
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRetracted Article: Long noncoding RNA HOTAIR promotes cell apoptosis by sponging miR-221 in Parkinson's disease†
Fan Zhou‡
a,
Sanping Xie‡ *a,
Juan Lib and
Shujie Duana
*a,
Juan Lib and
Shujie Duana
aDepartment of Neurology, The Central Hospital of Jingzhou, No. 1 Renmin Road, Jingzhou District, Jingzhou 434020, Hubei, China. E-mail: bdltdu@163.com; Tel: +86-0716-8435794
bDepartment of General Medicine, The Central Hospital of Jingzhou, Jingzhou, Hubei, China
First published on 18th September 2019
Abstract
Parkinson's disease (PD) is a common neurological disorder that is detrimental to the health of older people worldwide. Long noncoding RNAs (lncRNAs) have been reported to play essential roles in the pathogenesis and therapeutics of PD. LncRNA homeobox transcript antisense intergenic RNA (HOTAIR) is expressed in PD samples; however, the exact roles of HOTAIR and its mechanism remain largely unclear. Herein, the neurotoxins 1-methyl-4-phenylpyridine (MPP+) and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) were used to establish PD models in vitro and in vivo. The expressions of HOTAIR and microRNA-221 (miR-221) were measured by the quantitative real-time polymerase chain reaction (qRT-PCR). Cell viability and apoptosis were detected by the 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) assay and western blot or flow cytometry, respectively. The interaction between HOTAIR and miR-221 was explored by luciferase activity and RNA immunoprecipitation (RIP). The tyrosine hydroxylase (TH)-positive cells in MPTP-treated-mouse midbrains were analyzed by immunohistochemistry. The HOTAIR expression was up-regulated and that of miR-221 was down-regulated in the serum of PD patients and MPP+-treated SH-SY5Y cells. Overexpression of HOTAIR inhibited cell viability and promoted apoptosis in MPP+-treated SH-SY5Y cells. However, the down-regulation of HOTAIR showed an opposite effect. Moreover, miR-221 was validated to be bound to HOTAIR, and its addition reversed the regulatory effect of HOTAIR on cell viability and apoptosis in MPP+-treated SH-SY5Y cells. Moreover, the knockdown of HOTAIR attenuated the degree of PD and cell apoptosis by regulating miR-221 in MPTP-treated mice. In conclusion, HOTAIR contributed to cell apoptosis by sponging miR-221 in PD. This study elucidates a new mechanism for understanding the pathogenesis of PD and provides a promising target for the treatment of PD.
1. Introduction
Parkinson's disease (PD) is one of the most common neurodegenerative disorders, with increasing incidence in the elderly all over the world.1 It is characterized by classical features such as the formation of Lewy bodies and a decrease of dopaminergic neurons in the substantia nigra.2 Despite great advances in understanding the pathogenic mechanism and therapeutics of PD, effective strategies in this regard remain limited.3 Hence, the exploration of novel biomarkers is expected for the treatment of PD.Long noncoding RNAs (lncRNAs) have been reported to have potential in the prevention and therapeutics of PD.4 Moreover, lncRNAs play essential roles in regulating cell viability, mitochondrial function and tyrosine hydroxylase (TH) secretion in the SH-SY5Y cells.5 For example, lncRNA small nucleolar RNA host gene 1 (SNHG1) contributes to 1-methyl-4-phenylpyridine (MPP+)-induced cytotoxicity in PD by regulating cell viability, apoptosis and reactive oxygen species (ROS) production in the SH-SY5Y cells.6 LncRNA nuclear paraspeckle assembly transcript 1 (NEAT1) regulates cell viability and autophagy in MPP+-treated SH-SY5Y cells or 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-treated mice by mediating phosphatase and tensin homolog-induced putative kinase 1 (PINK1).7 LncRNA homeobox transcript antisense intergenic RNA (HOTAIR), a promising lncRNA, has been reported to play vital roles in regulating the progression of cancers such as gastric cancer, adrenocortical carcinoma, cervical cancer and glioblastoma.8–11 Moreover, HOTAIR is suggested to be expressed in brain tissues and associated with neurodegenerative disease.12 Notably, HOTAIR is up-regulated in PD and promotes PD progression by regulating leucine-rich repeat kinase 2 (LRRK2) expression.13 However, the underlying mechanism by which HOTAIR participates in PD progression remains poorly understood.
microRNAs (miRNAs), a class of small noncoding RNAs, play pivotal roles in the diagnosis, pathogenesis and therapeutics of PD.14 miR-221 has been suggested to be implicated in the development and progression of various cancers, such as ovarian cancer, hepatocellular carcinoma and oral cancer.15–17 Moreover, miR-221 is suggested to be down-regulated and to serve as a potential biomarker for PD.18 Additionally, miR-221 inhibits cell apoptosis and promotes neuronal survival in PD.19 Functional lncRNA is known to act as competing endogenous RNA (ceRNA) to sponge miRNA in many conditions. Moreover, the starBase online database predicted that there are putative binding sites between HOTAIR and miR-221. Hence, we hypothesized that miR-221 plays important roles in HOTAIR-mediated progression of PD. A previous study revealed various PD models in vitro or in vivo.20 In this study, we established PD models of MPP+-treated SH-SY5Y cells in vitro and MPTP-treated mice in vivo. Moreover, we investigated the effects of HOTAIR on cell apoptosis and explored the relationship between HOTAIR and miR-221.
2. Materials and methods
2.1. Participants and blood samples
A total of 24 patients with PD and 10 age-matched control subjects without neurological disorders were recruited from the Central Hospital of Jingzhou. All participants provided informed consent, and our study was accepted by the Research Ethics Committee of the Central Hospital of Jingzhou. Venous blood samples (5 mL) were collected from participants by vein puncture. Serum was collected by centrifugation at 2000 × g for 15 min after 30 min of clotting and then immediately stored at −80 °C until use.2.2. Cell culture and treatment
Human neuroblastoma cell line SH-SY5Y cells were purchased from American Tissue Culture Collection (ATCC, Manassas, VA, USA) and cultured in Dulbecco's modified Eagle's medium (Gibco, Carlsbad, CA, USA) containing 10% fetal bovine serum (Gibco), 100 U mL−1 penicillin and 100 μg mL−1 streptomycin (Invitrogen, Carlsbad, CA, USA) at 37 °C in a humidified atmosphere with 5% CO2 during the study. The cell line was validated by amplifying LINE-1 (a marker for species identification) (ESI Fig. 1†).21miR-221 mimic (miR-221), miRNA negative control (miR-NC), miR-221 inhibitor (anti-miR-221), inhibitor negative control (anti-miR-NC), pcDNA empty vector (vector), pcDNA-based HOTAIR overexpression vector (HOTAIR), small interfering RNA (siRNA) against HOTAIR (si-HOTAIR) and siRNA negative control (si-NC) were synthesized by Genepharma (Shanghai, China). Cell transfection was performed in SH-SY5Y cells for 48 h using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. The transfection efficacy was investigated by quantitative real-time polymerase chain reaction (qRT-PCR). After the transfection, cells were treated with MPP+ for further analyses. To establish a PD model in vitro, SH-SY5Y cells were treated with different concentrations (0, 0.25, 0.5, 1 and 2 mM) of MPP+ (Sigma, St. Louis, MO, USA) for 24 h or 1 mM MPP+ for various times (6, 12, 24 and 48 h).
2.3. qRT-PCR
Total RNA was isolated from serum, cells or tissues using TRIzol reagent (Invitrogen) according to the manufacturer's instructions. For measurement of HOTAIR expression, the complementary DNA (cDNA) was generated using an M-MLV Reverse Transcription Kit (Thermo Fisher, Wilmington, DE, USA) and then used for qRT-PCR using SYBR green (Applied Biosystems, Foster City, CA, USA) with the following amplification protocol: 95 °C for 5 min, 40 cycles of 95 °C for 15 s, and 60 °C for 1 min. For detection of miR-221 levels, cDNA was generated by a TaqMan microRNA Reverse Transcription Kit (Applied Biosystems). Every sample was prepared in triplicate, and the experiments were repeated three times. The expressions of HOTAIR and miR-221 were calculated using the 2−ΔΔCt method with GAPDH or U6 small RNA as endogenous controls, respectively.22 The primers were as follows: human: miR-221 (forward, 5′-CAGCATACATGATTCCTTGTGA-3′; reverse, 5′-CTTGGGTTTGAGATGTTTGG-3′), U6 (forward, 5′-GTGCTCGCTTCGGCAGCACATATAC-3′; reverse, 5′-AAAAATATGGAACGCTCACGAATTTG-3′), HOTAIR (forward, 5′-CAGTGGGGAACTCTGACTCG-3′; reverse, 5′-GTGCCTGGTGCTCTCTTACC-3′), GAPDH (forward, 5′-AACGGATTTGGTCGTATTGGG-3′; reverse, 5′-TCGCTCCTGGAAGATGGTGAT-3′); mouse: miR-221 (forward, 5′-GCTAAAGAGGGGGAGCAATC-3′; reverse, 5′-CTGCTCTTTGAGGGAGGACAA-3′), U6 (forward, 5′-CTCGCTTCGGCAGCACATA-3′; reverse, 5′-AAC GATTCACGAATTTGCGT-3′), HOTAIR (forward, 5′-AGGTCCCCAACATCGGTAGA-3′; reverse, 5′-GTTCCTTCCATCTGGACCCG-3′), GAPDH (forward, 5′-AGGTCGGTGTGAACGGATTTG-3′; reverse, 5′-TGTAGACCATGTAGTTGGGTCA-3′).2.4. Cell viability
The 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) assay was performed to measure the viability of SH-SY5Y cells. After adjusting the cell density to 5 × 104 mL−1, transfected SH-SY5Y cells were seeded into 96-well plates at a density of 1 × 104 cells per well and treated with 1 mM MPP+ for 24 h. Each group was prepared in triplicate. After the treatment, the cells were incubated with 0.5 mg mL−1 MTT solution (Thermo Fisher) for another 4 h. The formazan was dissolved with 100 μL of dimethylsulfoxide (DMSO, Thermo Fisher). The absorbance was measured at 490 nm using a microplate reader (Bio-Rad, Hercules, CA, USA).2.5. Western blot
Total protein was extracted from cells or tissues using RIPA lysis buffer (Beyotime Biotechnology, Shanghai, China) and then quantified by a BCA protein assay kit (Beyotime Biotechnology) after centrifugation at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g for 20 min at 4 °C. Subsequently, the proteins were denatured at 98 °C for 10 min, separated by SDS-PAGE gel electrophoresis and transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA, USA). The membranes were blocked with 5% non-fat milk for 1 h at room temperature and then incubated with primary antibodies overnight at 4 °C and secondary antibody conjugated with horseradish peroxidase (HRP) for 2 h at room temperature. The antibodies against B-cell lymphoma-2 (Bcl-2) (ab59348, 1:500 dilution), Bcl-2-associated x protein (Bax) (ab32503, 1:5000 dilution) and β-actin (ab8227, 1:5000 dilution) and the secondary antibody IgG (ab6721, 1:10000 dilution) were purchased from Abcam (Cambridge, UK). β-Actin was used as a loading control in this study. The protein signals were analyzed with Image Lab software (Bio-Rad) with abundance of β-actin as a loading control after interacting with an enhanced chemiluminescence (ECL) chromogenic substrate (Beyotime Biotechnology).
000 × g for 20 min at 4 °C. Subsequently, the proteins were denatured at 98 °C for 10 min, separated by SDS-PAGE gel electrophoresis and transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA, USA). The membranes were blocked with 5% non-fat milk for 1 h at room temperature and then incubated with primary antibodies overnight at 4 °C and secondary antibody conjugated with horseradish peroxidase (HRP) for 2 h at room temperature. The antibodies against B-cell lymphoma-2 (Bcl-2) (ab59348, 1:500 dilution), Bcl-2-associated x protein (Bax) (ab32503, 1:5000 dilution) and β-actin (ab8227, 1:5000 dilution) and the secondary antibody IgG (ab6721, 1:10000 dilution) were purchased from Abcam (Cambridge, UK). β-Actin was used as a loading control in this study. The protein signals were analyzed with Image Lab software (Bio-Rad) with abundance of β-actin as a loading control after interacting with an enhanced chemiluminescence (ECL) chromogenic substrate (Beyotime Biotechnology).
2.6. Cell apoptosis
Cell apoptosis was measured using an Annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) apoptosis detection kit (Sigma) by flow cytometry. After exposure to 1 mM MPP+ for 24 h, SH-SY5Y cells were washed with PBS and then resuspended in binding buffer. The cells were double stained with 10 μL Annexin V-FITC for 10 min and 5 μL PI for 10 min in the dark according to the manufacturer's instructions. The apoptotic cells were analyzed using a flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA). Every sample was prepared in triplicate.2.7. Luciferase activity assay
The putative binding sites of miR-221 and the sequences of HOTAIR were predicted using starBase. The sequences of HOTAIR containing wild-type (WT) or mutant-type (MUT) binding sites were amplified and then cloned into pGL3 vectors (Promega, Madison, WI, USA) to synthesize luciferase reporter vectors (HOTAIR-WT or HOTAIR-MUT), respectively. SH-SY5Y cells were co-transfected with 20 ng HOTAIR-WT or HOTAIR-MUT, 15 ng control vector and 40 nM miR-221 or miR-NC using Lipofectamine 2000 according to the manufacturer's protocols. The luciferase activity was measured using a luciferase assay kit (Promega) after transfection for 48 h according to the manufacturer's instructions.2.8. RNA immunoprecipitation (RIP)
The RIP assay was performed using an RNA-binding protein immunoprecipitation kit (Millipore) according to the manufacturer's protocols. In brief, SH-SY5Y cells transfected with miR-221 or miR-NC were lysed in RIP buffer and then interacted with magnetic beads bound with anti-Ago2. The enrichment of HOTAIR immunoprecipitated on the beads was measured by qRT-PCR.2.9. Animals and PD mouse model
C57BL/6 mice (male, ten weeks old) were obtained from Vital River Laboratory Animal Technology (Beijing, China) and were maintained in specific pathogen-free conditions with a 12 h light/dark cycle and free access to water and food. Mice were randomly divided into four groups: vehicle control (con), MPTP, MPTP + sh-NC and MPTP + sh-HOTAIR. Every group was assigned to minimize animals (n = 8 per group) under the approval of the Animal Research Committee of the Central Hospital of Jingzhou. MPTP hydrochloride (Sigma) was intraperitoneally injected at 2 h intervals at a dose of 20 mg kg−1 body weight. The con group was treated with equivalent volumes of sterile saline solution. The lentivirus vector of short hairpin RNA (shRNA) against HOTAIR (sh-HOTAIR) or shRNA negative control (sh-NC) (20 nM) constructed by GeneCopoeia (Rockville, MD, USA) was introduced into the midbrains of the mice 2 days before establishment of the MPTP-induced PD model. The mice were sacrificed 5 days after the last injection of MPTP, and midbrain samples were collected. Proportions of midbrains (n = 4 per group) were fixed with 10% formalin (Sigma), dehydrated and embedded for immunohistochemistry. The other samples were stored at −80 °C until use.2.10. Immunohistochemistry
Loss of TH is the key pathological feature of PD. TH+ neurons were analyzed by immunohistochemistry according to the manufacturer's protocols. In brief, the sections were treated using 3% H2O2 (Sigma) to block endogenous peroxidase and then incubated with antibody against TH (ab117112, 1:3000 dilution, Abcam) overnight at 4 °C and with HRP-conjugated anti-rabbit IgG for 30 min, followed by staining with diaminobenzidine (DAB) and hematoxylin (Sigma). The positive cells were detected with stereo investigator software (Microbright Field, Williston, VT, USA).
2.11. Statistical analysis
The data were presented as the mean ± standard deviation (SD) from three independent experiments. The relationship between the expression of miR-221 and HOTAIR was analyzed by Spearman rank correlation. Student's t test or one-way analysis of variance (ANOVA) was conducted to analyze statistical differences between groups using SPSS 18.0 software (SPSS, Inc., Chicago, IL, USA). P < 0.05 was regarded as statistically significant.3. Results
3.1. HOTAIR expression was increased in PD
Compared with the con group, the expression of HOTAIR was significantly enhanced in the serum of PD patients (Fig. 1A). This suggests that dysregulated HOTAIR plays important roles in PD progression. To analyze the potential role of HOTAIR, its expression was measured in an MPP+-treated PD model in vitro. The results showed that exposure to MPP+ led to a progressive increase of HOTAIR levels in a concentration-dependent manner in MPP+-treated SH-SY5Y cells (Fig. 1B). Moreover, the abundance of HOTAIR was abnormally elevated in MPP+-treated SH-SY5Y cells in a time-dependent manner (Fig. 1C). Hence, SH-SY5Y cells treated with 1 mM MPP+ for 24 h were used for further experiments. | ||
| Fig. 1 HOTAIR expression was up-regulated in PD. (A) The expression of HOTAIR was measured in the serum of patients with PD by qRT-PCR. (B) The levels of HOTAIR were detected in SH-SY5Y cells by qRT-PCR after treatment with different concentrations of MPP+ for 24 h. (C) The abundance of HOTAIR was examined in SH-SY5Y cells by qRT-PCR after exposure to 1 mM MPP+ for different treatment times. *P < 0.05, **P < 0.01, ***P < 0.001. | ||
3.2. HOTAIR induced cell apoptosis in MPP+-treated SH-SY5Y cells
To investigate the effects of HOTAIR on PD progression, SH-SY5Y cells were transfected with HOTAIR, vector, si-HOTAIR or si-NC and then treated with 1 mM MPP+ for 24 h. As a result, the abundance of HOTAIR was effectively enhanced in SH-SY5Y cells transfected with HOTAIR compared with that in the vector group, while it was obviously decreased in cells transfected with si-HOTAIR compared with that in the si-NC group (Fig. 2A). Treatment of MPP+ significantly inhibited the viability of SH-SY5Y cells, which was exacerbated by addition of HOTAIR but reversed by inhibition of HOTAIR (Fig. 2B). Moreover, decreased Bcl-2 protein levels and increased Bax protein expression were exhibited by SH-SY5Y cells after treatment with MPP+ (Fig. 2C). In addition, overexpression of HOTAIR notably enhanced the expression of Bax and impaired Bcl-2 abundance at protein level in MPP+-treated SH-SY5Y cells; its depletion caused an opposite effect (Fig. 2C). Similarly, the flow cytometry assay showed that treatment of MPP+ resulted in obvious apoptosis of SH-SY5Y cells, which was deteriorated by accumulation of HOTAIR but alleviated by exhaustion of HOTAIR (Fig. 2D).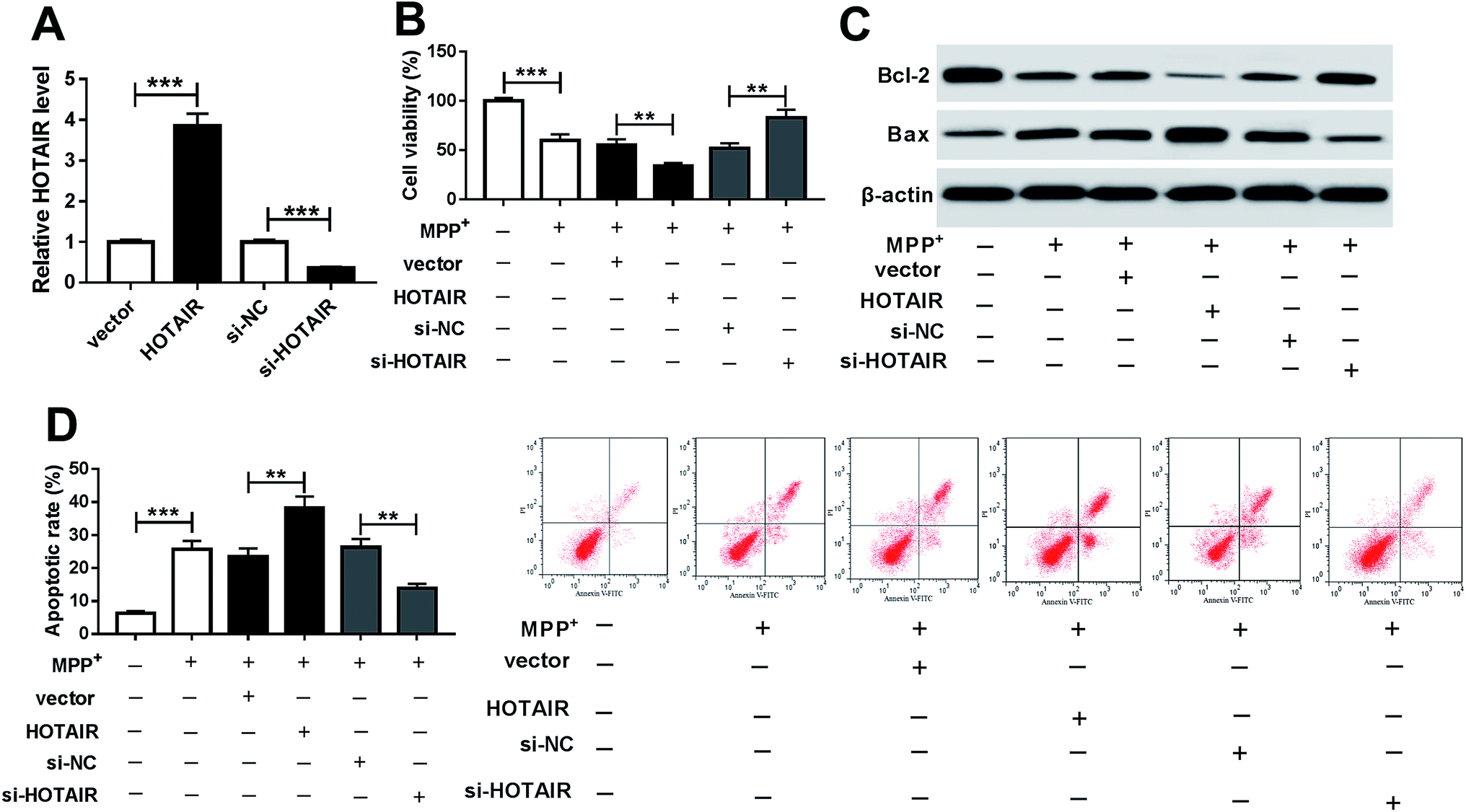 | ||
| Fig. 2 HOTAIR positively regulated cell apoptosis in MPP+-treated SH-SY5Y cells. (A) The expression of HOTAIR was measured in SH-SY5Y cells transfected with HOTAIR, vector, si-HOTAIR or si-NC by qRT-PCR. Cell viability (B), levels of Bcl-2 and Bax protein (C) and apoptosis (D) were detected in SH-SY5Y cells transfected with HOTAIR, vector, si-HOTAIR or si-NC after treatment with 1 mM MPP+ for 24 h by MTT, western blot or flow cytometry. **P < 0.01, ***P < 0.001. | ||
3.3. miR-221 was bound to HOTAIR
To explore the underlying mechanism by which HOTAIR participates in PD, potential miRNA was explored in SH-SY5Y cells. Bioinformatics analysis provided the putative binding sites of miR-221 and HOTAIR using starBase (Fig. 3A). To validate the prediction, the luciferase activity assay was conducted by constructing a luciferase reporter vector containing WT or MUT binding sites of HOTAIR (Fig. 3A). The results showed that overexpression of miR-221 led to great loss of luciferase activity in SH-SY5Y cells transfected with HOTAIR-WT, while its efficacy was lost in the HOTAIR-MUT group (Fig. 3B). Moreover, the enrichment of HOTAIR by RIP-Ago2 assay was significantly elevated in SH-SY5Y cells transfected with miR-221 compared with that of cells in the miR-NC group; however, there was little enrichment in the IgG group (Fig. 3C). Moreover, the expression of miR-221 was measured in the serum of PD patients and SH-SY5Y cells. The abundance of miR-221 was abnormally decreased in the serum of PD patients compared with that of the con group (Fig. 3D). Similarly, the expression of miR-221 was aberrantly inhibited in SH-SY5Y cells after exposure to MPP+ in a concentration and time-dependent manner (Fig. 3E and F). In addition, the level of miR-221 was negatively correlated with HOTAIR abundance in the serum of PD patients (R = −0.6571, P = 0.0005) (Fig. 3G). Additionally, the effects of HOTAIR on miR-221 expression were investigated in SH-SY5Y cells. The results showed that overexpression of HOTAIR significantly suppressed miR-221 levels, and its knockdown led to an obvious increase of miR-221 abundance (Fig. 3H).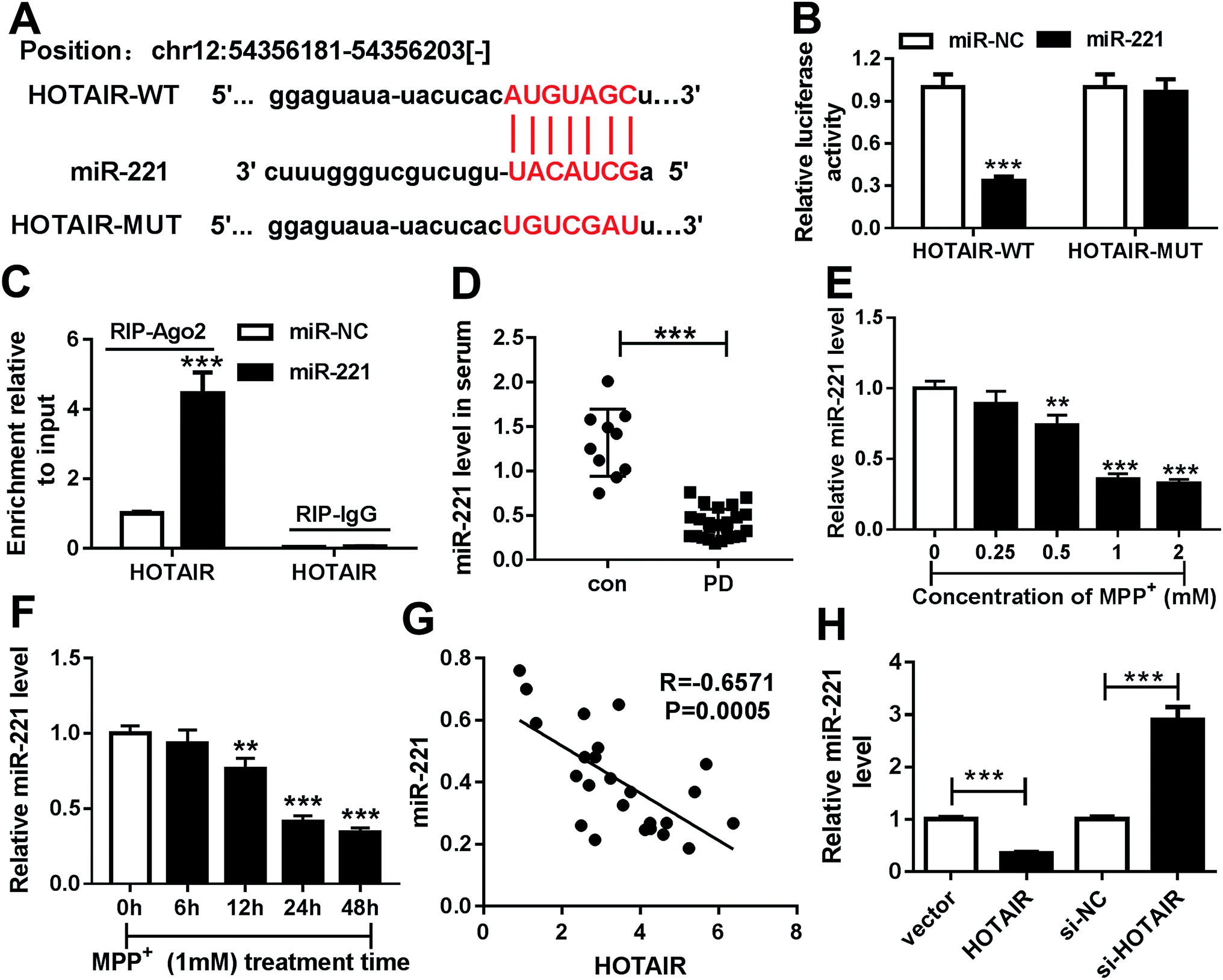 | ||
| Fig. 3 miR-221 was bound to HOTAIR. (A) The potential binding sites of miR-221 and HOTAIR were predicted by starBase. (B) Luciferase activity was measured in SH-SY5Y cells co-transfected with HOTAIR-WT or HOTAIR-MUT and miR-221 or miR-NC. (C) The enrichment of HOTAIR was measured in SH-SY5Y cells transfected with miR-221 or miR-NC after RIP assay by qRT-PCR. (D) The expression of miR-221 was detected in the serum of patients with PD or con by qRT-PCR. (E and F) The expression of miR-221 was measured in SH-SY5Y cells after treatment with various concentrations of MPP+ for 24 h or 1 mM MPP+ for different times by qRT-PCR. (G) The relationship between miR-221 level and HOTAIR abundance in patients with PD was investigated by Spearman rank correlation. (H) The abundance of miR-221 was detected in SH-SY5Y cells transfected with HOTAIR, vector, si-HOTAIR or si-NC by qRT-PCR. **P < 0.01, ***P < 0.001. | ||
3.4. HOTAIR regulated cell apoptosis by sponging miR-221 in MPP+-treated SH-SY5Y cells
To explore whether miR-221 is required for HOTAIR-mediated regulation of PD progression, SH-SY5Y cells were co-transfected with HOTAIR and miR-221 or miR-NC or si-HOTAIR and anti-miR-221 or anti-miR-NC. Addition of miR-221 reversed HOTAIR-mediated inhibition of cell viability in MPP+-treated SH-SY5Y cells (Fig. 4A). Knockdown of miR-221 ablated inhibition of HOTAIR-induced viability in MPP+-treated SH-SY5Y cells (Fig. 4B). Moreover, accumulation or depletion of miR-221 attenuated the regulatory effects of HOTAIR overexpression or knockdown on the expressions of Bcl-2 and Bax proteins in MPP+-treated SH-SY5Y cells (Fig. 4C and D). Additionally, introduction of miR-221 weakened HOTAIR-induced apoptosis of SH-SY5Y cells after MPP+ treatment, and its down-regulation alleviated the suppressive effects of HOTAIR down-regulation on apoptosis in MPP+-treated SH-SY5Y cells (Fig. 4E and F).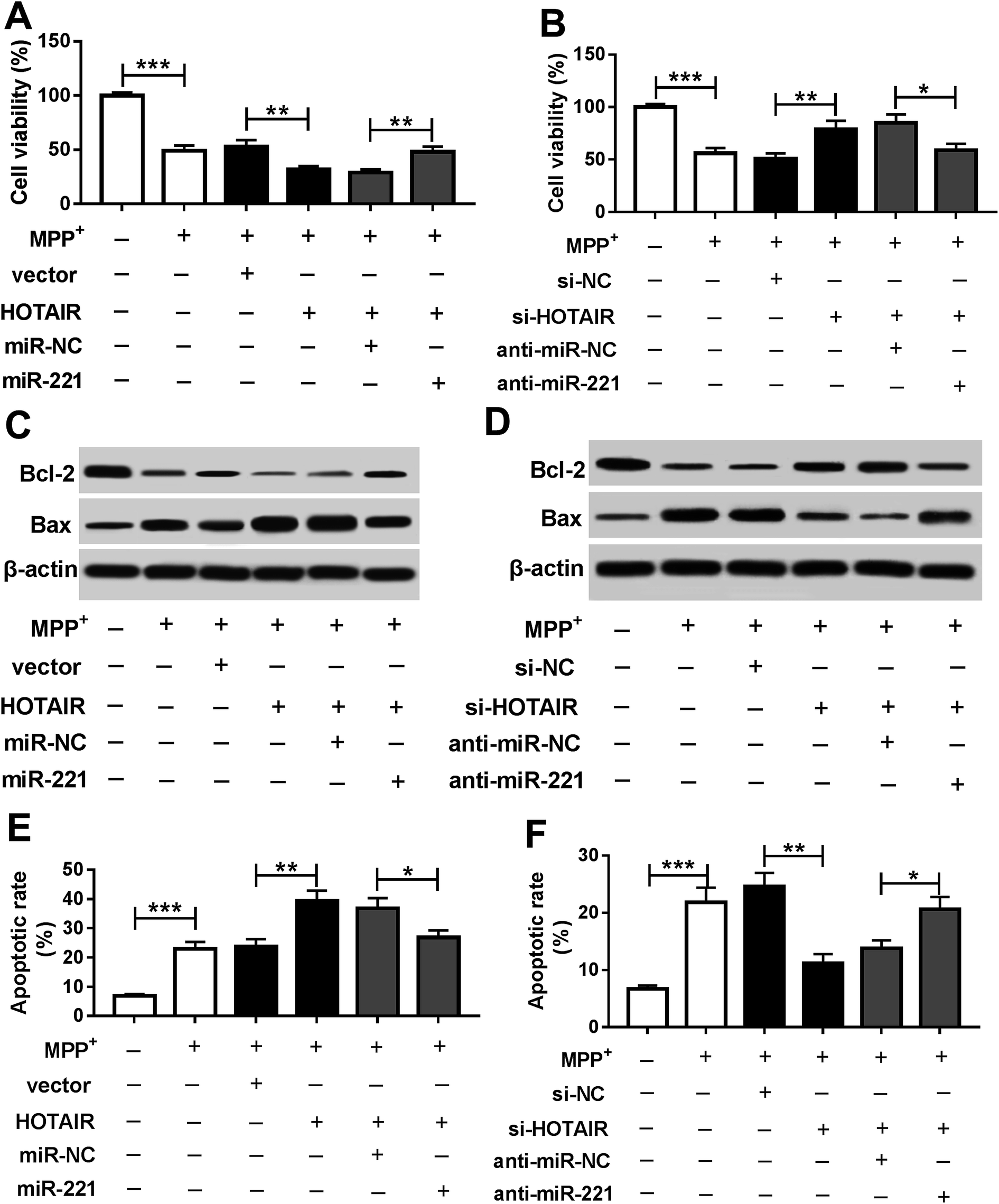 | ||
| Fig. 4 miR-221 reversed HOTAIR-mediated promotion of apoptosis in MPP+-treated SH-SY5Y cells. Cell viability (A and B), levels of Bcl-2 and Bax protein (C and D) and apoptosis (E and F) were detected in SH-SY5Y cells co-transfected with HOTAIR + miR-221, HOTAIR + miR-NC, si-HOTAIR + anti-miR-221 or si-HOTAIR + anti-miR-NC after treatment with 1 mM MPP+ for 24 h by MTT, western blot and flow cytometry, respectively. *P < 0.05, **P < 0.01, ***P < 0.001. | ||
3.5. Interference of HOTAIR protected against cell apoptosis by regulating miR-221 in MPTP-treated mice
To further analyze the effects of HOTAIR on PD progression, the role and mechanism of HOTAIR were explored in MPTP-treated mice. Treatment with MPTP resulted in great loss of TH positive cells, suggesting establishment of the PD model in vivo (Fig. 5A). However, knockdown of HOTAIR rescued TH+ neurons in MPTP-treated mice (Fig. 5A). Moreover, the expressions of HOTAIR, miR-221 and apoptosis-related proteins were measured in the midbrains of mice. The results showed that the abundance of HOTAIR was significantly enhanced in MPTP-treated mice, which was reversed by interference of HOTAIR (Fig. 5B). However, miR-221 levels were notably limited in mice after MPTP treatment, and absence of HOTAIR protected the abundance of miR-221 (Fig. 5C). Additionally, decreased Bcl-2 protein expression and increased Bax expression were displayed in MPTP-treated mice, which was counteracted by down-regulation of HOTAIR (Fig. 5D).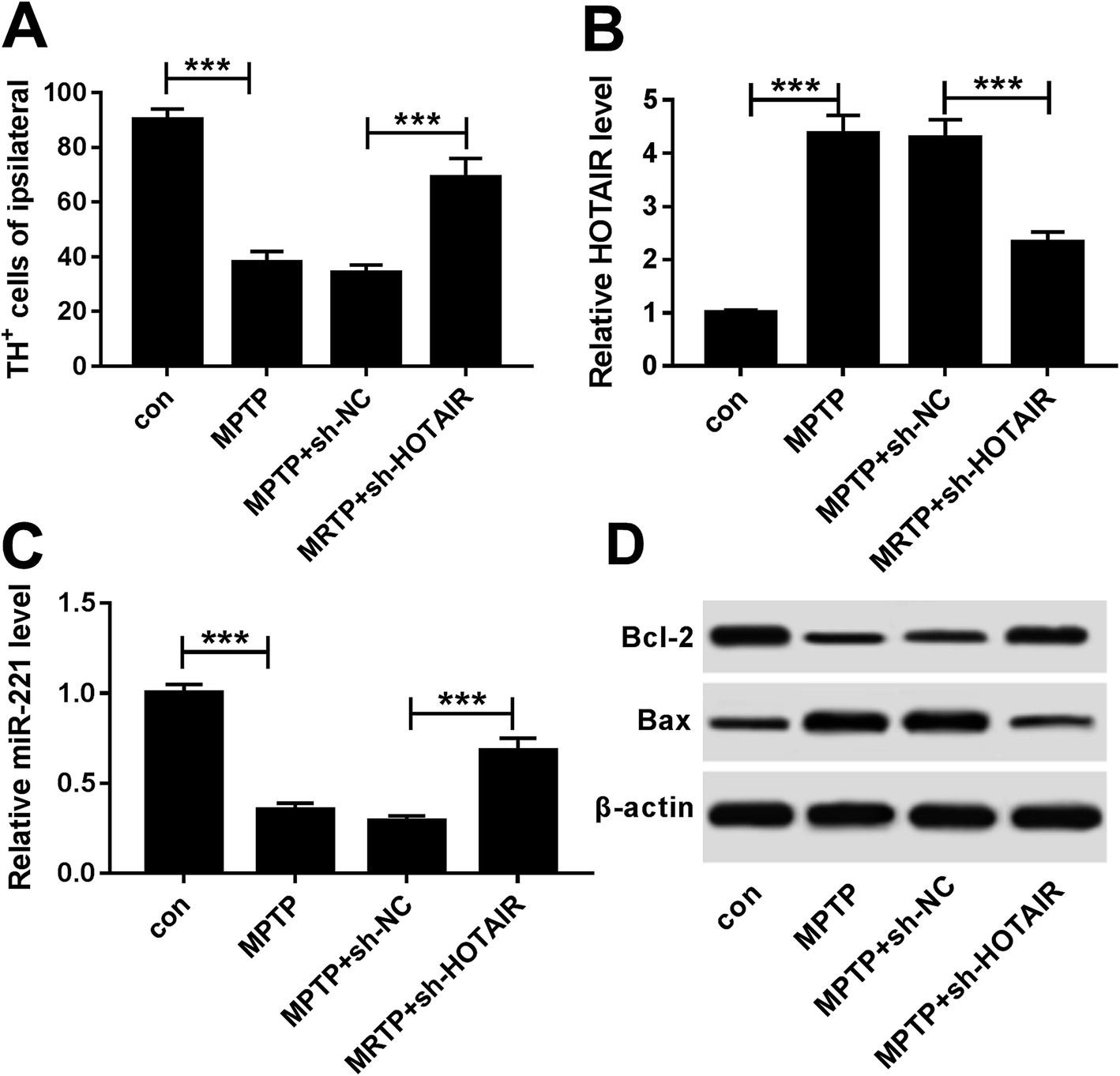 | ||
| Fig. 5 Knockdown of HOTAIR attenuated cell apoptosis by regulating miR-221 in MPTP-treated mice. (A) Ipsilateral TH+ cells were analyzed in the midbrains of mice in each group by immunohistochemistry. The expression levels of HOTAIR (B), miR-221 (C) and Bcl-2 and Bax proteins (D) were measured in the midbrains of each group by qRT-PCR and western blot, respectively. ***P < 0.001. | ||
4. Discussion
MPP+ and MPTP, as common neurotoxins, have been widely used to establish PD models.23,24 In this study, the PD model was also established by introduction of MPP+ and MPTP. We found that HOTAIR was highly expressed in PD, which is in agreement with previous work.25 However, the mechanism remains largely unknown. The available evidence indicates the importance of biological networks of lncRNAs and miRNAs in PD.26 Here, we propose for the first time that HOTAIR promotes apoptosis by sponging miR-221 in PD.A number of investigators have reported that HOTAIR exhibits pro-apoptotic roles under various conditions. For example, HOTAIR promoted apoptosis of chondrocytes through regulating miR-17-5p/fucosyltransferase 2 (FUT2)/β-catenin in osteoarthritis progression.27 Moreover, HOTAIR contributed to cell apoptosis and inflammatory injury via mediating protein kinase R (PKR) in keratinocytes upon exposure to ultraviolet B (UVB).28 In addition, HOTAIR promoted hippocampal neuronal cell apoptosis and ischemic infarct under hypoxia by regulating NADPH oxidase 2 (NOX2) expression.29 In this study, we also observed pro-apoptotic effects of HOTAIR in PD progression. HOTAIR inhibited cell viability and enhanced cell apoptosis in MPP+-treated SH-SY5Y cells, suggesting that HOTAIR exacerbates injury of PD. This is consistent with the results reported in a previous study.25 However, the mechanism of HOTAIR in PD progression remains an open question. The available evidence indicates that ceRNA networks are main pathways in the pathogenesis of cancers and other diseases, which suggests that lncRNA acts as an miRNA decoy.30 Increasing numbers of studies have reported that many miRNAs are sponged by HOTAIR under different conditions. For example, HOTAIR knockdown enhanced the sensitivity of breast cancer to radiation by activating miR-218.31 Furthermore, HOTAIR promoted cell proliferation, migration, and invasion and inhibited apoptosis by sponging miR-217 and regulating zinc finger E-Box binding homeobox 1 (ZEB1) in osteosarcoma.32 In the present study, we first validated the interaction between HOTAIR and miR-221 in SH-SY5Y cells by luciferase activity and RIP assays, which indicated that HOTAIR serves as a ceRNA for miR-221 in PD. Although interactions between HOTAIR and miR-221 have been reported in non-small cell lung cancer cells,33 it would be beneficial to directly confirm this target association in SH-SY5Y cells because of the alteration of the tumor microenvironment.
Here, we showed that miR-221 expression was down-regulated in PD, which is similar to a previous view that low expression of miR-221 can be regarded as a biomarker for PD.34 Moreover, miR-221 has been reported to alleviate inflammatory response and apoptosis of neuronal cells by targeting tumor necrosis factor α-induced protein 2 (TNFAIP2) in spinal cord ischemia-reperfusion.35 This revealed that miR-211 may play an anti-apoptotic role in neuronal cells under stress. Additionally, miR-221 exhibited a protective role in PD, as revealed by the enhanced proliferation and inhibited apoptosis in 6-hydroxydopamine (6-OHDA)-treated PC12 cells.36 Similarly, we found that introduction of miR-221 protected cell viability from HOTAIR in MPP+-treated SH-SY5Y cells. Moreover, we established a PD model in vivo to investigate the role of HOTAIR. The loss of TH is regarded as a main feature of PD.37 Hence, the expression of TH was measured in midbrain tissues, and the results showed that MPTP treatment led to a great decrease in TH-positive neurons, indicating successful establishment of the PD model. Then, we found that knockdown of HOTAIR inhibited apoptosis by regulating miR-221 in MPTP-treated mice. However, the detailed mechanism by which miR-221 participates in PD progression was not established in this study. Thus, the potential targets of miR-221 should be explored in future to better understand the molecular mechanism.
5. Conclusion
In conclusion, HOTAIR expression was enhanced and miR-221 was decreased in PD. Moreover, overexpression of HOTAIR enhanced apoptosis in MPP+-treated SH-SY5Y cells. miR-221 was bound to HOTAIR, and its introduction reversed HOTAIR-induced apoptosis in MPP+-treated SH-SY5Y cells. Additionally, HOTAIR knockdown suppressed apoptosis via up-regulating miR-221 in MPTP-treated mice. Collectively, HOTAIR contributed to apoptosis in PD by sponging miR-221. This may provide a novel avenue for treatment of PD.Conflicts of interest
The authors have no conflicts of interest to declare.References
- S. Przedborski, Nat. Rev. Neurosci., 2017, 18, 251–259 CrossRef CAS PubMed.
- L. Kalia and A. Lang, Lancet, 2015, 386, 896–912 CrossRef CAS.
- D. Charvin, R. Medori, R. Hauser and O. Rascol, Nat. Rev. Drug Discovery, 2018, 17, 804–822 CrossRef CAS PubMed.
- Z. Mortezaei, H. Lanjanian and A. Masoudi-Nejad, Genomics, 2017, 109, 158–164 CrossRef CAS PubMed.
- Y. Ni, H. Huang, Y. Chen, M. Cao, H. Zhou and Y. Zhang, Cell. Mol. Neurobiol., 2017, 37, 329–338 CrossRef CAS PubMed.
- N. Xie, J. Qi, S. Li, J. Deng, Y. Chen and Y. Lian, J. Cell. Biochem., 2019, 120, 5790–5801 CrossRef CAS PubMed.
- W. Yan, Z. Chen, J. Chen and H. Chen, Biochem. Biophys. Res. Commun., 2018, 496, 1019–1024 CrossRef CAS PubMed.
- Y. Song, R. Wang, L. Li, X. Liu, Y. Wang, Q. Wang and Q. Zhang, Int. J. Oncol., 2019, 54, 77–86 CAS.
- Z. Yan, L. He, J. Qiu, W. Deng, J. Lu, Z. Yuan, D. Liu, R. Zheng and W. Jiang, Eur. Rev. Med. Pharmacol. Sci., 2018, 22, 6640–6649 Search PubMed.
- X. Guo, H. Xiao, S. Guo, J. Li, Y. Wang, J. Chen and G. Lou, J. Cell. Physiol., 2019, 234, 3478–3489 CrossRef CAS PubMed.
- S. Tan, C. Pastori, C. Penas, R. Komotar, M. Ivan, C. Wahlestedt and N. Ayad, Mol. Cancer, 2018, 17, 74 CrossRef PubMed.
- M. Spreafico, B. Grillo, F. Rusconi, E. Battaglioli and M. Venturin, Int. J. Mol. Sci., 2018, 19, E2022 CrossRef PubMed.
- S. Wang, X. Zhang, Y. Guo, H. Rong and T. Liu, Oncotarget, 2017, 8, 24449–24456 Search PubMed.
- X. Lu, Z. Cui, S. Liu and F. Yin, Histol. Histopathol., 2018, 33, 447–453 CAS.
- X. Xie, Y. Huang, L. Chen and J. Wang, Oncol. Lett., 2018, 16, 6697–6704 Search PubMed.
- S. Huang, D. Zhou, Y. Li, Z. Ming, K. Li, G. Wu, C. Chen and Y. Zhao, J. Cell. Physiol., 2018, 234, 3500–3514 CrossRef PubMed.
- C. Lopes, L. Magalhães, C. Teófilo, A. Alves, R. Montenegro, M. Negrini and Â. Ribeiro-Dos-Santos, BMC Cancer, 2018, 18, 721 CrossRef PubMed.
- W. Ma, Y. Li, C. Wang, F. Xu, M. Wang and Y. Liu, Cell Biochem. Funct., 2016, 34, 511–515 CrossRef CAS PubMed.
- S. Oh, H. Park, L. He, C. Skibiel, E. Junn and M. Mouradian, Redox Biol., 2018, 19, 62–73 CrossRef CAS PubMed.
- V. Dung and D. Thao, Adv. Exp. Med. Biol., 2018, 1076, 41–61 CrossRef CAS PubMed.
- T. Jiang and H. Wang, Hum. Cell, 2014, 27, 176–178 CrossRef PubMed.
- K. J. Livak and T. D. Schmittgen, Methods, 2001, 25, 402–408 CrossRef CAS PubMed.
- W. Eum, M. Shin, C. Lee, H. Yeo, E. Yeo, Y. Choi, H. Kwon, D. Kim, O. Kwon, K. Lee, K. Han, J. Park, D. Kim and S. Choi, Biochimie, 2018, 156, 158–168 CrossRef PubMed.
- L. Wang, H. Yang, Q. Wang, Q. Zhang, Z. Wang, Q. Zhang, S. Wu and H. Li, Toxicol. Lett., 2018, 291, 11–28 CrossRef CAS PubMed.
- S. Liu, B. Cui, Z. Dai, P. Shi, Z. Wang and Y. Guo, Curr. Neurovasc. Res., 2016, 13, 115–120 CrossRef CAS PubMed.
- P. Chatterjee, D. Roy, M. Bhattacharyya and S. Bandyopadhyay, BMC Genomics, 2017, 18, 721 CrossRef PubMed.
- J. Hu, Z. Wang, Y. Shan, Y. Pan, J. Ma and L. Jia, Cell Death Dis., 2018, 9, 711 CrossRef PubMed.
- G. Liu and W. Zhang, Braz. J. Med. Biol. Res., 2018, 51, e6896 Search PubMed.
- L. Yang and Z. Lu, Biochem. Biophys. Res. Commun., 2016, 479, 186–191 CrossRef CAS PubMed.
- J. Chan and Y. Tay, Int. J. Mol. Sci., 2018, 19, E1310 CrossRef PubMed.
- X. Hu, D. Ding, J. Zhang and J. Cui, Biosci. Rep., 2019, 39, BSR20181038 CrossRef CAS PubMed.
- B. Wang, X. Qu, J. Liu, J. Lu and Z. Zhou, J. Cell. Physiol., 2019, 234, 6173–6181 CrossRef CAS PubMed.
- Y. J. Sun, J. Li and C. H. Chen, Eur. Rev. Med. Pharmacol. Sci., 2019, 23, 4226–4233 Search PubMed.
- H. Ding, Z. Huang, M. Chen, C. Wang, X. Chen, J. Chen and J. Zhang, Park. Relat. Disord., 2016, 22, 68–73 CrossRef PubMed.
- D. Zhao, S. Deng, Y. Ma, Y. Hao and Z. Jia, NeuroReport, 2018, 29, 655–660 CAS.
- L. Li, J. Xu, M. Wu and J. Hu, Bratisl. Lek. Listy, 2018, 119, 22–27 CAS.
- E. Fernández-Espejo and C. Bis-Humbert, NeuroToxicology, 2018, 67, 178–189 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra06107j |
| ‡ These authors contribute to this work equally as co-first authors. |
| This journal is © The Royal Society of Chemistry 2019 |