
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComment on ‘Solution growth and thermal treatment of crystals lead to two new forms of 2-((2,6-dimethylphenyl)amino)benzoic acid’ by R. Hu, Y. Zhoujin, M. Liu, M. Zhang, S. Parkin, P. Zhou, J. Wang, F. Yu and S. Long, RSC Adv., 2018, 8, 15459†
Paulo R. S. Salbego *,
Tainára Orlando and
Marcos A. P. Martins*
*,
Tainára Orlando and
Marcos A. P. Martins*
Núcleo de Química de Heterociclos (NUQUIMHE), Department of Chemistry, Federal University of Santa Maria (UFSM), 97105-900, Santa Maria, RS, Brazil. E-mail: paulosalbego@gmail.com; marcos.nuquimhe@gmail.com
First published on 11th September 2019
Abstract
The study mentioned in the title of this comment article reports on two new polymorphic forms of 2-((2,6-dimethylphenyl)amino)benzoic acid, one with new X-ray diffraction data showing its crystal structure, and the other without. However, our investigation suggests that the newly reported crystal structure (Form II) is in fact the same structure previously reported (Form I), differing in how the structure refinement was performed. Herein, we demonstrated from the raw crystallographic data of Form I that both structures are the same, which invalidates the discovery of the new polymorph. In this comment article, we discuss point by point the findings reported by Long et al., as well as the fact that the report of the other polymorph (Form III) could present a broader investigation in order to assess its existence.
Introduction
Herein, we discuss the results regarding the finding of new polymorphic forms of 2-((2,6-dimethylphenyl)amino)benzoic acid (HDMPA) by Long et al.1 The authors presented several techniques, such as single-crystal X-ray diffraction, FT-IR, Raman spectroscopy, powder X-ray diffraction (PXRD), and theoretical calculations to characterize the supposed polymorphs. However, some problems became evident during the discussion of Long's article, which will be discussed here.The main issue observed is regarding the crystalline structure presented as proof of the existence of the new polymorph (Form II).1 The authors credit the formation of the new polymorph by obtaining a different X-ray measurement after refinement (different unit cell data) when compared with the structure previously reported by Kovala-Demertzi et al.2 (Form I).
However, when investigating the two crystalline forms, we observed a similarity in the crystalline lattice that attracted our attention. New refinement of the structure of Form I2 led us to obtain the same structure reported by Long et al.1 Nowadays, this type of problem, that is, report of new structures as polymorphs – while in fact they are the result of refinement choices made by the authors – has already been discussed by Steed and Steed3 in a recent review. In this sense, an erroneous choice of refinement was observed in the study that reported Form I. In a second moment, Form II with correct refinement was reported as a new polymorph, with all discussion comparing both forms. However, the two reported structures are in fact the same one. The authors of the mentioned article1 consider that the conformational flexibility led to the formation of the polymorph. However, in this case, this phenomenon may be refuted. Additionally, we discussed some points reported by Long et al.1 regarding the structure of Form III.
Discussion
Our first questions regarding the new polymorph reported by Long et al.1 (Form II) relied on the supramolecular analysis of the supposed new polymorph and the earlier structure reported by Kovala-Demertzi et al.2 (Form I). After growing the supramolecular cluster4–6 of both structures and applying an overlay – by using the molecule M1 as reference – high resemblance between the crystalline structures was observed. This indicated the possibility of being either the same crystalline structure (with a different refinement) or the phenomenon of quasi-isostructural polymorphism.7 The first step to assess this doubt was to attempt a new refinement for the raw data reported by Kovala-Demertzi et al.2Refinement
The new refinement from original X-ray diffraction files of Form I was performed. A cell transformation was suggested to reduce the unit cell by the PLATON software.8 The transformation was carried out, leading to a unit cell with new cell parameters. From this, the structure was solved by the SIR92,9 which is the same as the reported Form II by Long et al.1 More detailed explanation about refinement process are in the ESI of this comment article.Cell parameters of Form I reported by Kovala-Demertzi et al.,2 Form II reported by Long et al.1 as the new polymorph, and Form I with the new refinement performed by our group based on Kovala-Demertzi et al.2 original data, herein after referred to as ‘Form I re-refined’, are shown in Table 1 of this comment article. All data were acquired from measurements at 296 K. Some alerts can be observed in the refined structure, but these are alerts do not compromise the quality of the structure data. Additionally, these alerts cannot be resolved in our new refinement since we do not have access to the required data. With these data comparison (Table 1 of this comment), we can suggest that the two reported structures are in fact the same one, only differing in how the refinement was performed.
| Form I2 | Form II1 | Form I re-refineda | |
|---|---|---|---|
| a This investigation; performed from the original data of Form I reported by Kovala-Demertzi et al.2 | |||
| CCDC | 233330 | 1590161 | 1861886 |
| Crystal system | Triclinic | Triclinic | Triclinic |
| Space group | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P |
| a (Å) | 15.8375(16) | 7.524(2) | 7.5311(7) |
| b (Å) | 7.5311(7) | 8.103(2) | 8.0960(9) |
| c (Å) | 11.1845(12) | 11.191(3) | 11.1845(12) |
| α (°) | 83.728(9) | 72.582(4) | 72.496(10) |
| β (°) | 104.806(9) | 83.740(4) | 83.728(9) |
| γ (°) | 79.038(8) | 73.854(4) | 73.792(17) |
| V (Å3) | 1248.56 | 625.071 | 624.275 |
| Z | 4 | 2 | 2 |
| Z′ | 2 | 1 | 1 |
| R-factor (%) | 5.53 | 4.74 | 3.66 |
This piece of information refutes the possibility of quasi-isoestrutural polymorphism7 or any other phenomenon. Additional crystallographic data can be obtained in the Cambridge Crystallographic Data Centre under the number 1861886. We have no doubts about the good crystallographic data reported by Long et al.,1 in which presents the right refinement when compared to the first refinement reported. Nevertheless, the problem lies in the incorrect attribution of a new polymorphic form for the HDMPA measured crystals when compared with previous data by Kovala-Demertzi et al.2 From this moment, all the topics of the commented article were discussed considering the existence and comparison of these two polymorphs.
Long et al.1 demonstrated conformational variability by a superposition of all three experimental conformations. However, with the new refinement of Form I, no variation at the molecular level (Fig. 1 of this comment) between the independent molecules of each measurement was observed.
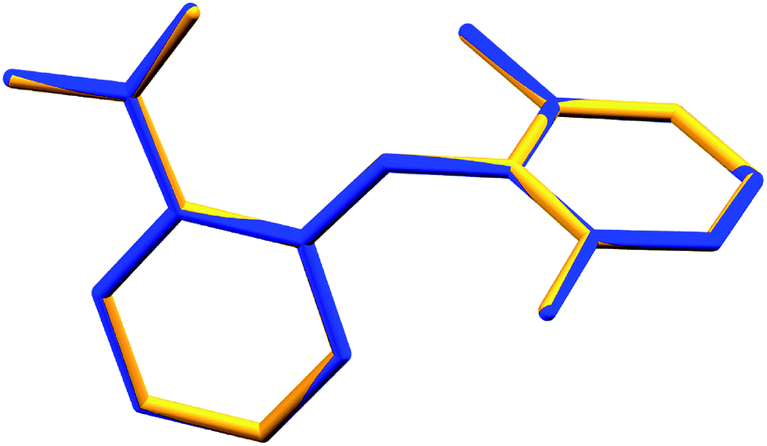 | ||
| Fig. 1 Superposition of the molecules in the asymmetric unit of Form II1 (blue) and Form I re-refined (yellow). All non-hydrogenated atoms were used for the overlay (RMS = 0.00966). | ||
Additionally, Long et al.1 reported in the investigation other molecular data, such as the “bond length of C1–N7 and N7–C8 in the conformers of HDMPA” and the “torsion angle of the conformers” and compared these with the previous Form I. After our new refinement of Form I, virtually the same values were achieved when compared with the supposed new Form II (Tables S1 and S2 in the ESI† of this comment). Data from Tables 2 and 6 of the original study were affected by this new result. In a supramolecular perspective, a supramolecular cluster overlay (Fig. 2 of this comment) was carried out between Form II1 and the re-refined Form I, and no difference in their geometric structure was observed.
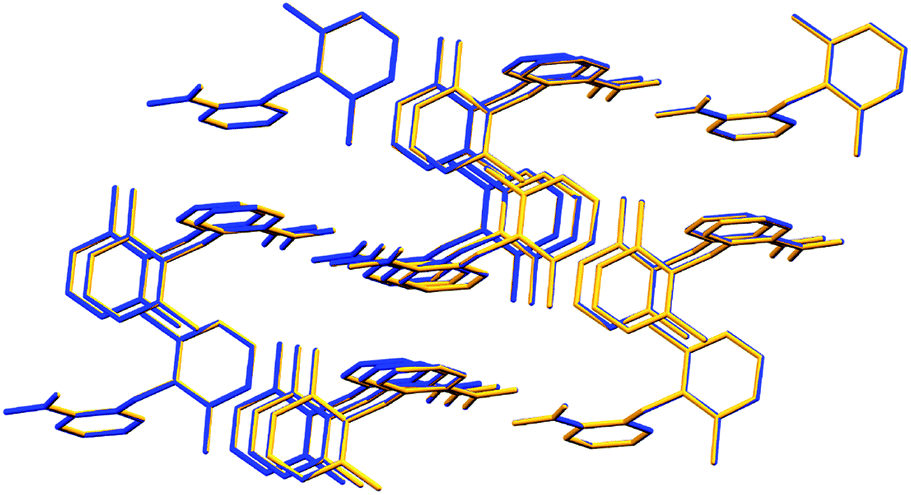 | ||
| Fig. 2 Supramolecular cluster overlay of Forms II1 (blue) and I re-refined (yellow). | ||
Long et al.1 reported that an exhaustive polymorph screening was performed to obtain the literature Form I,2 yet only the Form II was generated. Because of this, Long et al.1 questioned the authenticity of the previous literature form. However, it is clear now that there are no doubts regarding the authenticity of Form I reported by Kovala-Demertzi et al.,2 since both studies are dealing with the same crystalline phase with different refinement performed. In general, for studies in this field, a serendipitous10 behavior may be a perfect guide, as mentioned in the good work of Alexander Rulev10 (2017), especially when comparing the ‘Curious’ vs. the ‘Purposeful’. This also may assist the researcher to interpret data more ‘open-mindedly’.
SEM, synthon analysis, and PXRD
Long et al.1 reported that Form II1 crystals were grown as “colorless blocks from (but not limited to) acetone, and Form III was generated from thermal treatment of Form II samples” and mentioned the Fig. 2 of the original article to demonstrate the SEM images. However, the SEM images did not supply absolute data that can prove the existence of any crystalline phase, since the crystalline habit or color did not indicate with certainty this information. The SEM images for Form II should now be considered as a characterization of the original Form I already reported by Kovala-Demertzi et al.2The synthon images presented (Fig. 4 of the mentioned article) and discussed by the authors to distinguish the polymorphs should be disregarded. This discussion concerning the synthons caught our attention and the way this discussion is presented requires caution on the part of the reader, since the synthons presented by the authors as different are actually present in both crystalline lattices, and the images were arranged in different crystallographic axes views, which gave the impression of being distinct.
Long et al.1 also performed the overlay of the simulated PXRD patterns and identifying that they were almost identical between Forms I and II, in addition to arguing that disappearing polymorphism may be the reason of the absence of Form I. At this point, this data should have drawn the authors' attention to a simpler question, which is that it was more likely to be the same crystal structure. Since the PXRD reflects the diffraction pattern of all crystalline phase not only at unit cell level, the two samples presenting identical simulated patterns may indicate this direction. Under these circumstances, the results seen in Fig. 7 of the original study were affected. The discussion regarding the section ‘Spectroscopic characteristics’ of the commented article can be observed in the ESI.†
Thermal properties
Long et al.1 reported differential scanning calorimetry investigation (DSC). The discussion of this topic should now be considered as a characterization of Form I already reported in 2004 by Kovala-Demertzi et al.,2 in which already reported a melting temperature of 209–210 °C.2 Consequently, the results in Fig. 5 of the mentioned article are affected.The discussion regarding the conversion of Form II into a third form (Form III) should be approached with care and, therefore, be now considered/called Form II, if this form is proven in further investigations. The reported new Form III has only the endothermic peak as evidence, since IR and Raman analyses presented only subtle differences in the values when compared with ‘Form II’. The observation of the two endothermic peaks by DSC analysis should undergo further investigation, and it would be interesting to have them analyzed under lower heating rates (e.g., 2 or 5 °C min−1). Additionally, a triplicated analysis may be carried out to help to confirm this experiment and furnish a standard deviation of the values.
Long et al.,1 also presented a discussion on the number of molecules in the asymmetric unit and the structure stability while arguing that structures with Z′ = 1 are commonly more stable than structures with higher Z′, which indicates the unlikely conversion from Form II to I. However, in this case, this discussion is unnecessary, since it contests the same structure with Z′ = 1.
Computational results and Hirshfeld surface analysis
All data presented by Long et al.1 in this section should be disregarded since the two crystalline structures used are, in fact, the same. One of the points that the authors used to prove that the system indicates to be one of conformational polymorphism is the conformation scan analysis, although some inconsistencies were observed. Long et al.1 reported that “the global minima of τ are identified at ±78.5° and ±98.0° due to the symmetry of the benzene ring. The conformers of both forms are located near the minima, but in two different energy valleys”. Nevertheless, when we observe the scan reported in Fig. 10 of the mentioned study, the two valleys are actually in a range of torsion values of around 75 to 110° and the other in −75 to −110°. Additionally, it would be important to furnish additional data regarding the scan steps used. The Hirshfeld surface analysis should be disregarded, since the data presented will be for the comparison of the same structure (Fig. 11 of the original article is affected).Conclusions
Despite the different techniques used and the experimental effort in the crystallization trials using different solvents, the data presented by Long et al. do not reflect the main proposal of the study, that is, report a new polymorph. In this comment article, we presented crystallographic evidence which sustains that the new crystalline form (II) should be disregarded as a new polymorph. As a result, all content regarding this form (II), presented in the mentioned article, is affected (all figures and tables). It is worth noting that we cannot exclude the possibility of other polymorphs for the compound HDMPA; however, the current data reported by the authors cannot sustain this possibility. Regarding the new Form III, we believe that caution must be taken before affirming the existence of this polymorph, since the presented evidence showed subtle differences or qualitative data thus requiring further investigation to confirm this result.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge the research fellowships from CAPES/FAPERGS (P. R. S. S., process number 18/2551-0000562-2; DOCFIX), CAPES (T. O.) and CNPq (M. A. P. M.), as well as Dr Jerry P. Jasinski for kindly sending the original X-ray diffraction files of Form I.Notes and references
- R. Hu, Y. Zhoujin, M. Liu, M. Zhang, S. Parkin, P. Zhou, J. Wang, F. Yu and S. Long, RSC Adv., 2018, 8, 15459–15470 RSC.
- V. Dokorou, K.-D. Dimitra, P. J. Jerry, G. Angeliki and a D. Mavroudis, Helv. Chim. Acta, 2004, 87, 1940–1950 CrossRef CAS.
- K. M. Steed and J. W. Steed, Chem. Rev., 2015, 115, 2895–2933 CrossRef CAS.
- M. A. P. Martins, C. P. Frizzo, A. C. L. Martins, A. Z. Tier, I. M. Gindri, A. R. Meyer, H. G. Bonacorso and N. Zanatta, RSC Adv., 2014, 4, 44337–44349 RSC.
- M. A. P. Martins, P. R. S. Salbego, G. A. de Moraes, C. R. Bender, P. J. Zambiazi, T. Orlando, A. B. Pagliari, C. P. Frizzo and M. Hörner, CrystEngComm, 2018, 20, 96–112 RSC.
- P. R. S. Salbego, C. R. Bender, M. Hörner, N. Zanatta, C. P. Frizzo, H. G. Bonacorso and M. A. P. Martins, ACS Omega, 2018, 3, 2569–2578 CrossRef CAS.
- D. Dey, S. P. Thomas, M. A. Spackman and D. Chopra, Chem. Commun., 2016, 52, 2141–2144 RSC.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- A. Altomare, G. Cascarano, C. Giacovazzo, A. Guagliardi, M. C. Burla, G. Polidori and M. Camalli, J. Appl. Crystallogr., 1994, 27, 435 Search PubMed.
- A. Y. Rulev, New J. Chem., 2017, 41, 4262–4268 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1861886. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra07188h |
| This journal is © The Royal Society of Chemistry 2019 |