
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOn-demand radiosynthesis of N-succinimidyl-4-[18F]fluorobenzoate ([18F]SFB) on an electrowetting-on-dielectric microfluidic chip for 18F-labeling of protein†
Hee-Kwon
Kim
 abcd,
Muhammad Rashed
Javed
abe,
Supin
Chen
f,
Kirstin A.
Zettlitz
abi,
Jeffrey
Collins
ab,
Anna M.
Wu
abi,
Chang-Jin “C. J.”
Kim
gh,
R.
Michael van Dam
abg and
Pei Yuin
Keng
*abf
abcd,
Muhammad Rashed
Javed
abe,
Supin
Chen
f,
Kirstin A.
Zettlitz
abi,
Jeffrey
Collins
ab,
Anna M.
Wu
abi,
Chang-Jin “C. J.”
Kim
gh,
R.
Michael van Dam
abg and
Pei Yuin
Keng
*abf
aDepartment of Molecular and Medical Pharmacology, University of California, Los Angeles, Los Angeles, CA 90095, USA
bCrump Institute for Molecular Imaging, University of California, Los Angeles, Los Angeles, CA 90095, USA
cDepartment of Nuclear Medicine, Molecular Imaging & Therapeutic Medicine Research Center, Chonbuk National University Medical School and Hospital, Jeonju, 54907, Republic of Korea
dResearch Institute of Clinical Medicine of Chonbuk National University, Biomedical Research Institute of Chonbuk National University Hospital, Jeonju, 54907, Republic of Korea
eDepartment of Chemical Engineering, University of Wah, Quaid Avenue, Wah Cantt, Pakistan
fDepartment of Materials Science and Engineering, National Tsing Hua University, No. 101, Section 2, Guangfu Road, Hsinchu, Taiwan. E-mail: keng.py@mx.nthu.edu.tw; Fax: +886-3-5722366; Tel: +886-3-5715131 ext. 33884
gBioengineering Department, University of California, Los Angeles, Los Angeles, CA 90095, USA
hMechanical and Aerospace Engineering Department, University of California, Los Angeles, Los Angeles, CA 90095, USA
iDepartment of Molecular Imaging and Therapy, Beckman Research Institute, City of Hope, Duarte, CA 91010, USA
First published on 9th October 2019
Abstract
An all-electronic, droplet-based batch microfluidic device, operated using the electrowetting on dielectric (EWOD) mechanism was developed for on-demand synthesis of N-succinimidyl-4-[18F]fluorobenzoate ([18F]SFB), the most commonly used 18F-prosthetic group for biomolecule labeling. In order to facilitate the development of peptides, and proteins as new diagnostic and therapeutic agents, we have diversified the compact EWOD microfluidic platform to perform the three-step radiosynthesis of [18F]SFB starting from the no carrier added [18F]fluoride ion. In this report, we established an optimal microliter droplet reaction condition to obtain reliable yields and synthesized [18F]SFB with sufficient radioactivity for subsequent conjugation to the anti-PSCA cys-diabody (A2cDb) and for small animal imaging. The three-step, one-pot radiosynthesis of [18F]SFB radiochemistry was adapted to a batch microfluidic platform with a reaction droplet sandwiched between two parallel plates of an EWOD chip, and optimized. Specifically, the ratio of precursor to base, droplet volume, reagent concentration, reaction time, and evaporation time were found be to be critical parameters. [18F]SFB was successfully synthesized on the EWOD chip in 39 ± 7% (n = 4) radiochemical yield in a total synthesis time of ∼120 min ([18F]fluoride activation, [18F]fluorination, hydrolysis, and coupling reaction, HPLC purification, drying and reformulation). The reformulation and stabilization step for [18F]SFB was important to obtain a high protein labeling efficiency of 33.1 ± 12.5% (n = 3). A small-animal immunoPET pilot study demonstrated that the [18F]SFB-PSCA diabody conjugate showed specific uptake in the PSCA-positive human prostate cancer xenograft. The successful development of a compact footprint of the EWOD radiosynthesizer has the potential to empower biologists to produce PET probes of interest themselves in a standard laboratory.
1. Introduction
Despite the efficacy of positron emission tomography (PET) in medicine and life sciences, the majority of PET imaging is performed using a single probe, 2-[18F]fluoro-2-deoxyglucose ([18F]FDG), while there are more than 1800 F-18 labelled PET probes that have been identified.1 In addition to small molecule PET probes, biomolecules such as peptides, proteins and antibodies as diagnostic and therapeutic agents have received increasing interest due to their high specificity and affinity in targeting distinct cellular antigens in vivo.2,3 For clinical application, the radioisotope F-18 is most attractive for PET imaging due to the moderate half-life (109.7 min), high positron yield (96.7%), higher resolution image due to the short positron trajectory (0.5 mm mean), high specific activity, wide availability, and the ability to transport the radioisotope from a network of cyclotrons to nearby imaging sites.4 To enable biologists to label and image a specific biologically important mechanism, such as receptor-specific interactions of peptides, proteins, hormones, antibodies and antibody fragments, a new labeling technology platform to advance medicine and life sciences is critically needed. Previously, our group has demonstrated the first proof-of-principle batch microfluidic radiosynthesizer for the radiosynthesis of [18F]FDG, [18F]fallypride and [18F]FLT with reliable yield and with sufficient radioactivity for imaging multiple mice.5–7 Our group has also successfully demonstrated the synthesis of high molar activity radiotracers using this all-electronic microfluidic radiosynthesizer.8 In this report, we further expand the applications from 2-step nucleophilic fluorination of aliphatic substrates to a more challenging and complicated reaction, which involves a 3-step nucleophilic fluorination of an aromatic substrate. [18F]fluoroarenes, such as 6-[18F]fluoro-3,4-dihydroxy-L-phenylalanine ([18F]FDOPA), N-succinimidyl-4-[18F]fluorobenzoate ([18]SFB), [18F]altanserin, [18F]setoperone and others are commonly found in radiopharmaceuticals currently used in the clinic due to their high metabolic stability in vivo.9 However, no carrier added (n.c.a) aromatic nucleophilic fluorination reaction involves high temperature synthesis, toxic reagents, long reaction time and often suffers from low yield.10 Successful demonstration of this class of PET probes on a microfluidic radiosynthesizer would further demonstrate the versatility of the EWOD radiosynthesizer for diverse PET probes synthesis needed in the clinic and in preclinical research. Furthermore, the development of such microdroplet radiochemistry methodology can be translated among other droplet-based radiochemistry platforms including an emerging ultra-compact droplet-based microfluidic radiosynthesizer developed by one of us.1118F-labeling of peptides and biomolecules is typically performed via a 2-step methodology: (1) n.c.a. [18F]fluoride ion labeling of a prosthetic group at high temperature and (2) conjugation of the 18F-labelled prosthetic group to the protein of interest under physiological conditions.12N-Succinimidyl-4-[18F]fluorobenzoate ([18F]SFB) is one of the most commonly used prosthetic groups for labeling protein due to its versatility in labeled amine groups, which often doesn't require the introduction of special handles into the protein, as well as its in vivo stability.4,13 While we demonstrated the synthesis of [18F]SFB as a proof of concept, this technology could also be applied to synthesize many other prosthetic groups to prepare a wide range of labelled proteins for PET imaging. The multistep synthesis of [18F]SFB has been reported in conventional macroscale radiosynthesizers with radiochemical yields (RCYs) ranging from 20–77% within 30–90 min.14–16 Over the years, many research studies have been devoted to improving the laborious multistep and multi-pot12,13,16 synthesis of [18F]SFB, ultimately leading to a convenient and shorter three-step, one-pot procedure.14,15,17 Following the fluorination step, an organic base is used in the hydrolysis step to yield the fluorobenzoic acid ([18F]FBA) intermediate. Due to the sensitivity of the final coupling reaction with the acylation agent (TSTU or HSTU), an additional azeotropic evaporation is performed at the end of the hydrolysis step to achieve a high coupling efficiency.18–21 The improved synthesis has been implemented on conventional automated radiosynthesizers (e.g., IBA Synthera and Sofie ELYXYS FLEX/CHEM) with 69–85% RCY.14,22
Though [18F]SFB can be convenient reacted with peptides or proteins to produce imaging probes, it is not widely available due to insufficient demand. The radiosynthesis of [18F]SFB can only be performed in a specialized radiochemistry laboratory and requires an expensive automated synthesizer, and a hot cell for operation of the synthesizer. Due to the high capital investment needed for radiochemistry, [18F]SFB is limited to a relatively small number of groups that have access to a nearby radiochemistry laboratory.23 Thus, a new microfluidic platform for radiochemistry that was compact and self-shielded, so that it could be used in any standard laboratory, has the potential to overcome this challenge and enable wide availability of [18F]SFB and thus enable 18F-labeling of a wide range of biomolecules for PET imaging.
Leveraging advancements in microfabrication and microelectronics, microfluidic technology has the potential of integrating the entire PET radiochemistry operation, which includes synthesis, purification and quality control modules into a compact and inexpensive radiosynthesizer.24 Microfluidic devices capable of manipulating liquid in nanoliter to microliter scales allow precise control over reaction conditions and the rapid mixing, heat and mass transfer enable quicker reactions that provide distinct advantages for PET probes radiochemistry.25–28 The small volume is commensurate to the picomole to nanomole quantities of PET probes needed for preclinical and clinical PET imaging. Recently, Kimura and his colleagues in Japan optimized the three-steps radiosynthesis of [18F]SFB into a single continuous flow microfluidic chip with 64% radiochemical yield.29 One major downside of such flow-through radiosynthesizer is in their inability to perform solvent exchange reactions, and so part of the process must be carried out in conventional, macroscale apparatus. Bejot and co-workers have demonstrated the integration of solvent evaporation steps and achieved the synthesis of [18F]SFB with 64% radiochemical yield within 25 min in a 60 μL batch microreactor with integrated valves and mechanical actuators to control the flow of reagents in and out of the reaction vessel.21
Previously, our group has develop a dedicated EWOD microfluidic radiosynthesizer capable of performing all unit operations involved in the multistep synthesis of [18F]FDG, [18F]FLT and [18F]fallypride such as solvent exchange, fluorination and hydrolysis on a single chip in even smaller reaction volumes (1–16 μL) without the need for bulky mechanical actuators needed in other microfluidic systems.5–7,30 Herein, we report on two main advancements over our previous publications: (1) we expanded the capability of the EWOD microfluidic synthesizer to perform aromatic nucleophilic fluorination and (2) we established several critical microscale chemical rule sets to achieve a facile radiosynthesis of [18F]SFB in high and reliable yield. We further show that [18F]SFB synthesized on the EWOD chip could be conjugated to the anti-prostate stem cell antigen (PSCA) cys-diabody (A2cDb),31,32 resulting in sufficient specific activity of the 18F-labeled PSCA diabody (2.9 μCi μg−1 protein) to conduct a pilot immunoPET study of a prostate cancer xenograft bearing mouse (Fig. 1). While this paper is focused on the development of the droplet radiochemistry on the EWOD chip, our collaborators are also currently developing an automated, self-shielded prototype droplet radiosynthesizer equipped with a radioactivity concentration, reagent delivery, product collection and purification platforms.
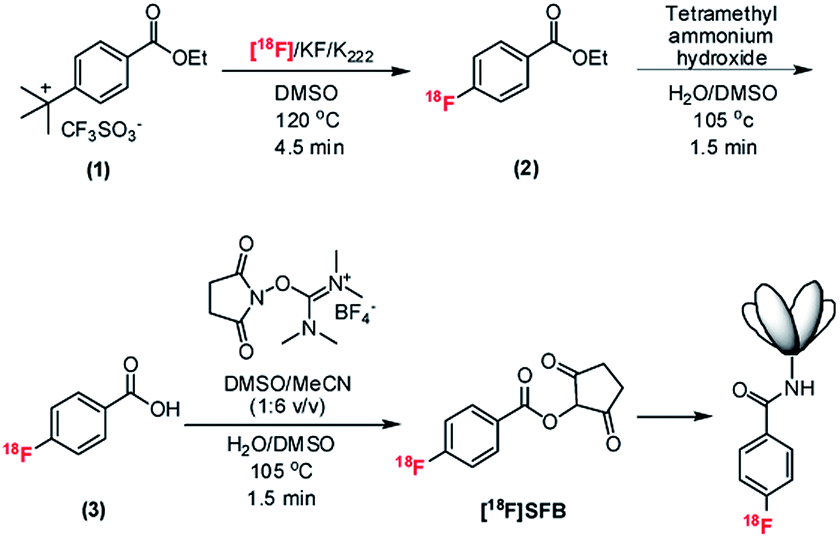 | ||
| Fig. 1 Overall radiosynthetic scheme of [18F]SFB on the EWOD chip and the conjugation of the 18F-labeled prosthetic group to the PSCA-diabody. The conjugation is performed in sodium-borate buffer (SBB, 50 μmol L−1, pH 8.7) and incubated for 10 min at 34 °C. Cartoon of diabody is not to scale. | ||
2. Materials and methods
2.1 EWOD chip
Potassium carbonate (K2CO3), potassium hydrogen carbonate (KHCO3), 4,7,13,16,21,24-hexaoxa-1,10-diazabicyclohexacosane (Kryptofix 222), tetrapropylammonium hydroxide solution (1 M; NPr4OH), O-(N-succinimidyl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HSTU), anhydrous acetonitrile (MeCN, 99.8%), anhydrous dimethyl sulfoxide (DMSO, 99.9%), hexanes, ethyl acetate, 1 M acetic acid solution, trifluoroacetic acid, and methanol were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO). 4-(Ethoxycarbonyl)-N,N,N-trimethylbenzenaminium triflate (FB precursor) and the N-succinimidyl 4-fluorobenzoate (SFB) standard compound was purchased from ABX Advanced Biochemical Compounds (Radeberg, Germany) and used as received.No-carrier-added [18F]fluoride ion was obtained from the UCLA Crump Institute Cyclotron and Radiochemistry Technology Center by irradiation of 97% 18O-enriched water with an 11 MeV proton beam using an RDS-111 cyclotron (Siemens Medical Solution, Knoxville, TN). Radioactivity was determined using a calibrated ion chamber (Capintec CRC-15R, Ramsey, NJ). A radio-thin layer chromatography (radio-TLC) scanner (MiniGITA star; Raytest USA, Inc, Wilmington, NC) was used to analyze fluorination efficiency, and analytical high performance liquid chromatography (HPLC; Knauer, Germany) equipped with a degasser (Model 5050), pump (Model 1000), a variable wavelength UV detector (254 nm) and a radiometric detector (Eckert & Ziegler, Washington DC, USA) was used to measure chemical and radiochemical purity. The HPLC was equipped with HPLC analysis was carried out using a Phenomenex Luna reversed-phase C-18 column (250 × 4.6 mm) (Torrance, CA). The purification of the final product [18F]SFB was performed on a similar analytical HPLC system with an additional SecurityGuard C18 column (Phenomenex) (Torrance, CA).
2.2 Radiosynthesis of [18F]SFB on EWOD chip
Kryptofix 2.2.2 (12 mg; 0.032 mmol) and potassium carbonate (2.4 mg; 0.017 mmol) were dissolved in water (120 μL). An aliquot of the phase transfer catalyst mixture (7 μL) was mixed with no-carrier-added [18F]fluoride/[18O]H2O (50 μL; ∼185 mBq to generate the [18F]KF/K222 complex). Separately, the FB precursor (1.2 mg, 3.40 mmol) was dissolved in 40 μL of anhydrous DMSO, and HSTU (5 mg; 0.14 mmol) was dissolved in 50 mL of anhydrous MeCN. A stock solution of 160 mM NPr4OH was prepared by diluting 16 mL of the 1 M NPr4OH in 100 mL of H2O/DMSO (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) mixture.
:1 v/v) mixture.
Six droplets (2 μL each) of the [18F]KF/K222 complex were loaded onto the EWOD chip using a micropipette through one of the dedicated loading edges. Each droplet was transported from the loading edge to the reaction site via EWOD actuation. One MeCN droplet (3 μL) was then added to the reaction site. The resulting mixture was heated to 105 °C and held at this temperature for 1 min to remove the water. Subsequently, one cycle of azeotropic distillation was performed by adding 3 droplets (3 μL each) of MeCN to the dried residue and heating at 105 °C for 1 min. To the dried [18F]KF/K222 complex, one 2 μL droplet of the FB precursor (compound 1) (84 mM) was added. The reaction mixture was gradually heated to 120 °C and held at the set temperature for 4.5 min to perform the fluorination reaction. After the fluorination reaction, two droplets (3 μL each) of the NPr4OH solution (160 mM) in 3:1 mixture of H2O/DMSO was added to the ethyl-[18F]fluorobenzoate intermediate (compound 2). The saponification reaction was performed by heating the reaction mixture at 105 °C for 1.5 min. After the saponification reaction, two droplets (3 μL each) of the HSTU solution (280 mM) in MeCN was directly added to the crude [18F]fluorobenzoic acid intermediate (compound 3) for the final esterification step. The reaction mixture was heated to 105 °C and held at the set temperature for 3.5 min to yield [18F]SFB, the final product. The crude reaction product was extracted with 40 μL MeCN and 20 μL MeOH. A small aliquot of the crude product was removed for radio-TLC and radio-HPLC analyses, while the remainder of the crude product was subjected to HPLC purification.
2.3 [18F]SFB purification and reformulation
About 60 μL of crude product collected from the EWOD chip was loaded onto the analytical radio-HPLC equipped with a 100 μL sample loop. The product was separated via an isocratic elution of MeCN/H2O/TFA 50:50:0.1 (v/v/v) at a flow rate of 1 mL min−1. The [18F]SFB fraction eluted between 15 and 16 min and the product was collected into a vial containing 3 μL of 1 M acetic acid. The HPLC mobile phase was evaporated to dryness at 77 °C under a constant nitrogen flow for 10 min to obtain final compound for the subsequent radiolabeling reaction. The dried [18F]SFB was transferred from the scintillation vial into a polypropylene Eppendorf tube (200 μL) using ∼200 μL of MeCN to minimize loss of product on the sides of the wall of the larger volume scintillation vial. Then the MeCN was evaporated to dryness in a water bath heated at 77 °C. The dried and formulated [18F]SFB was obtained with 39 ± 7% (n = 4) radiochemical yield within 120 min. The average radio chemical purity was 98%.
2.4 Quality control
The radio-fluorination, hydrolysis and esterification efficiencies of the respective intermediate product and [18F]SFB were determined by radio-TLC on silica gel plates (60 F254 with aluminum backing), with 1:1 v/v ethyl acetate/hexane mixture as the mobile phase. The radioactivity distribution was scanned with a radio-TLC scanner. The retention factors (Rf) of the [18F]fluoride ion, [18F]fluorobenzoic acid, [18F]SFB and ethyl-4-[18F]fluorobenzoate were 0.00, 0.00, 0.32, and 0.57, respectively (Fig. S-1†). Radiochemical purity of the final [18F]SFB product was analyzed on the Phenomenex Luna reversed-phase C-18 column (250 × 4.6 mm) with isocratic elution of MeCN/H2O/TFA 50:50:0.1 (v/v/v) at a flow rate of 0.5 mL min−1. The identity of the final [18F]SFB product was confirmed by comparing to a chromatogram of SFB reference standard SFB. A representative HPLC chromatogram (UV and γ-detection) of the purified and formulated [18F]SFB is shown in Fig. S-2.†
2.5 Conjugation of [18F]SFB with PSCA-diabody
[18F]SFB was resuspended in sodium borate buffer (33 MBq) in 25 μL SBB, 50 μM, pH 8.7 and incubated with A2cDb (50 μg in 50 μL SBB) for 10 min at 34 °C. Conjugated [18F]FB-A2cDb was purified using a Micro Bio-Spin size exclusion columns (BioRad) that was pre-blocked with PBS, 1% fetal bovine serum (FBS) as previously described.33 Labelling efficiency and radiochemical purity were determined using ITLC strips (Biodex Medical Systems) with saline as solvent and analyzed by gamma counting (Wizard 3′ 1480 Automatic Gamma Counter, PerkinElmer). The immunoreactive fraction of radiolabeled A2cDb was measured by incubation with excess PSCA-positive cells (22Rv1-PSCA) and control cells (22Rv1) for 1 h at room temperature. Supernatant and cell-bound fractions were analyzed by gamma counting as previously described.342.6 Small animal imaging
A male nude mouse (Jackson Laboratories, JAX002019 NU/J) bearing subcutaneous 22Rv1-PSCA (right shoulder) and 22Rv1 (left shoulder) xenografts was injected intravenously with 10 μg [18F]FB-A2cDb (30 μCi/1.1 MBq) in 100 μL saline. PET/CT imaging was performed under 2% isoflurane anesthesia. A two-hour dynamic PET acquisition (Inveon, Siemens) was followed by a CT scan (MicroCAT, Siemens). PET images were reconstructed as filtered back projections (FBP), and PET/CT images are presented as maximum intensity projection (MIP) overlays using AMIDE software.35Ex vivo biodistribution was performed by weighing and gamma counting tissues and calculating % ID/g values based on 1% of the injected dose. All procedures performed in studies involving animals were in accordance with the Animal Welfare Act and Regulations (USDA) and the PHS Policy (NIH/OLAW) and were approved by the University of California Los Angeles (UCLA) Chancellor's Animal Research Committee (ARC), Los Angeles, USA.3. Results
3.1 Droplet radiosynthesis of [18F]SFB
Based on the one-pot synthetic methodologies that have been reported for [18F]SFB using conventional macroscale radiosynthesizer,15 we have further optimized the reaction conditions on the EWOD chip to achieve a reliable yield. All processes, which include [18F]fluoride complex drying, solvent exchange, fluorination, hydrolysis and coupling reaction, were performed on a single EWOD chip. The optimal reaction condition of the fluorination step used 167 nmol of precursor, 216 nmol of K2CO3 and 390 nmol of K2.2.2 in 2 μL of DMSO. Reacting at 120 °C for 4.5 min yielded an average fluorination efficiency of 93 ± 1% (n = 6). The saponification of 4-[18F]fluoroethylbenzoate was carried out at 105 °C for 1.5 min using NPr4OH (167 mM) in a 3:1 v/v mixture of H2O/DMSO. Lastly, the coupling step was performed using 1670 nmol of HSTU in a mixture of MeCN and DMSO (6:1 v/v) at 105 °C for 3.5 min. The RCY of [18F]SFB after HPLC purification was 39 ± 7% (n = 4), The total synthesis time was ∼120 min, including the on-chip [18F]fluoride ion concentration, radiofluorination, saponification, coupling reaction, analytical-scale HPLC purification, and followed by solvent removal to complete dryness (to facilitate subsequent conjugation reaction). In a typical radiosynthesis on the EWOD chip, ∼222 MBq of [18F]fluoride/[18O]H2O was loaded on the chip and ∼37 MBq [18F]SFB was collected after the HPLC purification. Upon removal of the HPLC mobile phase and reformulation, ∼33 MBq of [18F]SFB was used for the subsequent protein labeling and mice imaging experiment. Even under the optimized drying condition, ∼5% of the radioactivity was lost during the solvent removal process, which resulted in only 33 MBq used for subsequent labeling experiment.
3.2 Radiolabeling of A2cDb using [18F]SFB
[18F]SFB radiolabeling of A2cDb and purification of the resulting [18F]FB-A2cDb conjugate was completed within 30 min with a radiolabeling efficiency of 33 ± 13% (n = 3) and radiochemical purity of 94.6 ± 2.1% (n = 3) as determined by instant thin layer chromatography (ITLC). [18F]FB-A2cDb retained antigen-specific binding to PSCA-expressing cells (immunoreactive fraction 75.6 ± 6.5%, n = 2).3.3 [18F]FB-A2cDb immunoPET imaging of prostate cancer xenograft bearing mouse
[18F]FB-A2cDb (10 μg/1.1 MBq) was injected i.v. into a male nude mice bearing PSCA-positive and PSCA-negative prostate cancer xenografts and a dynamic PET scan (0–120 min) was acquired. Antigen-specific accumulation in the PSCA-positive tumor (right shoulder) could be observed as early as 30 min post injection and increased up to 2 h post injection. A small amount of activity was seen in the PSCA-negative tumor (left shoulder), but this is potentially blood pool activity as the diabody is not completely cleared from the blood at 120 post injection. The [18F]FB-A2cDb showed rapid clearance from the blood and renal secretion of activity into the urine.4. Discussion
4.1 EWOD chip for radiosynthesis
Droplets sandwiched between the two plates of the EWOD chip are moved by electrowetting force when an electrical potential is applied to electrodes on the base plate to accomplish operations such as droplet generation, transport, splitting and mixing.36 The EWOD electrodes at the reaction site are capable of resistive heating and thermistic temperature sensing in addition to droplet movement (Fig. 2).37 The reaction site electrodes were configured as four concentric rings (maximum size 12 mm diameter) to center droplets and accurately monitor and control their temperature as they shrink. Each of the four rings is independently capable of feedback temperature control, which prevents the regions of faster droplet evaporation from excessive heating.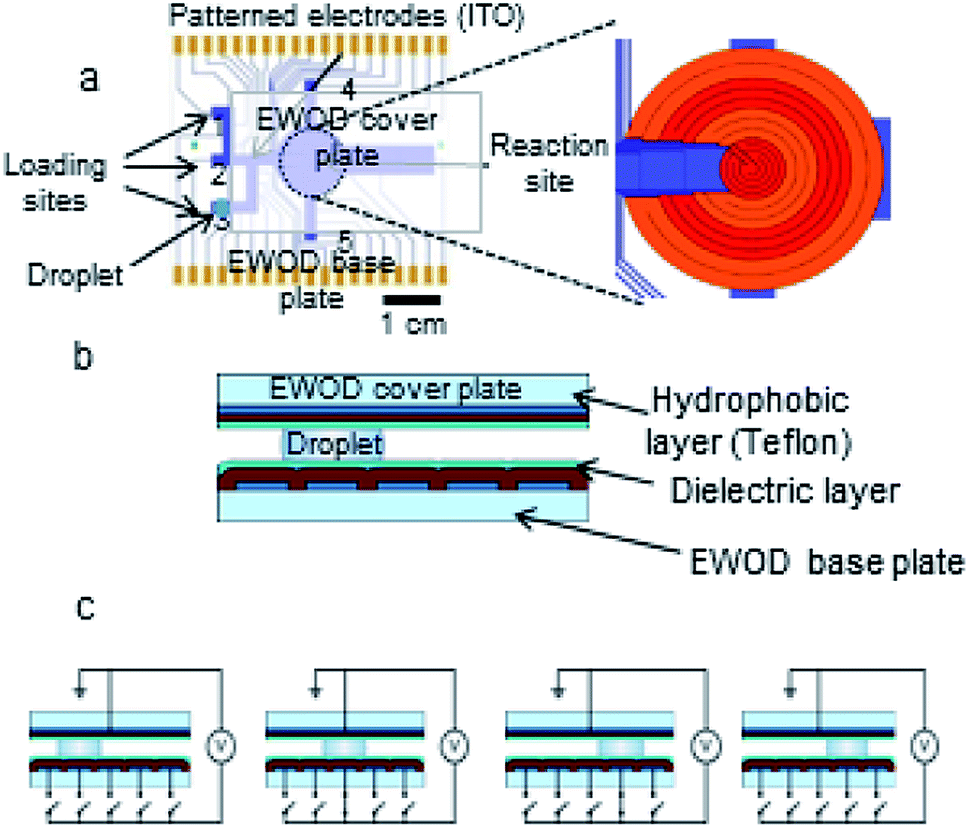 | ||
| Fig. 2 (a) EWOD chip base plate design. Inset shows detail of the multifunctional electrowetting, heating, and temperature sensing electrodes at the reaction site. (b) Cross-sectional view of EWOD chip. (c) Droplet movement on chip by sequential connection of EWOD electrodes to actuation voltage. | ||
4.2 Radiosynthesis of [18F]SFB synthesis on the EWOD chip
Leveraging our previous work5–7 in developing an all-electronic, compact, easy to use and reliable EWOD microfluidic radiosynthesizer for F-18 PET radiochemistry, in this report we demonstrated a three-step, one-pot radiosynthesis of [18F]SFB on the EWOD chip. Similar to our previous reports, we found that the conditions established in the macroscale are not suitable for the microdroplet radiosynthesis. In particular, we observed low fluorination and radiochemical yields when simply down-scaling the reagent volume and amount used in macroscale reactions. The focus of this work was to establish a reliable droplet radiochemistry method for aromatic nucleophilic fluorination and the subsequent functional group transformation by investigating the effect of reagent concentrations, reagent ratios, reaction times, and droplet sizes. During this development stage, the majority of the operation was performed manually, which limited the radioactivity used in this work. While this report focused on the development of a reliable microscale radiochemistry on the EWOD chip, we are also currently developing an automated reagent delivery, product collection and purification system38 towards a fully automated microfluidic radiosynthesizer.4.2.1.1 Precursor to base ratio. Our group adapted the synthetic methodology developed by Tang and Kabalka et al. in the microdroplet radiosynthesis on EWOD chip.15 Briefly, the ethyl 4-(trimethylammonium triflate)benzoate (FB precursor) was radiofluorinated using the activated complex of [18F]KF/K2.2.2 to yield ethyl-4-[18F]fluorobenzoate intermediate. High throughput optimization studies were first performed using the lower cost and easily prepared glass-Teflon substrates, which mimic the sandwiched configuration of the EWOD chip.5 The optimized conditions found on the glass-Teflon substrates were then translated onto the EWOD chip. Such workflow enabled large numbers of optimization experiments in the shortest time period and at the lowest cost. Based on our first optimization study, we found that the highest fluorination yield was obtained by using the phase transfer catalyst (PTC) complex (K+/K2.2.2) to precursor ratio of 2.3 (Fig. 3). The number of moles of the PTC complex is equal to the number of moles of K2.2.2; the number of moles of K2CO3 is fixed at 0.5× the amount of K2.2.2. In the macroscopic conditions, the ratios of K+/K2.2.2 complex to precursor used in the fluorination of the FB precursor typically ranged between 4
:1 to 6:1. As shown in Fig. 3, fluorination efficiency drastically decreases at molar ratios above 3 in the microdroplet synthesis.
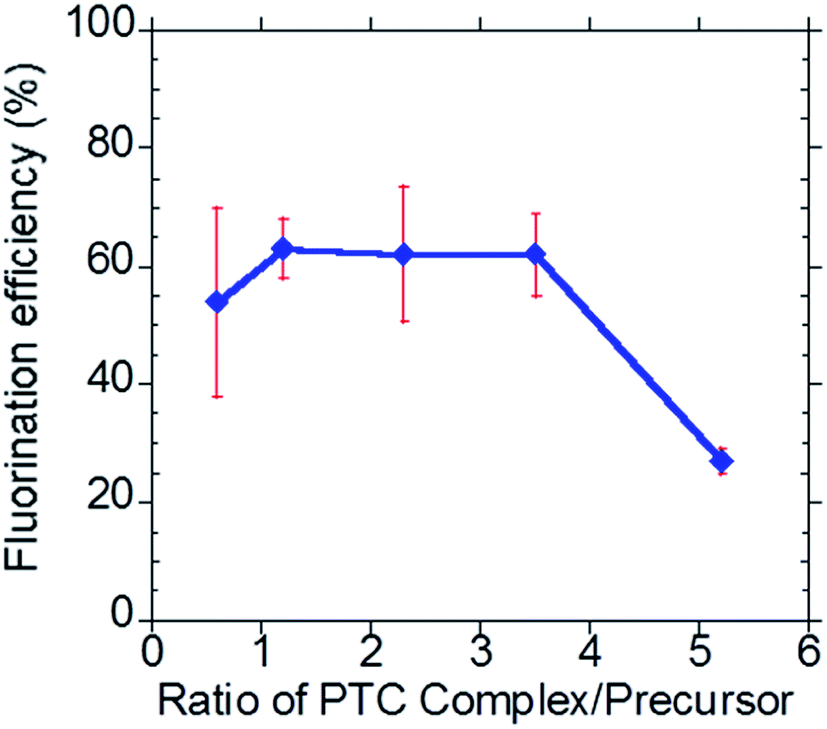 | ||
| Fig. 3 Investigation of the effect of the phase transfer catalyst complex (K+/K2.2.2) to precursor ratio. | ||
4.2.1.2 Concentration and reaction time. Capitalizing the flexibility of the four concentric multifunctional heaters, the reaction volume at each step can be varied between 1–17 μL (for the current chip design and setup) to achieve the optimal reaction conditions. In this example, the EWOD radiochemistry platform can efficiently accommodate larger volume of the [18F]fluoride radioisotope (12 μL) to produce sufficient radioactivity of [18F]SFB for protein conjugation and micro-PET imaging of mice, while performing other reactions in smaller volumes (i.e., 2 μL) to increase the overall reagent concentrations and thus the reaction kinetics. Furthermore, by decreasing the reaction droplet size, only a minute amount of reagent is needed to achieve the same reagent concentration, a significant advantage when using scarce and expensive reagents. More importantly, the higher concentration of reaction does not affect the downstream HPLC purification because the total amount of reagents remains in the nanomole range and does not saturate the analytical HPLC column. We have recently demonstrated the effect of droplet size and reagent concentration in enhancing the microdroplet radiosynthesis of [18F]FLT on the EWOD chip.6 As a first step to improve upon our previous results, we reduced the reaction volume from 6 μL to 2 μL with a concurrent increase in the reagent concentration. Using the 2 μL reaction volumes, we next investigated the reaction kinetics of the fluorination reaction (4.5, 7 and 10 min) on the Teflon-glass substrate in the presence of 7 mM precursor, and 18 mM PTC complex (i.e. in optimal PTC
:precursor ratio of 2.3 as discussed previously) at 120 °C. The fluorination conversion was lower at 4.5 min than longer reaction times and increased with increasing reaction time as anticipated (Fig. 4, red trace). Upon increasing the reagent concentrations by 4×, the fluorination efficiency in the 4.5 min reaction increased from ∼40% to ∼75% (Fig. 4, square green trace). Further enhancement of the fluorination efficiency to 81% was observed by increasing the reagent concentration by 12× (Fig. 4, blue trace). Prolonging the reaction time to 7, 10 and 12 min did not further improve the conversion. In contrast, upon lowering the fluorination time from 4.5 to 3.0 min, a drastic reduction in the fluorination conversion (27 ± 13% (n = 6)) was observed (Fig. 4, blue trace). Therefore, we chose the following optimal fluorination conditions: 4.5 min reaction, 84 mM precursor, 108 mM K2CO3 and 195 mM K2.2.2 in DMSO.
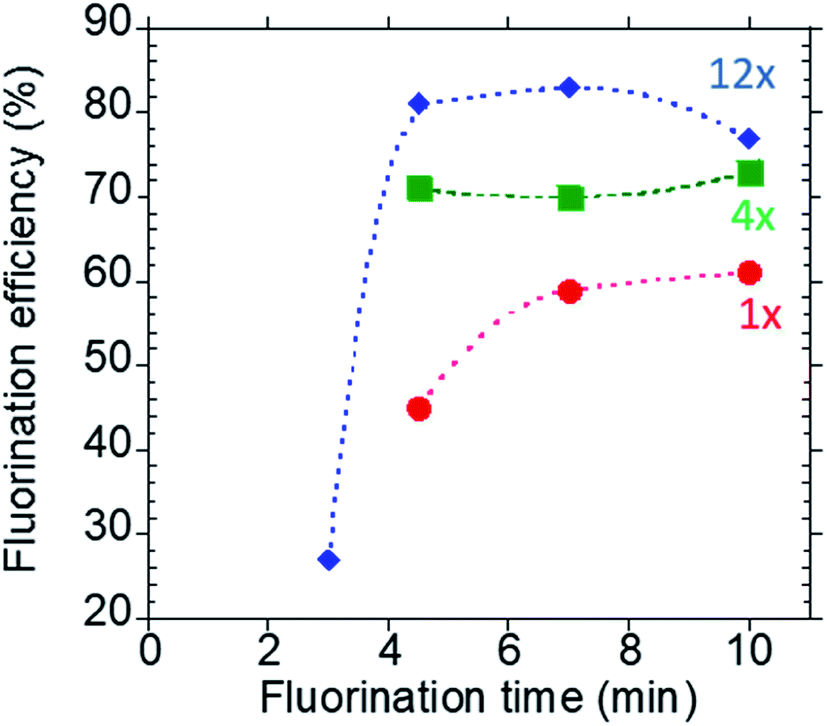 | ||
| Fig. 4 Kinetics of the fluorination reaction at different reagent concentrations. Reactions were performed in 2 μL of DMSO at 120 °C. The blue trace (diamond marker) represents the optimized concentration, i.e. 12× the concentration used in the macroscale reaction; the green trace (square marker) represents 4× concentrations and the red trace (circular marker) represents the same concentration as used in the macroscale. | ||
After the substitution reaction using n.c.a [18F]fluoride ion, the ethyl ester protecting group is typically removed by hydrolysis using an aqueous base (e.g.: NaOH) or acid (e.g.: HCl) to form the 4-[18F]fluorobenzoic acid ([18F]FBA) intermediate product (8, 9). However, this method requires an intermediate purification step to remove the water prior to the final coupling step. In this work, we adapted the method developed by the Kabalka group and the Shen group, which used an organic base for the deprotection reaction.15,39 This method eliminates the need for the intermediate purification step and thus reduces the overall radiosynthetic time. On the EWOD chip, the ethyl 4-[18F]fluorobenzoate intermediate (Compound 2) was hydrolyzed using NPr4OH in a mixture of DMSO and water (3:1 v/v). Initially, we found that residual DMSO from the fluorination step resulted in a low and unreliable performance of the hydrolysis step, perhaps due to dilution of the reaction, which reduces the overall concentration of reagents. However, upon optimization of the fluorination reaction using smaller droplet size (i.e. 2 μL instead of 6 μL), minimal residual DMSO remained after fluorination and the hydrolysis performed well. In comparison to the 2 μL reaction droplet, a large pool of DMSO condensates remained on the heater site using 6 μL of reaction droplet, which necessitates an additional drying step before proceeding to the hydrolysis step (Fig. S-3†). Furthermore, the additional heating time to remove the DMSO condensates after the fluorination step resulted in a large loss of radioactivity and was time consuming.
Based on the new method, we investigated two reaction temperatures (95 °C and 105 °C) and the reaction times needed to achieve quantitative conversion to the [18F]fluorobenzoic acid intermediate product (Compound 3). We found a high deprotection efficiency at 105 °C, while no significant conversion was observed at lower temperature (95 °C) despite prolonging the reaction time up to 25 min. At 105 °C, the hydrolysis was quantitatively completed in less than 2 min as confirmed by radio-TLC.
In the final reaction step, the crude tetra-n-propylammonium 4-[18F]fluorobenzoate salt (Compound 3) in a mixture of NPr4OH in H2O/DMSO was directly treated with HSTU without removing the residual water. It is worth noting that conventional macroscopic method typically requires a time-consuming azeotropic drying step or solid phase extraction (SPE) purification step in between the hydrolysis and the esterification step due to the sensitivity of the coupling agent (i.e.: HSTU) to water. In the microdroplet reaction, we found that the coupling reaction of HSTU and the [18F]fluorobenzoate salt proceed with quantitative yield in a mixture of solvents (MeCN/DMSO 1/6 v/v) on EWOD chip, which is consistent with an earlier report by Bannwarth.40 Similar to the previous optimization studies, the reaction kinetics for the ester formation was first investigated on Teflon-glass substrates, and then the optimal conditions were later translated to the EWOD chip. Based on the kinetic studies, the optimal reaction time was found to be 3.5 min at 105 °C (Fig. S-2†).
Table 1 summarizes the optimal microdroplet reaction condition for [18F]SFB on EWOD to achieve an overall radiochemical yield of 39 ± 7% (n = 4). The lower radiochemical yield of [18F]SFB in comparison to the macroscale synthesizer is presumably due to the volatilization of the radioactive [18F]fluoromethane41,42 (bp: −78 °C) and/or the [18F]fluoro-4-ethylbenzoate (bp: 210 °C). These losses was suspected to occur in the first step of the [18F]SFB synthesis. Despite the lower radiochemical yield, the EWOD platform and the chemistry reported here reliably produced ∼33 MBq of [18F]SFB starting from 222 MBq of [18F]fluoride, which is sufficient for routine biomolecule labeling and mice imaging. Although the reaction time for each reaction step on EWOD chip is significantly faster in comparison to macroscopic module, the overall synthesis time is only comparable with the conventional system. During this developmental stage, many operations were performed manually such as reagent loading, activating droplet operation, heating and product collection. Our group is currently developing novel engineering methods for automating the reagent loading and product extraction, which could reduce the overall synthesis time to ∼80 min.
| Step | Time (min) | Reagents (nmol) | Droplet volume (μL) | |
|---|---|---|---|---|
| a Some variable steps such as reagent loading, radioactivity measurement, configuring the EWOD software and etcetera are not included in the above table. | ||||
| 1 | Load [18F]KF/K2.2.2 | 12 | ||
| EWOD actuation | 6 | K2CO3 (216) | ||
| Heat to 105 °C | 8 | K2.2.2 (390) | ||
| 2 | Azeotropic drying | MeCN | 9 | |
| EWOD actuation | 1 | |||
| Heat to 105 °C | 2 | |||
| 3 | Fluorination | Precursor (167) | 2 | |
| Load precursor by EWOD | 1 | |||
| Reaction at 120 °C | 4.5 | |||
| Total heating time | 5 | |||
| 4 | Hydrolysis | NPr4OH (960) | 6 | |
| Load NPr4OH by EWOD | 2 | |||
| Reaction at 105 °C | 1.5 | |||
| Total heating time | 7 | |||
| 5 | Esterification | HSTU (1668) | 6 | |
| Load HSTU by EWOD | 2 | |||
| Reaction at 105 °C | 3.5 | |||
| Total heating time | 6 | |||
| 6 | Product extraction | 10 | ||
| 7 | HPLC purification | 10 | n/a | 2500 |
| 8 | Drying and formulation | 20 | n/a | n/a |
4.3 Purification, formulation and quality control
Due to the minute amount of reagents used in the [18F]SFB synthesis on the EWOD chip, we utilized the analytical column HPLC for the purification step to achieve >95% radiochemical purity. Using the analytical HPLC for the final purification is highly desirable as the smaller diameter column provides a better separation and results in a lower volume of mobile phase in comparison to the semi-preparative scale purification typically used in macroscale radiosynthesis. The purified [18F]SFB was collected in ∼2.5 mL and the mobile phase was gradually evaporated to dryness in a hot water bath under a steady flow of nitrogen stream. The water bath temperature was set to be below the boiling point of MeCN (bp: 82 °C) to avoid bumping and splashing of the solvent mixture. In a typical evaporation process, ∼5% of radioactivity was lost. After reformulating in 50 μM SBB, a small aliquot of the [18F]SFB was injected into an analytical HPLC to determine the chemical and radiochemical purity. In literature, [18F]SFB is known to be stable under acidic conditions and degrades at pH of 10 or higher.13 In this study, we observed rapid degradation of the [18F]SFB during the drying and reformulation process. Attempts to use the resulting 90–95% pure [18F]SFB for conjugation with the diabody resulted in an extremely poor conjugation efficiency (1–2%). To stabilize the purified [18F]SFB during the drying and reformulation processes, a small amount of aqueous acetic acid (bp: 118 °C) was added to the HPLC collection vial. Due to the high boiling point of acetic acid, the overall pH of the final mixture remains slightly acidic throughout the entire evaporation process and the resulting pH can be easily buffered with 3 μL of 1 M acetic acid to achieve the desired pH needed in the subsequent conjugation reaction. Using the stabilized [18F]SFB with 95% radiochemical purity, the anti-PSCA-diabody was labeled with 33.1 ± 12.5% efficiency and radiochemical purity of 94.6 ± 2.1% (n = 3) as determined by instant thin layer chromatography (ITLC).4.4 Radiolabeling and PET imaging of [18F]FB-A2cDb
The anti-PSCA A2cDb was successfully radiolabeled using [18F]SFB synthesized from the EWOD microfluidic radiosynthesizer. Here, labeling to lysine residues using [18F]SFB and size exclusion purification of the radiotracer was executed within 30 min and resulted in radiolabeled [18F]FB-A2cDb with sufficient purity for preclinical in vivo immunoPET imaging. [18F]FB-A2cDb specifically targets human PSCA-expressing prostate cancer cells in vivo and rapid blood clearance results in high-contrast PET images at early time points (Fig. 5). The pharmacokinetic properties of the 50 kDa cys-diabody format (t1/2 = 2–5 h in mice) make it a good match to the half-life of F-18, enabling same-day imaging.43,44 Further advantages of using a short-lived radionuclide in combination with an antibody fragment is the reduced radiation dose and exposure for the patients compared to using radiolabeled full-length antibodies conjugated to longer-lived isotopes.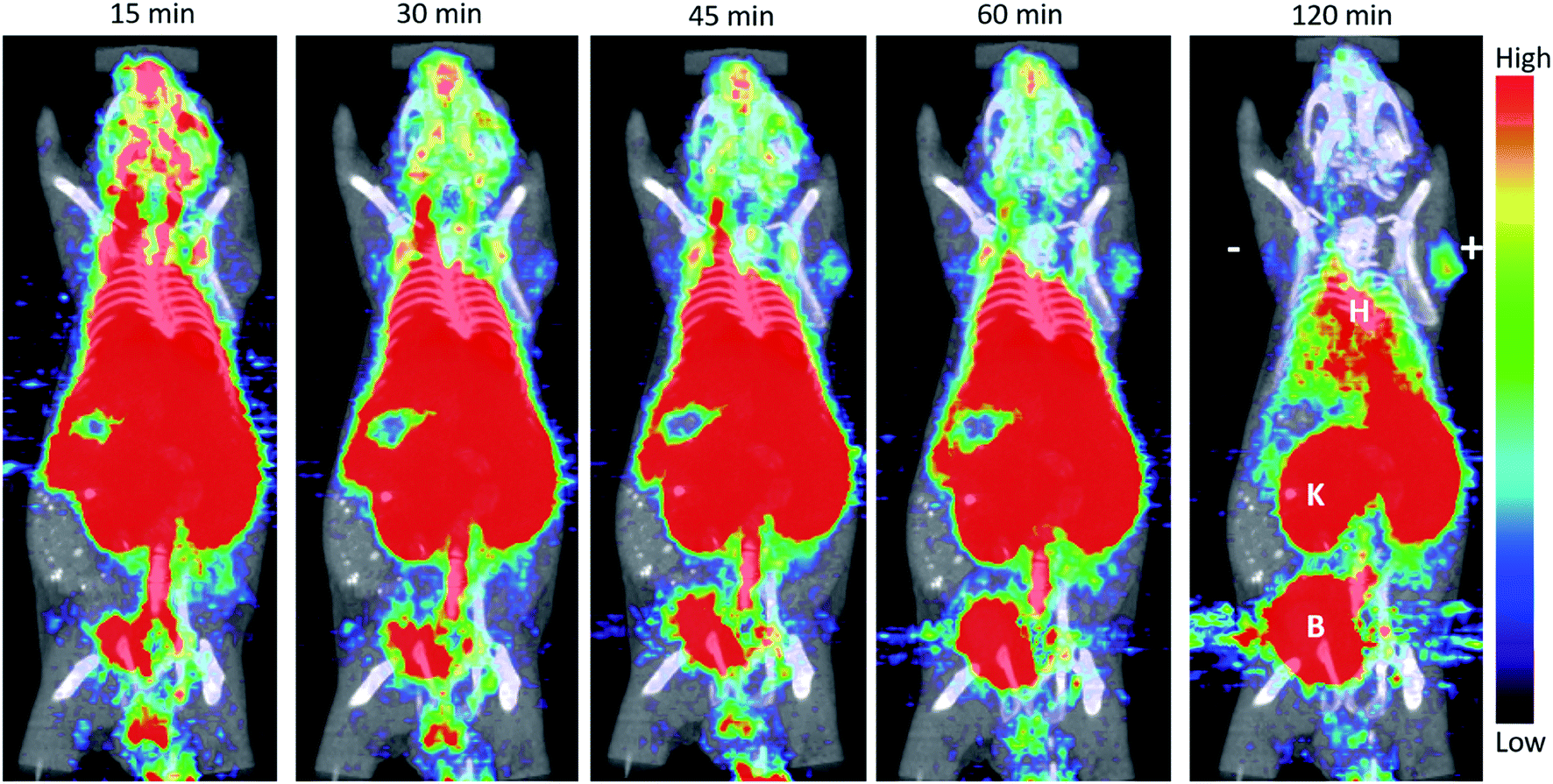 | ||
| Fig. 5 ImmunoPET imaging of a mouse bearing 22Rv1 (left; “−”) and 22Rv1-PSCA (right; “+”) subcutaneous tumors. [18F]FB-A2cDb (10 μg/1.11 MBq) was injected via the tail vein. Shown are 10 min frames of a 120 min dynamic scan as 5 mm MIP PET/CT overlay. B = bladder; K = kidney; H = heart. | ||
5. Conclusion
We have demonstrated the capability of the EWOD microfluidic radiosynthesizer to perform a sophisticated 3-step, one-pot synthesis, which involved a nucleophilic aromatic fluorination reaction, saponification and an esterification followed by off-chip HPLC purification and reformulation in 39 ± 7% (n = 4) radiochemical yield with a total synthesis time of ∼120 min. This report builds on earlier work on the EWOD radiosynthesizer, exemplifying the flexibility of performing different classes of 18F-radiochemical reactions, including both aliphatic and aromatic nucleophilic fluorinations. Furthermore, we showed that sufficient amount of [18F]SFB was produced for PSCA-diabody labeling and in vivo imaging. The compact EWOD chip radiosynthesizer could potentially be self-shielded and automated to enable imaging scientists to routinely synthesize [18F]SFB in their own laboratories or clinics for labeling a plethora of biomolecules of interest.Conflicts of interest
Drs van Dam and Keng are consultants to Sofie Biosciences. The Regents of the University of California have licensed technology to Sofie Biosciences that was invented by Drs van Dam, and Keng, and have taken equity in Sofie Biosciences as part of the licensing transaction.Acknowledgements
This work is supported by funds provided in part by the Department of Molecular and Medical Pharmacology and the UCLA Foundation from a donation made by Ralph and Marjorie Crump for the Crump Institute for Molecular imaging. We thank Dr David Stout at the Crump Cyclotron and Radiochemistry Technology Center and Dr Saman Sadeghi and the staff of the UCLA Biomedical Cyclotron facility for providing [18F]fluoride ion for these studies. We thank Dr Jia Wang for assistance with performing synthesis optimization studies.References
- Radiosynthesis Database of PET Probes (RaDaP), http://www.nirs.qst.go.jp/research/division/mic/db2/, accessed May 8, 2017 Search PubMed.
- K. E. McCabe and A. M. Wu, Cancer Biother. Radiopharm., 2010, 25, 253–261 CrossRef CAS.
- A. C. Freise and A. M. Wu, Mol. Immunol., 2015, 67, 142–152 CrossRef CAS PubMed.
- O. Jacobson, D. O. Kiesewetter and X. Chen, Bioconjugate Chem., 2015, 26, 1–18 CrossRef CAS.
- P. Y. Keng, S. Chen, H. Ding, S. Sadeghi, G. J. Shah, A. Dooraghi, M. E. Phelps, N. Satyamurthy, A. F. Chatziioannou, C.-J. Kim and R. M. van Dam, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 690–695 CrossRef CAS.
- M. R. Javed, S. Chen, H.-K. Kim, L. Wei, J. Czernin, C.-J. Kim, R. M. van Dam and P. Y. Keng, J. Nucl. Med., 2014, 55, 321–328 CrossRef CAS.
- M. R. Javed, S. Chen, J. Lei, J. Collins, M. Sergeev, H.-K. Kim, C.-J. Kim, R. M. van Dam and P. Y. Keng, Chem. Commun., 2014, 50, 1192–1194 RSC.
- M. Sergeev, M. Lazari, F. Morgia, J. Collins, M. R. Javed, O. Sergeeva, J. Jones, M. E. Phelps, J. T. Lee, P. Y. Keng and R. M. van Dam, Commun. Chem., 2018, 1, 10 CrossRef.
- M. Tredwell, S. M. Preshlock, N. J. Taylor, S. Gruber, M. Huiban, J. Passchier, J. Mercier, C. Génicot and V. Gouverneur, Angew. Chem., 2014, 126, 1–6 CrossRef.
- S. Preshlock, M. Tredwell and V. Gouverneur, Chem. Rev., 2016, 116, 719–766 CrossRef CAS.
- J. Wang, P. H.-S. Chao and R. M. van Dam, Lab Chip, 2019, 19, 2415–2424 RSC.
- S. M. Okarvi, Eur. J. Nucl. Med. Mol. Imaging, 2001, 28, 929–938 CrossRef CAS.
- G. Vaidyanathan and M. R. Zalutsky, Nat. Protoc., 2006, 1, 1655–1661 CrossRef CAS.
- U. Ackermann, S. D. Yeoh, J. I. Sachinidis, S. S. Poniger, A. M. Scott and H. J. Tochon-Danguy, J. Labelled Compd. Radiopharm., 2011, 54, 671–673 CrossRef CAS.
- G. Tang, W. Zeng, M. Yu and G. Kabalka, J. Labelled Compd. Radiopharm., 2008, 51, 68–71 CrossRef CAS.
- F. Wüst, C. Hultsch, R. Bergmann, B. Johannsen and T. Henle, Appl. Radiat. Isot., 2003, 59, 43–48 CrossRef.
- J. Marik and J. L. Sutcliffe, Appl. Radiat. Isot., 2007, 65, 199–203 CrossRef CAS.
- G. Tang, W. Zeng, M. Yu and G. Kabalka, J. Labelled Compd. Radiopharm., 2008, 51, 68–71 CrossRef CAS.
- G. Tang, X. Tang and X. Wang, J. Labelled Compd. Radiopharm., 2010, 53, 543–547 CrossRef CAS.
- P. Mäding, F. Füchtner and F. Wüst, Appl. Radiat. Isot., 2005, 63, 329–332 CrossRef PubMed.
- R. Bejot, A. M. Elizarov, E. Ball, J. Zhang, R. Miraghaie, H. C. Kolb and V. Gouverneur, J. Labelled Compd. Radiopharm., 2011, 54, 117–122 CrossRef CAS.
- M. Lazari, J. Collins, B. Shen, M. Farhoud, D. Yeh, B. Maraglia, F. T. Chin, D. A. Nathanson, M. Moore and R. M. van Dam, J. Nucl. Med. Technol., 2014, 42, 203–210 CrossRef.
- P. Y. Keng, M. Esterby and R. M. van Dam, in Positron Emission Tomography - Current Clinical and Research Aspects, ed. C.-H. Hsieh, InTech, Rijeka, Croatia, 2012, pp. 153–182 Search PubMed.
- M. Brivio, W. Verboom and D. N. Reinhoudt, Lab Chip, 2006, 6, 329–344 RSC.
- P. D. I. Fletcher, S. J. Haswell, E. Pombo-Villar, B. H. Warrington, P. Watts, S. Y. F. Wong and X. Zhang, Tetrahedron, 2002, 58, 4735–4757 CrossRef CAS.
- P. Watts, G. Pascali and P. A. Salvadori, J. Flow Chem., 2012, 2, 37–42 CrossRef CAS.
- V. Arima, G. Pascali, O. Lade, H. R. Kretschmer, I. Bernsdorf, V. Hammond, P. Watts, F. D. Leonardis, M. D. Tarn, N. Pamme, B. Z. Cvetkovic, P. S. Dittrich, N. Vasovic, R. Duane, A. Jaksic, A. Zacheo, A. Zizzari, L. Marra, E. Perrone, P. A. Salvadori and R. Rinaldi, Lab Chip, 2013, 13, 2328–2336 RSC.
- A. M. Elizarov, Lab Chip, 2009, 9, 1326–1333 RSC.
- H. Kimura, K. Tomatsu, H. Saiki, K. Arimitsu, M. Ono, H. Kawashima, R. Iwata, H. Nakanishi, E. Ozeki, Y. Kuge and H. Saji, PLoS One, 2016, 11, e0159303 CrossRef.
- S. Chen, M. R. Javed, H.-K. Kim, J. Lei, M. Lazari, G. J. Shah, M. van Dam, P. Y. Keng and C.-J. Kim, Lab Chip, 2014, 14, 902–910 RSC.
- E. J. Lepin, J. V. Leyton, Y. Zhou, T. Olafsen, F. B. Salazar, K. E. McCabe, S. Hahm, J. D. Marks, R. E. Reiter and A. M. Wu, Eur. J. Nucl. Med. Mol. Imaging, 2010, 37, 1529–1538 CrossRef CAS.
- G. A. Sonn, A. S. Behesnilian, Z. K. Jiang, K. A. Zettlitz, E. J. Lepin, L. A. Bentolila, S. M. Knowles, D. Lawrence, A. M. Wu and R. E. Reiter, Clin. Cancer Res., 2016, 22, 1403–1412 CrossRef CAS.
- K. A. Zettlitz, W.-T. K. Tsai, S. M. Knowles, N. Kobayashi, T. R. Donahue, R. E. Reiter and A. M. Wu, J. Nucl. Med., 2018, 59, 1398–1405 CrossRef CAS.
- K. A. Zettlitz, R. Tavaré, S. M. Knowles, K. K. Steward, J. M. Timmerman and A. M. Wu, Clin. Cancer Res., 2017, 23, 7242–7252 CrossRef CAS.
- A. M. Loening and S. S. Gambhir, Mol. Imaging, 2003, 2, 131–137 CrossRef.
- S. K. Cho, H. Moon and C.-J. Kim, J. Microelectromech. Syst., 2003, 12, 70–80 CrossRef.
- R. M. van Dam, C. J. Kim, S. Chen, H. J. Ding, G. J. Shah and P. Y. Keng, US Pat. 9193640 B2, 2018.
- G. J. Shah and C. -J. Kim, Lab Chip, 2009, 9, 2402–2405 RSC.
- S. Olma and K.-F. Shen, PCT WO/2010/033196 A2, 2010.
- W. Bannwarth and R. Knorr, Tetrahedron Lett., 1991, 32, 1157–1160 CrossRef CAS.
- W. R. Banks, M. R. Satter and D.-R. Hwang, Appl. Radiat. Isot., 1994, 45, 69–74 CrossRef CAS.
- H. Sun and S. G. DiMagno, J. Fluorine Chem., 2007, 128, 806–812 CrossRef CAS.
- T. Olafsen and A. M. Wu, Semin. Nucl. Med., 2010, 40, 167–181 CrossRef.
- K. A. Zettlitz, R. Tavaré, W.-T. K. Tsai, R. E. Yamada, N. S. Ha, J. Collins, R. M. van Dam, J. M. Timmerman and A. M. Wu, Eur. J. Nucl. Med. Mol. Imaging, 2019, 46(2), 489–500 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra06158d |
| This journal is © The Royal Society of Chemistry 2019 |