphate buffer solution (PBS) with 0.2 mM H2O2 (pH = 7.15). (b) DPVs of the CuxONPs60/GF300, CuxONPs60/GF400 and CuxONPs60/GF500 in 0.1 M PBS with 0.2 mM H2O2 (pH = 7.15). (c) Cyclic voltammograms (CVs) of the bare GF and the CuxONPs60/GF400 electrodes in 0.1 mM PBS (pH = 7.15) without and with 0.2 mM H2O2. | ||
A further increase of electrodeposition self-assembly time causes severe agglomeration of CuxONPs with a reduction in the number of active sites (Fig. S4e–h†) resulting in a decrease in the current response. The annealing temperature also has a significant effect on the size of CuxONPs and performance of the final CuxONPs/GF electrode. For comparison, a series of CuxONPs/GF electrodes were prepared by annealing the precursors of HNPs/GF derived from the optimal electrodeposition time at different temperatures (300 °C and 500 °C, which were denoted CuxONPs60/GF300 and CuxONPs60/GF500, respectively). Apparently, the current response signal of the CuxONPs60/GF400 electrode is obviously higher than that of the CuxONPs60/GF300 and the CuxONPs60/GF500 electrodes (Fig. 4b). This may be attributed to the incomplete decomposition of the HNPs60 precursor for the CuxONPs60/GF300 electrode (Fig. S5a†). The residual non-conductive HNPs60 inhibit electron transport on the surface of GF, leading to poor electrocatalytic activity towards H2O2 sensing. For the CuxONPs60/GF500 electrode, an increase of CuxONPs60 size can be observed as shown in Fig. S5b,† hence reducing the utilization of active sites and leading to a decrease in electrocatalytic performance.32
As a result, compared with the bare GF (no redox peaks, Fig. 4c), the CuxONPs60/GF400 electrode displays a couple of sharp redox peaks, indicating an excellent electrocatalytic activity of the CuxONPs60/GF400 electrode towards H2O2 detection. Meanwhile, the CuxONPs60/GF400 electrode shows an obvious reduction peak at pH = 7.15 (Fig. S6†), which means that the electrode can be applied to a physiological environment. The low detection limit and the linear range of the CuxONPs60/GF400 electrode for H2O2 were measured by differential pulse voltammetry (DPV). As shown in Fig. 5a, the CuxONPs60/GF400 electrode displays two linear responses for H2O2 detection with a good sensitivity of 56.25 μA mM−1 cm−2 and a low detection limit of 0.023 μM in the range of 0.07–1.13 μM (Fig. 5b), whilst displaying an ultrahigh sensitivity of 3437.5 μA mM−1 cm−2 in the range of 1.20–133 μM (Fig. 5c). The reason for two linear relationships herein was probably caused by the different H2O2 absorption and activation on the CuxONPs60/GF400 electrode catalyst under different H2O2 concentrations.32 At extremely low H2O2 concentration, the electrocatalytic process is dominated by H2O2 absorption, but at high H2O2 concentration, the process is dominated by H2O2 activation. Accordingly, the low detection limit obtained at the CuxONPs60/GF400 electrode was estimated to be 0.023 μM (S/N = 3). As a contrast, the sensing performances of recently reported H2O2 sensors based on copper-based electrodes are shown in Table S2.†32–41 Detection limit of 0.023 μM and linear range of 0.07–133 μM achieved by using the CuxONPs60/GF400. The overall performance of CuxONPs60/GF400 exceed the most of copper-based electrodes, which benefiting from the unique microfiber architecture and synergetic effect as well as strong coupling between components with countless active sites and boosted electrons transport. In addition, few group attempt for electrochemical detection of H2O2 on fiber electrode.
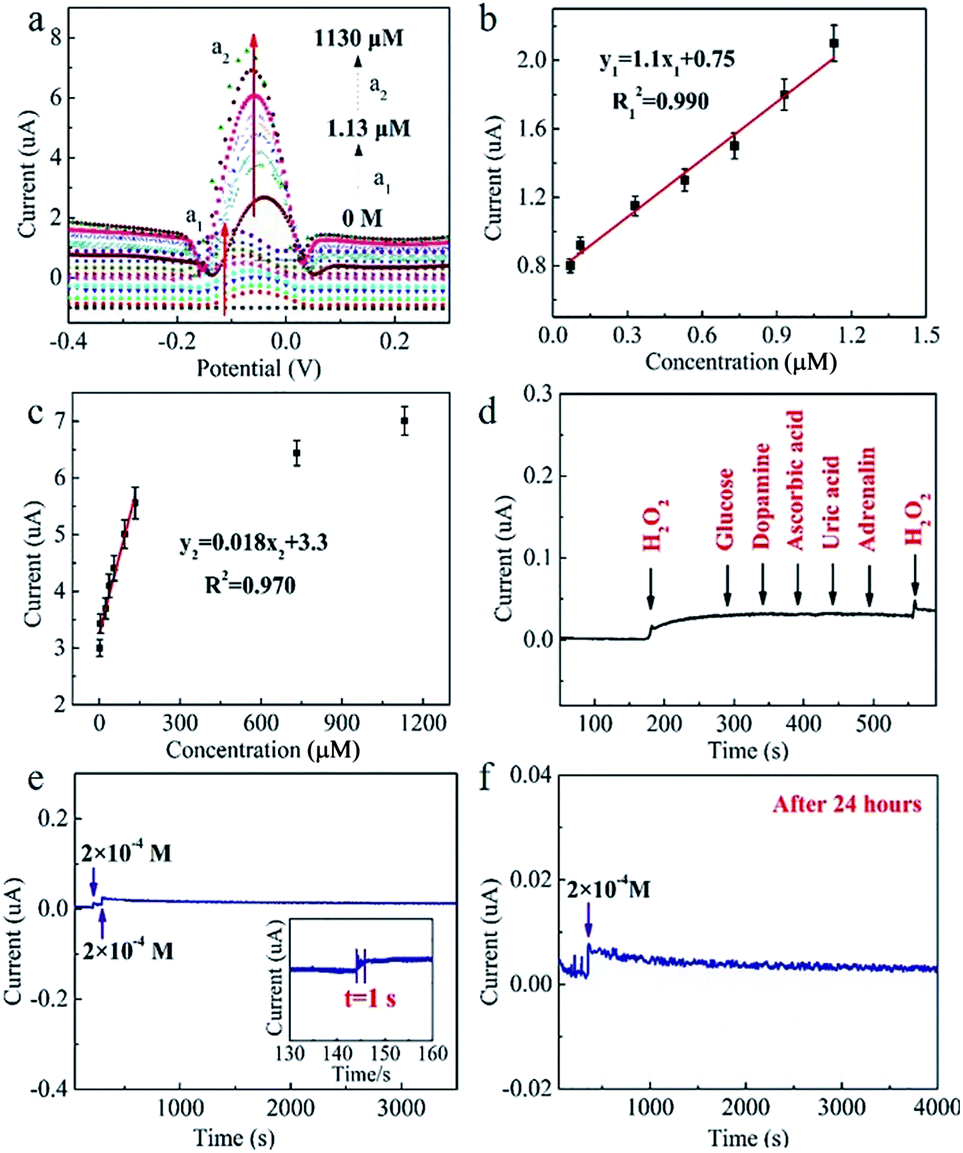 | ||
| Fig. 5 (a) DPVs of the CuxONPs60/GF400 electrode in PBS (pH = 7.15) of various H2O2 concentrations. The concentration of H2O2 is from 0.07 μM to 1.13 mM, scan rate: 50 mV s−1. (b) The fitting curves of reductive current vs. concentration of H2O2 (0.07, 0.09, 0.11, 0.13, 0.33, 0.53, 0.73, 0.93, 1.13 μM). (c) The fitting curve of current response vs. H2O2 concentration (1.2, 5.2, 9.2, 13.2, 53.2, 93.2, 133, 733, 1130 μM). (d) Amperometric responses of CuxONPs60/GF400 electrode at potential of −0.15 V in the 0.1 M PBS with 0.2 mM H2O2, 0.04 mM glucose, 0.04 mM dopamine, 0.04 mM ascorbic acid, 0.04 mM uric acid and 0.04 mM adrenalin. (e) Amperometric curve of the CuxONPs60/GF400 consistently adding 0.2 mM H2O2 under gentle agitation. (f) The amperometric curve of the same CuxONPs60/GF400 after 24 h with addition of 0.2 mM H2O2. | ||
The selectivity test of the CuxONPs60/GF400 electrode was carried out in 0.1 M phosphate buffer solution (PBS). 0.2 mM H2O2 was added, followed by 0.04 mM glucose, 0.04 mM dopamine, 0.04 mM ascorbic acid, 0.04 mM uric acid and 0.04 mM adrenalin, respectively. As shown in Fig. 5d, after adding 0.2 mM H2O2, a distinct current response was observed, and there was almost no change in the peak current in the presence of the interferents. In addition, when a further 0.2 mM H2O2 was added, the current signal showed the same response as after the first H2O2 addition, indicating the excellent selectivity of the CuxONPs60/GF400 electrode. The CuxONPs60/GF400 electrode shows a response time of H2O2 detection within 1 s, indicating it tracks very quickly (Fig. 5e inset). The peak current barely changed after 3500 s (Fig. 5e), and after storing at room temperature for 24 h, the response signal of the CuxONPs60/GF400 electrode remains 98% of its original response for H2O2 detection (Fig. 5f), revealing a good operational stability of the CuxONPs60/GF400 electrode.
Detection of H2O2 in milk and human serum
The CuxONPs60/GF400 electrode was used for the determination of 0.47, 0.60, 5.30, and 94.10 μM H2O2 in diluted milk and diluted human serum samples (Table 1 and Fig. S7 and S8†), respectively. The recovery rates calculated according to the calibration curves (Fig. 5b and c) were 104.3%, 96.7%, 101.7% and 100.8% for milk, and 102.1%, 95.0%, 115.3% and 101.0% for human serum, respectively, the relative standard deviation were less than 4.10% for n = 3, indicating that the CuxONPs60/GF400 electrode is effective for H2O2 detection in a biological system.| Sample | Added (μM) | Founded (μM) | Recovery (%) | Repeatability (% RSD) |
|---|---|---|---|---|
| Milk | 0.47 | 0.49 | 104.3 | 4.01 |
| 0.60 | 0.58 | 96.7 | 3.15 | |
| 5.30 | 5.39 | 101.7 | 3.08 | |
| 94.10 | 94.83 | 100.8 | 2.15 | |
| Human | 0.47 | 0.48 | 102.1 | 3.34 |
| Serum | 0.60 | 0.57 | 95.0 | 2.08 |
| 5.30 | 6.11 | 115.3 | 4.10 | |
| 94.10 | 95.06 | 101.0 | 4.05 |
Conclusions
In summary, we have fabricated a graphene fiber electrode modified with well-dispersed and superfine copper oxide nanoparticles derived from electrodeposition self-assembly of a copper-based metal organic frameworks. The as-prepared CuxONPs/graphene fiber electrode exhibits a high number of active sites and enhanced electrode exhibits a high number of active sites and enhanced electron-transfer ability. It shows highly efficient electro-catalytic activity for nonenzymatic electrochemical H2O2 detection, greater than any previously reported CuxONPs-based electrodes. This work provides a simple method for the rapid production of a novel fiber electrode for electrochemical sensor applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NSFC (21575014, 21905025), Beijing Natural Science Foundation (2184122), the Fundamental Research Funds for the Central Universities (2018CX01017), Beijing Institute of Technology Research Fund Program for Young Scholars, the project of State Key Laboratory of Explosion Science and Technology (Beijing Institute of Technology, YBKT18-03), and Analysis & Testing Center, Beijing Institute of Technology.References
- J. Zhang, J. Han, Z. Shi, Y. Ju, Z. Zhang and M. Gu, Appl. Surf. Sci., 2019, 465, 357–361 CrossRef CAS.
- L. Gao, J. Zhuang, L. Nie, J. Zhang, Y. Zhang, N. Gu, T. Wang, J. Feng, D. Yang, S. Perrett and X. Yan, Nat. Nanotechnol., 2007, 2, 577 CrossRef CAS PubMed.
- Z. Zhang, J. Hao, W. Yang, B. Lu, X. Ke, B. Zhang and J. Tang, ACS Appl. Mater. Interfaces, 2013, 5, 3809–3815 CrossRef CAS PubMed.
- F. Natalio, R. André, A. F. Hartog, B. Stoll, K. P. Jochum, R. Wever and W. Tremel, Nat. Nanotechnol., 2012, 7, 530 CrossRef CAS PubMed.
- P. Pengo, S. Polizzi, L. Pasquato and P. Scrimin, J. Am. Chem. Soc., 2005, 127, 1616–1617 CrossRef CAS PubMed.
- W. Shi, X. Zhang, S. He and Y. Huang, Chem. Commun., 2011, 47, 10785–10787 RSC.
- W. Chen, J. Chen, A. L. Liu, L. M. Wang, G. W. Li and X. H. Lin, ChemCatChem, 2011, 3, 1151–1154 CrossRef CAS.
- X. X. Wang, Q. Wu, Z. Shan and Q. M. Huang, Biosens. Bioelectron., 2011, 26, 3614–3619 CrossRef CAS PubMed.
- P. Roy, Z. H. Lin, C. T. Liang and H. T. Chang, Chem. Commun., 2012, 48, 4079–4081 RSC.
- Z. Chen, J. J. Yin, Y. T. Zhou, Y. Zhang, L. Song, M. Song, S. Hu and N. Gu, ACS Nano, 2012, 6, 4001–4012 CrossRef CAS PubMed.
- M. Baghayeri, H. Alinezhad, M. Tarahomi, M. Fayazi, M. Ghanei-Motlagh and B. Maleki, Appl. Surf. Sci., 2019, 478, 87–93 CrossRef CAS.
- J. Cai, S. Ding, G. Chen, Y. Sun and Q. Xie, Appl. Surf. Sci., 2018, 456, 302–306 CrossRef CAS.
- K. Zhang, N. Zhang, H. Cai and C. Wang, Microchim. Acta, 2012, 176, 137–142 CrossRef CAS.
- L. Lu and X. Huang, Microchim. Acta, 2011, 175, 151–157 CrossRef CAS.
- J. Huang, Y. Zhu, H. Zhong, X. Yang and C. Li, ACS Appl. Mater. Interfaces, 2014, 6, 7055–7062 CrossRef CAS PubMed.
- L. Zhou, L. Kuai, W. Li and B. Geng, ACS Appl. Mater. Interfaces, 2012, 4, 6463–6467 CrossRef CAS PubMed.
- R. Agarwal, K. Verma, N. K. Agrawal, R. K. Duchaniya and R. Singh, Appl. Therm. Eng., 2016, 102, 1024–1036 CrossRef CAS.
- C. Yang, X. Su, F. Xiao, J. Jian and J. Wang, Sens. Actuators, B, 2011, 158, 299–303 CrossRef CAS.
- X. Zhang, S. Sun, J. Lv, L. Tang, C. Kong, X. Song and Z. Yang, J. Mater. Chem. A, 2014, 2, 10073–10080 RSC.
- C. Kong, L. Tang, X. Zhang, S. Sun, S. Yang, X. Song and Z. Yang, J. Mater. Chem. A, 2014, 2, 7306–7312 RSC.
- Y. Sun, M. Luo, Y. Qin, S. Zhu, Y. Li, N. Xu, X. Meng, Q. Ren, L. Wang and S. Guo, ACS Appl. Mater. Interfaces, 2017, 9, 34715–34721 CrossRef CAS PubMed.
- Y. Hu, Z. Zhang and C. Yang, Anal. Chim. Acta, 2007, 601, 95 CrossRef CAS.
- X. Zhu, X. Niu, H. Zhao and M. Lan, Sens. Actuators, B, 2014, 195, 274–280 CrossRef CAS.
- M. Zhang, Y. Wang, L. Huang, Z. Xu, C. Li and G. Shi, Adv. Mater., 2016, 27, 6708–6713 CrossRef PubMed.
- V. Gupta, P. Mahbub, P. N. Nesterenko and B. Paull, Anal. Chim. Acta, 2018, 1005, 81–92 CrossRef CAS PubMed.
- A. A. M. Abdurhman, Y. Zhang, G. Zhang and S. Wang, Anal. Bioanal. Chem., 2015, 407, 8129–8136 CrossRef CAS PubMed.
- M. L. Huffman and B. J. Venton, Analyst, 2009, 134, 18–24 RSC.
- Y. T. Liao, Y. Y. Huang, H. M. Chen, K. Komaguchi, C. H. Hou, J. Henzie, Y. Yamauchi, Y. Ide and K. C. W. Wu, ACS Appl. Mater. Interfaces, 2017, 9, 42425–42429 CrossRef CAS PubMed.
- M. Zhang, Y. Wang, L. Huang, Z. Xu, C. Li and G. Shi, Adv. Mater., 2015, 27, 6708–6713 CrossRef CAS.
- T. Baran, S. Wojtyła, C. Lenardi, A. Vertova, P. Ghigna, E. Achilli, M. Fracchia, S. Rondinini and A. Minguzzi, ACS Appl. Mater. Interfaces, 2016, 8, 21250–21260 CrossRef CAS PubMed.
- X. Sun, S. Guo, Y. Liu and S. Sun, Nano Lett., 2012, 12, 4859–4863 CrossRef CAS PubMed.
- W. Meng, S. Xu, L. Dai, Y. Li, J. Zhu and L. Wang, Electrochim. Acta, 2017, 230, 324–332 CrossRef CAS.
- F. Xu, M. Deng, G. Li, S. Chen and L. Wang, Electrochim. Acta, 2013, 88, 59–65 CrossRef CAS.
- H. Song, C. Ma, L. You, Z. Cheng, X. Zhang, B. Yin, Y. Ni and K. Zhang, Microchim. Acta, 2015, 182, 1543–1549 CrossRef CAS.
- B. B. Jiang, X. W. Wei, F. H. Wu, K. L. Wu, L. Chen, G. Z. Yuan, C. Dong and Y. Ye, Microchim. Acta, 2014, 181, 1463–1470 CrossRef CAS.
- M. Liu, R. Liu and W. Chen, Biosens. Bioelectron., 2013, 45, 206–212 CrossRef CAS PubMed.
- C. Zhang, M. Wang, L. Liu, X. Yang and X. Xu, Electrochem. Commun., 2013, 33, 131–134 CrossRef CAS.
- S. Li, Y. Zheng, G. W. Qin, Y. Ren, W. Pei and L. Zuo, Talanta, 2011, 85, 1260–1264 CrossRef CAS PubMed.
- Y. K. Hsu, Y. C. Chen and Y. G. Lin, Appl. Surf. Sci., 2015, 354, 85–89 CrossRef CAS.
- A. Gu, G. Wang, J. Gu, X. Zhang and B. Fang, Electrochim. Acta, 2010, 55, 7182–7187 CrossRef CAS.
- J. Huang, Y. Zhu, H. Zhong, X. Yang and C. Li, ACS Appl. Mater. Interfaces, 2014, 6, 7055–7062 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional Fig. S1–S7, Tables S1 and S2, and more discussions. See DOI: 10.1039/c9ra03802g |
| This journal is © The Royal Society of Chemistry 2019 |