
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDroplet-based in situ X-ray absorption spectroscopy cell for studying crystallization processes at the tender X-ray energy range†
Jacinta Xto *ab,
Reto Wettera,
Camelia N. Borcaa,
Christophe Frieha,
Jeroen A. van Bokhovenab and
Thomas Huthwelker*a
*ab,
Reto Wettera,
Camelia N. Borcaa,
Christophe Frieha,
Jeroen A. van Bokhovenab and
Thomas Huthwelker*a
aPaul Scherrer Institut, 5232 Villigen, Switzerland. E-mail: Thomas.huthwelker@psi.ch
bInstitute for Chemical and Bioengineering, ETH Zürich, 8093 Zürich, Switzerland
First published on 24th October 2019
Abstract
The understanding of nucleation and crystallization is fundamental in science and technology. In solution, these processes are complex involving multiple transformations from ions and ion pairs through amorphous intermediates to multiple crystalline phases. X-ray absorption spectroscopy (XAS), which is sensitive to liquid, amorphous and crystalline phases offers prospects of demystifying these processes. However, for low Z elements the use of in situ X-ray absorption spectroscopy requires the tender X-ray range, which is often limited by vacuum requirements thereby complicating these measurements. To overcome these challenges, we developed a versatile and user-friendly droplet-based in situ X-ray absorption spectroscopy cell for studying crystallization processes. Time-resolved in situ experiments under ambient conditions are carried out in the cell whilst the cell is mounted in the vacuum chamber of a tender X-ray beamline. By following changes in the Ca K-edge X-ray absorption near edge structure (XANES), we captured in situ the intermediate phases involved during calcium carbonate crystallization from aqueous solutions. In addition, through linear combination fitting it was possible to qualitatively observe the evolution of each phase during the reaction demonstrating the potential of the cell in studying complex multiphase chemical processes.
1. Introduction
Liquid phase chemical processes are important in a myriad of scientific fields. Key areas of interest include material synthesis,1 photo-chemical reactions2 and catalytic processes.3 Of particular importance are phase transitions involved in the nucleation of crystals from solution. These processes are ubiquitous in nature and central to scientific advancement in the fields of materials, drug and environmental research. A particular example of such a process is the nucleation and crystallization of calcium carbonate. Calcium carbonate, is abundant and one of the most dynamic and fascinating minerals widely utilized by organisms to develop functional materials.4–6 Despite its simple structure consisting primarily of calcium and carbonate ions, the mechanism of its nucleation and crystallization is quite complicated. One of the major challenges faced in elucidating the mechanism is the fact that the nucleation and crystallization process involves multiple phase transformations starting from dissolved ions in aqueous solution, which convert via amorphous intermediates into different crystalline phases.7–10 The lifetimes of such intermediates range from seconds to minutes depending on the thermodynamic conditions.11,12 Understanding these mechanisms is vital for the design and fabrication of calcium carbonate-based biomimetic materials.XAS is a powerful element-specific probe of the chemical environment of the atom of interest.13 As monochromatic X-rays are required, most XAS experiments are performed at large-scale synchrotron facilities. In a typical experiment, monochromatic photons are shot onto the sample, and the absorption coefficient is deduced either by direct measurement of the transmitted X-rays, or from the fluorescent photons. The XAS spectrum is the absorption coefficient as function of the incoming photon energy, at energies above the threshold for creation of photoelectrons. The spectrum consists of two main regions, the XANES which includes the pre-edge, the edge and the near edge regions and the Extended X-ray Absorption Fine Structure (EXAFS).14,15 The XANES region is characterized by both electronic transitions from the core level to higher unoccupied orbitals and by scattering of photoelectrons in the surrounding of the probed atom, providing information on oxidation state and local geometry.16 The EXAFS region, which is dominated by photoelectron scattering from the nearest atoms, gives information on the coordination number, distances and the type of nearest neighbour.
The distinctive strength of XAS is its element specificity and its applicability to any sample environment be it liquid, gas or solid. In contrast to diffraction, XAS probes the short-range order, and is one of the very few techniques that probes both amorphous and crystalline matter with time resolutions ranging from milliseconds to hours. Through the development of in situ cells, XAS has been used to study aqueous chemical reactions,17–19 to demystify electrochemical reactions,13,20,21 to understand catalytic processes22–24 and to study micrometre sized environmental materials in X-ray scanning transmission microscopy.25 The study of low Z elements such as calcium, magnesium, or the L edges of transition metals by XAS, requires the use of tender X-rays (0.5–5 keV). Due to the low transmission of tender X-rays in air such experiments require specialized experimental chambers maintained under vacuum or helium environment. To achieve this, a variety of liquid cells have been designed which often consist of one or two X-ray transparent windows (silicon nitride) in which the sample (thin liquid film) is sandwiched between the windows or flows through them.26–31
While these cells address homogeneous systems, the study of precipitation reactions poses additional challenges; once a solid precipitate forms, clogging of tubing in flow through cells or sedimentation at the bottom of the reactor is inevitable hampering the complete analysis of the reaction post nucleation, simply because the precipitate is not within the probing beam. To overcome these challenges that are not only unique to calcium carbonate but also to a variety of other nucleation processes involving low Z elements, we developed a versatile droplet-based in situ environmental cell to study precipitation reactions under controlled conditions. Through the efficient control of the gas phase composition, the chemical composition and supersaturation within the droplets can be set. The compact and vacuum tight design of the cell makes it possible to maintain ambient environmental conditions in the reactor independent of the vacuum chamber of the synchrotron endstation. The main advantage of the droplet-based cell is that the entire droplet is probed, minimizing pitfalls caused by sedimentation of precipitates out of the probing beam. Herein, we demonstrate the potential of the cell in studying environmentally relevant nucleation processes involving low Z elements by following the crystallization of calcium carbonate in situ.
2. Experimental
2.1. Materials and method
2.2. Design of the in situ cell
With an enclosed sample environment of about 50 mm3, the cell consists of four main pieces (Fig. 1a). Piece (1), made of stainless steel, is the window holder, which hosts the X-ray transparent window and the connection to the gas inlet and outlet tubing. Piece (2), which is also made of stainless steel, hosts the sample holder-piece (3). Piece (4), which is made of copper, is the back plate and is used for vacuum tight sealing of the cell and for mounting the cell onto the sample manipulator in the beamline. The cross-section drawing of the cell in Fig. 1b shows that the window holder (piece 1) has a conically shaped opening to offer a sufficiently large solid angle for the incoming and outgoing beam. At the base of the cone, there is a circular opening where a detachable X-ray transparent window is attached. Here a 500 nm thick silicon nitride window from Silson (wafer size 10 × 10 mm2, window size 5 × 5 mm2 or 6 × 3 mm2) was used.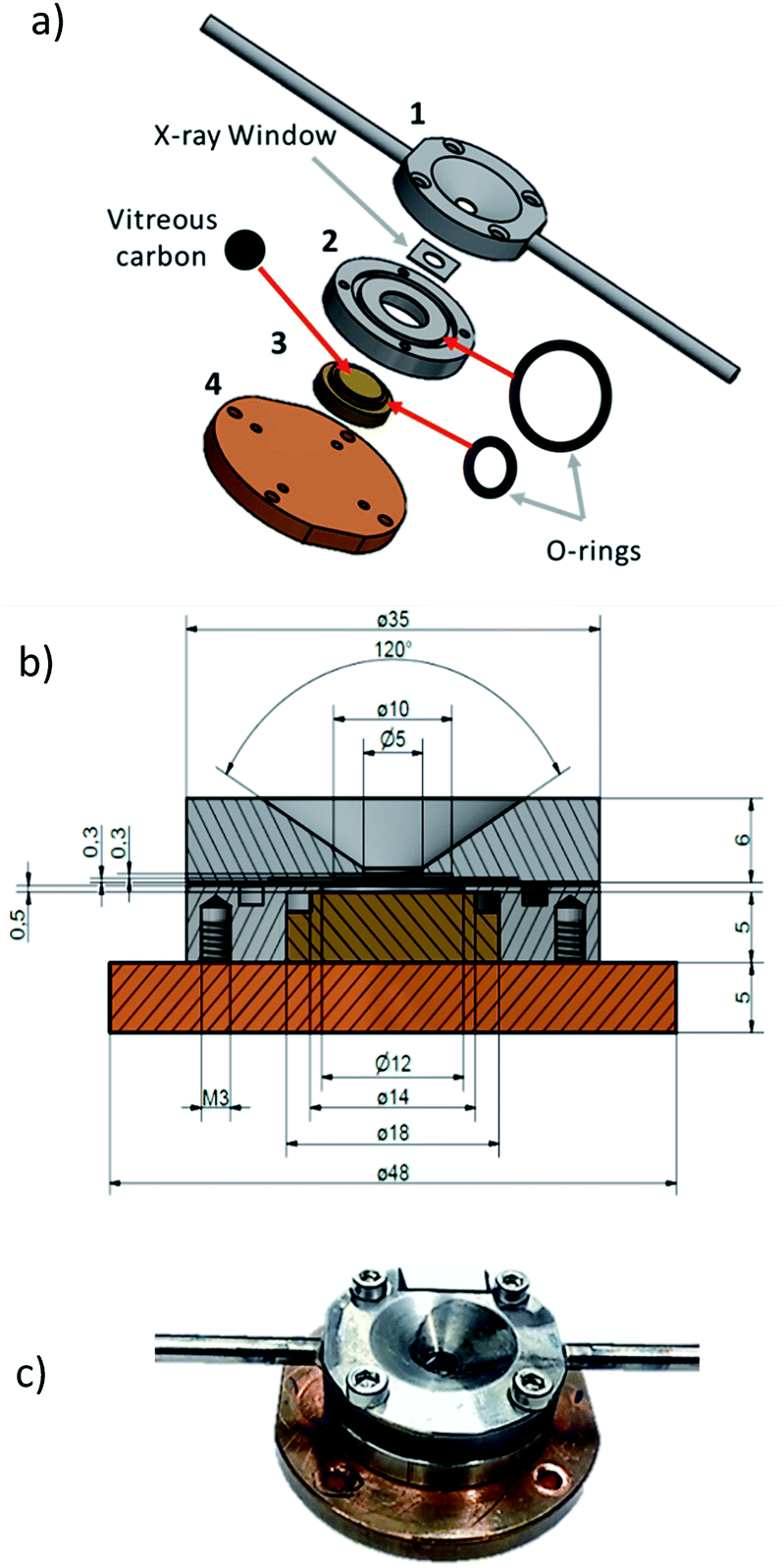 | ||
| Fig. 1 (a) Schematic diagram of the individual cell parts of the in situ cell, (b) cross-section of the cell with dimensions in mm and (c) picture of the cell when closed and ready for mounting in the vacuum chamber. | ||
The window was attached to the base of the cone using ecobond wax. The main advantage of a detachable window is that it can easily be replaced with any other suitable window e.g. a thinner/thicker silicon nitride window, diamond or Kapton window. Ecobond was used as the bonding agent because it is relatively inert and easy to use, however any other adhesive can be used depending on the specific requirements. On both sides of the window holder, gas inlet and outlet channels are drilled and at the exit of each channel, a steel tube (3 mm outer diameter) is soldered to connect the cell to standard tube fittings. Piece (2) houses the sample holder (3) and connects the window holder (1) with the base plate (4). It has an O-ring groove scooped on the surface to ensure vacuum tight fastening with the window holder. The sample holder (3), is tightly inserted into piece (2) and can be easily replaced or adjusted depending on the needs of the experiment. It is made of copper to ensure good heat contact to the backplate (4). At the top of the sample holder, there is a groove for fitting an O-ring, which seals the interface between sample holder and piece (2) vacuum tight. The sample holder is made of copper for good thermal contact to the backplate to allow sample heating or cooling via the back plate. The sample holder has a shallow trench where a removable vitreous carbon support is mounted. Vitreous carbon is preferred because it is chemically inert. Secondly, its electric conductivity prevents ejection of nano particulate matter upon possible charging by the intense synchrotron X-rays. Thirdly, a vitreous carbon sample holder is advantageous for measurements of X-ray fluorescence, as there is only infinitesimal fluorescence background for energies sufficiently above the carbon edge. The scheme in Fig. 1b shows the dimensions of the cell whereas the picture in Fig. 1c shows a picture of the compact cell when completely assembled and ready for mounting in the vacuum chamber.
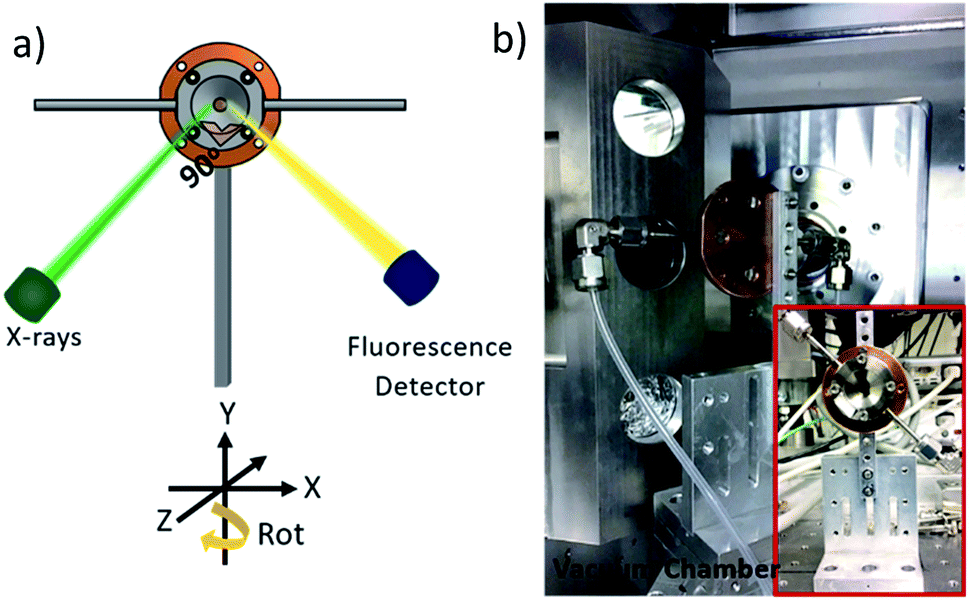 | ||
| Fig. 2 (a) Schematic representation of the sample mounting geometry with respect to the incoming X-rays and the fluorescence detector and (b) picture of the in situ cell mounted in the vacuum chamber of the PHOENIX beamline at the Swiss Light Source (inset front view of the cell before mounting). | ||
The gas supply is attached by standard fittings to 3 mm steel tubes, which are welded to the window holder (1) of the cell. For connection to the gas flow system PFA tubes are used, which are guided via vacuum flanges to the gas flow system, located outside the vacuum chamber.
2.3. Gas flow system
Fig. 3 shows a schematic of the gas flow system, which consists of a humidifying system, connected to mass flow controllers, which regulate the gas flow rates and tune the humidity through the mixing flow of dry and humid gases. Humidity and temperature sensors are positioned at the inlet and outlet of the in situ cell to ascertain the relative humidity in the cell. A pressure sensor is also attached to the flow system. Using a computer-controlled regulation valve (Pfeiffer EVR 116), the pressure in the gas flow system can be set to pressures between 1 bar and ∼30 mbar, just above the vapour pressure of water in the humidifier. Running the system at lower pressures has the advantage of minimizing the pressure difference on the silicon nitride window, and allows doing experiments with thinner windows, which are not stable enough to stand 1 bar pressure difference. The pressure measurement also makes it possible to determine whether the cell is vacuum tight. A gas drying/scrubbing system and a membrane pump coupled to a pressure head are also included in the flow system. All the input and output signals of the flow system are calibrated to a 0–10 V signal making it possible to remotely control and monitor the entire flow system using LabVIEW and EPICS control system enabling automated time-resolved experiments which can last several days.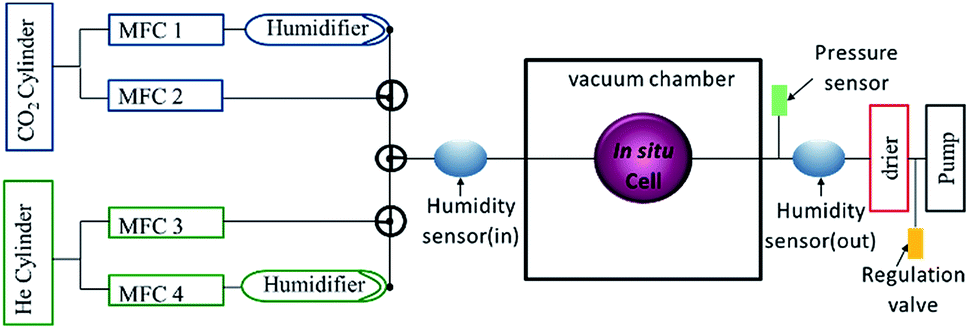 | ||
| Fig. 3 Schematic representation of the gas flow system consisting of four mass flow controllers (MFC). | ||
3. Results and discussion
3.1. In situ study of calcium carbonate crystallization from solution using the droplet-based in situ cell
To test the capability of the cell, freshly prepared calcium carbonate droplets were mounted into the cell using a micropipette. Approximately 0.1 μl sized droplets were deposited on the vitreous carbon sample support and loaded within 20 s in the in situ cell. Calcium K-edge X-ray absorption near edge spectroscopy (XANES) was performed on the droplets to elucidate the structural changes occurring in the droplet solution, while changing the gas phase composition in the cell. An X-ray beam size of 0.6 × 0.6 mm2, slightly larger than the size of one droplet was used for the experiments to ensure that the entire droplet was measured.To monitor and quantify the phase evolution in the droplets, calcium carbonate reference spectra of calcite (Sigma Aldrich), aragonite (synthesized), vaterite (synthesized), ACC (synthesized) and 10 mM calcium chloride solution were measured. The solid reference samples were first characterized by X-ray powder diffraction (Fig. S1a†). For XANES measurements, the powder reference samples were pressed on a conducting carbon tape, which was attached to an electrically insulated copper plate to measure the total electron yield (TEY) signal. The liquid sample was measured in a flow through liquid cell. For the solid samples, measurements were done in both TEY and fluorescence modes whereas the liquid reference sample was only measured in fluorescence mode. Fig. 4a shows that the calcium K-edge XANES spectrum of the solvated calcium reference (10 mM calcium chloride) has a single intense pre-peak (1s → 3d) centred at ∼4039.0 eV and an intense Ca–O scattering white line peak centred at 4050.0 eV. The amorphous calcium carbonate reference is very similar to it, except that the white line peak is slightly broader and shifted by about 1 eV to 4049.0 eV. The calcite and vaterite XANES reference spectra are distinctively different from each other and from the amorphous calcium carbonate spectrum: The vaterite spectrum has an intense single pre-peak centred at 4039.0 eV (1s → 3d), a shoulder peak at 4045.0 eV (1s → 4p), a doubly split white line with the first peak centred at 4048.0 eV and the 2nd peak at 4050.0 eV and a further post edge feature centred at 4055.0 eV. In contrast, calcite, which is centrosymmetric, displays a weak doubly split pre-peak (Fig. S1b†) centred at 4039.0 eV (1s → 3d), a shoulder peak at 4045.0 eV (1s → 4p), a doubly split white line with the first peak centred at 4047.4 eV and the 2nd peak at 4049.3 eV and a further post edge feature centred at 4060.0 eV.
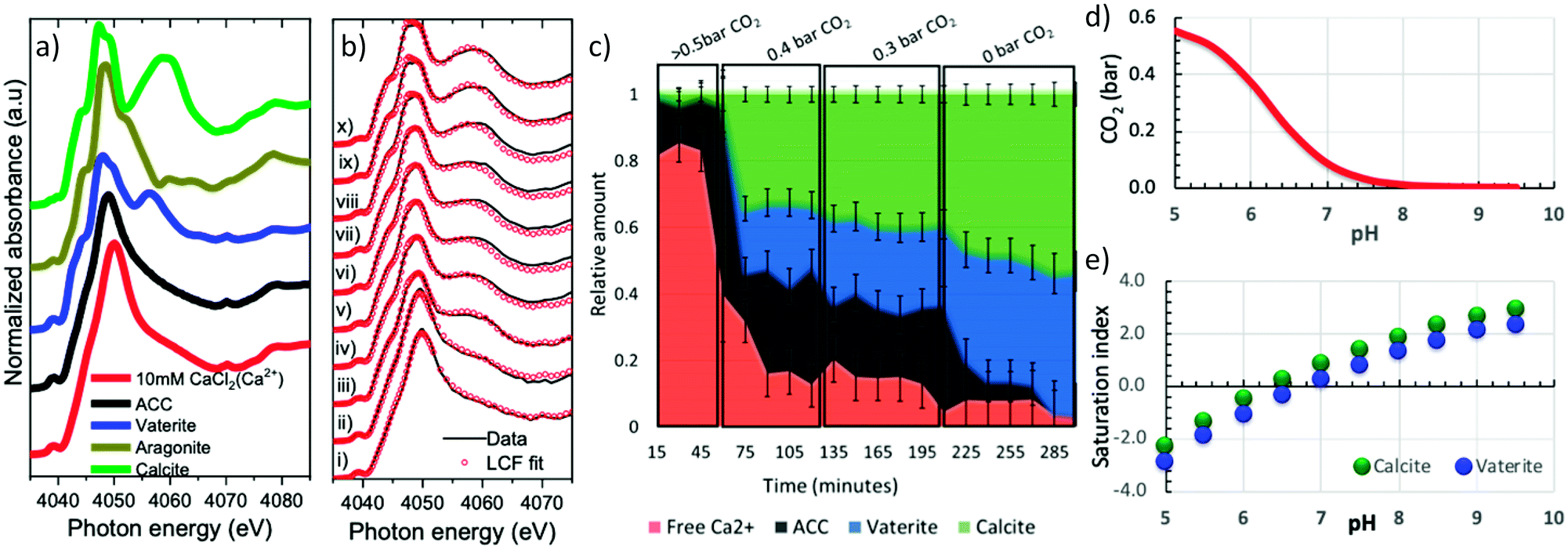 | ||
| Fig. 4 (a) Ca K-edge XANES of solvated calcium (Ca2+) (10 mM CaCl2), ACCs, vaterite, aragonite and calcite reference spectra, (b) Ca K-edge XANES spectra of the droplet during the reaction at very high relative humidity 90 ± 5% at (i) >0.5 bar (ii–iv) 0.4 bar, (v–vii) 0.3 bar and (viii–x) 0 bar carbon dioxide partial pressure, (c) ratio of each species in the cell as determined by linear combination fitting of the measured spectra with reference spectra, (d) Visual Minteq calculation of the pH of a 10 mM calcium chloride solution as a function of carbon dioxide partial pressure and (e) Visual Minteq simulation of the calcite and vaterite saturation index as function of pH. | ||
Fig. 4b shows a series of calcium K-edge XANES spectra (black lines) of the droplet measured whilst changing the carbon dioxide partial pressure in the cell at a constant relative humidity of 90 ± 5%. The fraction of ACC, vaterite, and calcite in the sample was determined based on Linear Combination Fitting (LCF) of the reference spectra (Fig. 4a) to each of the measured spectra (red dots in Fig. 4b) using Athena software.34 The plot in Fig. 4c shows the determined fraction of each component in the droplet as function of time and carbon dioxide partial pressure. At an initial carbon dioxide partial pressure of 0.5 bar, the XANES spectrum in Fig. 4b(i) resembles the reference spectrum of solvated Ca2+ in Fig. 4a. It displays a pre-peak feature at 4039.0 eV and single white line at 4049.9 eV. The LCF results in Fig. 4c show that ∼82% of the calcium is solvated, while ∼18% is bound in ACC.
Decreasing the carbon dioxide partial pressure results in an immediate shift of white line position to 4049.4 eV in Fig. 4b(ii). A corresponding sharp decrease in solvated calcium to 40% and an increase in the amorphous calcium carbonate to 58% is observed in Fig. 4c. New features associated with crystalline calcium carbonate begin to emerge in the XANES spectra with time in Fig. 4b(ii–iv): A shoulder peak appears at 4045.0 eV, the white line position shifts to lower energy and a post edge peak appears at ∼4060.0 eV. A further decrease in the carbon dioxide partial pressure drives the system towards crystallization in which the fraction of vaterite and calcite increases whereas the fraction of solvated calcium and ACC decreases. When all the humid carbon dioxide is fully replaced with humid nitrogen, all the ACC is consumed (Fig. 4c) and only about 3% solvated calcium ions are present with everything else is crystalline calcium carbonate in the form of vaterite 44% or calcite 53%.
The observed formation of ACC, carbonate and vaterite as intermediate phases during calcium carbonate precipitation from a saturated solution is consistent with findings reported in literature, which are performed using other analytical techniques such as time-resolved small and wide angle scattering,12,35,36 electron microscopy37 and isotope tracing.38 In this work, aragonite formation was not detected at any stage of the crystallization process because the experiments were done in big droplets, at room temperature and without any aragonite-promoting additive such as magnesium ions. This is consistent with the literature, where at room temperature aragonite formation has been observed to occur either in confinement ∼25 nm pores39,40 or in solutions containing inorganic additives such as magnesium ions41–48 or organic additives/solvents.49–51
Carbon dioxide has a strong influence on the pH of aqueous solutions because it readily dissolves in water to form carbonic acid, which dissociates to bicarbonate and carbonate ions respectively (eqn (1)).
| CO2(g) + H2O(l) ⇆ H2CO3(aq) ⇆ HCO3(aq)− + H+ ⇆ CO3(aq)2− + H+ | (1) |
From a thermodynamic point of view, setting the gas phase composition explicitly sets the free energy of water and carbon dioxide in the equilibrated solution. This is because the chemical potential μi of species i (with i = water or carbon dioxide) relates to the partial pressure pi by the simple relation μi = kT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(pi/pi0), where pi0 is the partial pressure at an appropriately chosen reference state. The pH speciation in a 10 mM calcium carbonate solution as a function of carbon dioxide partial pressure calculated using visual minteq52 in Fig. 4d shows that a decrease in carbon dioxide partial pressure increases the pH. Fig. 4e further shows that the consequence of increased pH is that the saturation index of both calcite and vaterite increases. (The saturation index is the term S = logIAP − logKs where IAP is the ion activity product and Ks is the equilibrium solubility constant.) A negative value of S implies undersaturation (i.e. IAP is lower than the solubility constant) and a positive value implies supersaturation and precipitates can form. Above pH 6, Fig. 4e shows that the solution is supersaturated with respect to vaterite and calcite and therefore crystallization occurs.
ln(pi/pi0), where pi0 is the partial pressure at an appropriately chosen reference state. The pH speciation in a 10 mM calcium carbonate solution as a function of carbon dioxide partial pressure calculated using visual minteq52 in Fig. 4d shows that a decrease in carbon dioxide partial pressure increases the pH. Fig. 4e further shows that the consequence of increased pH is that the saturation index of both calcite and vaterite increases. (The saturation index is the term S = logIAP − logKs where IAP is the ion activity product and Ks is the equilibrium solubility constant.) A negative value of S implies undersaturation (i.e. IAP is lower than the solubility constant) and a positive value implies supersaturation and precipitates can form. Above pH 6, Fig. 4e shows that the solution is supersaturated with respect to vaterite and calcite and therefore crystallization occurs.
The presence of ACC despite the high carbon dioxide partial pressure of 0.5 bar (Fig. 4c) in the cell is likely due to undesired precipitation when loading the droplets into the in situ cell. During loading, the carbon dioxide saturated solution gets in contact with air and carbon dioxide can exsolve from the droplet, causing supersaturation and precipitation on the droplets surface. We also note that the observed start of further crystallization at carbon dioxide pressure above 0.3 bar is not fully consistent with the thermodynamic predictions as shown in Fig. 4e.
This deviation between the experimental observation and thermodynamic calculations may be due to, uncertainties in the exact droplet concentrations due to handling or beam induced evaporation, uncertainties in the in carbon dioxide partial pressure and temperature of the sample. However, the observed trends are in qualitative agreement with the thermodynamic predictions of precipitation of calcium carbonates with decreasing carbon dioxide partial pressure.
Dark field microscopy images in Fig. 5 of a saturated calcium carbonate droplet solution imaged inside the in situ cell using a Leica DM 4000M research microscope summarizes the reaction in the cell. Fig. 5a shows a freshly loaded droplet, maintained at 90 ± 5% relative humidity and 0.5 bar carbon dioxide partial pressure, which does not have any evident precipitates. Changing the gas phase environment of the droplet by decreasing the carbon dioxide partial pressure as illustrated in Fig. 5b increases the pH and consequently the supersaturation in the droplet inducing precipitation in the droplet as shown in Fig. 5c (some precipitated regions are circled in red).
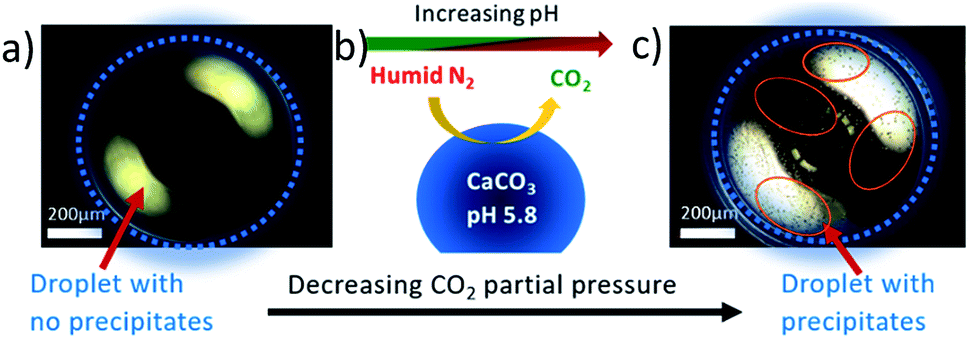 | ||
| Fig. 5 (a) Dark field microscopy image of a calcium carbonate rich droplet in the in situ cell under 0.5 bar carbon dioxide partial pressure and high relative humidity 90 ± 5%, (b) a schematic representation of the reaction in the cell and (c) dark field microscope image of calcium carbonate rich droplet while decreasing the carbon dioxide partial pressure to 0 bar shows (precipitates can be seen as dark spots in the droplet). | ||
3.2. Capabilities and limitations of the droplet-based in situ cell
The key advantage of the droplet-based cell is its capability of handling multiphase systems (liquid, amorphous and crystalline) within a confined static volume of a droplet. In standard liquid cells, this is often challenging due to sedimentation of the precipitates at the onset of nucleation resulting in fluctuations in sample amount in the probed region, making it challenging to follow multistep reactions. Flow through cells on the other hand have the drawback of clogging of tubes due to precipitation on the walls and fluctuations in sample volume arising from the pressure induced deformation of the thin windows used to enclose the solution in a vacuum tight cell.53 Liquid-jet configurations are attractive alternatives that do not suffer from drawbacks associated with windows; however, they require large sample volumes and are susceptible to clogging due to precipitation in the feeding tubes.34,54Compared to other droplet-based methods, the main advantage of this cell is that it is simple to use and can be implemented for standard XAS measurements. While aerodynamic levitation offers a windowless and substrate-free technique of elucidating crystallization processes, it is a highly specialized technique, which requires immense effort to generate and stabilize the droplet hence it is still not feasible as a standard XAS technique.55,56 The sessile droplet on the other hand is also challenging to stabilize in vacuum and difficult to set the thermodynamic conditions in the droplet because it is in contact with a solution in the syringe.54
The cell is limited to reactions that are controllable via the gas phase because the mixing of different liquids in situ is not yet feasible. Continuous refreshing of sample is also not possible, hence beam induced damage on the sample can occur. Multiple droplets can however be loaded and measured independently to overcome this challenge. Due to the small sample volume, it is also challenging to incorporate pH and temperature sensors in the sample for accurate determination of the sample environment during the reaction. In addition, the cell requires an X-ray transparent window to maintain the reaction environment when mounted in the vacuum chamber of the synchrotron beamline.
4. Conclusions
We introduce a new in situ cell for X-ray absorption spectroscopy at the tender X-ray range (0.3–8 keV). With a compact and vacuum tight design, the cell can be used to elucidate time-resolved in situ reactions involving aerosols and/or liquid droplets using X-ray absorption spectroscopy. The time-resolved observation and quantification of intermediate phases involved during calcium carbonate formation from solution demonstrates the potential of the cell in understanding mineralization processes. In addition, the cell has also been used to study the nucleation/precipitation kinetics of baryte for application to radioactive waste disposal57 further demonstrating its potential in studying environmentally relevant processes. This new inexpensive and simple to use in situ cell presents new opportunities for exploiting tender X-rays for elucidating multiphase chemical processes encountered in environmental chemistry, biomineralization, geochemistry and material research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the Swiss National Science Foundation (SNSF) for funding the study under project number 157148. The authors would also like to thank the Swiss Light Source at Paul Scherrer Institut, Villigen PSI, Switzerland for the award of beam time at the PHOENIX beamline.Notes and references
- Y. Marcus and G. Hefter, Chem. Rev., 2008, 106, 4585–4621 CrossRef.
- Y. Ogi, Y. Obara, T. Katayama, Y. I. Suzuki, S. Y. Liu, N. C. M. Bartlett, N. Kurahashi, S. Karashima, T. Togashi, Y. Inubushi, K. Ogawa, S. Owada, M. Rubešová, M. Yabashi, K. Misawa, P. Slavíček and T. Suzuki, Struct. Dyn., 2015, 2, 1–19 CrossRef.
- H. Shi, J. A. Lercher and X. Y. Yu, Catal. Sci. Technol., 2015, 5, 3035–3060 RSC.
- Y. Ma, B. Aichmayer, O. Paris, P. Fratzl, A. Meibom, R. A. Metzler, Y. Politi, L. Addadi, P. U. P. A. Gilbert and S. Weiner, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 6048–6053 CrossRef CAS PubMed.
- I. Polishchuk, A. A. Bracha, L. Bloch, D. Levy, S. Kozachkevich, Y. Etinger-Geller, Y. Kauffmann, M. Burghammer, C. Giacobbe, J. Villanova, G. Hendler, C.-Y. Sun, A. J. Giuffre, M. A. Marcus, L. Kundanati, P. Zaslansky, N. M. Pugno, P. U. P. A. Gilbert, A. Katsman and B. Pokroy, Science, 2017, 358, 1294–1298 CrossRef CAS.
- D. M. Duffy, Science, 2017, 358, 1254–1255 CrossRef CAS.
- M. Albéric, L. Bertinetti, Z. Zou, P. Fratzl, W. Habraken and Y. Politi, Adv. Sci., 2018, 5, 1701000 CrossRef.
- D. Gebauer, P. N. Gunawidjaja, J. Y. P. Ko, Z. Bacsik, B. Aziz, L. Liu, Y. Hu, L. Bergström, C. W. Tai, T. K. Sham, M. Edén and N. Hedin, Angew. Chem., Int. Ed., 2010, 49, 8889–8891 CrossRef CAS.
- F. Konrad, F. Gallien, D. E. Gerard and M. Dietzel, Cryst. Growth Des., 2016, 16, 6310–6317 CrossRef CAS.
- C. C. Tester, C.-H. Wu, S. Weigand and D. Joester, Faraday Discuss., 2012, 159, 345 RSC.
- A. V Radha, T. Z. Forbes, C. E. Killian, P. U. P. A. Gilbert and A. Navrotsky, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 16438–16443 CrossRef.
- J. D. Rodriguez-Blanco, K. K. Sand and L. G. Benning, in New Perspectives on Mineral Nucleation and Growth, ed. A. E. S. Van Driessche, M. Kellermier, L. G. Benning and D. Gebauer, Springer, 2017, pp. 95–110 Search PubMed.
- J. A. van Bokhoven and C. Lamberti, Emission Spectroscopy X-Ray Absorption and X-Ray Theory and Applications, John Wiley & Sons, Ltd Registered, 1st edn, 2016 Search PubMed.
- M. Newville, Rev. Mineral. Geochem., 2014, 78, 33–74 CrossRef CAS.
- J. E. Penner-Hahn, X-Ray Absorption Spectroscopy, Elsevier Inc., 2nd edn, 2003, vol. 2 Search PubMed.
- G. S. Henderson, F. M. F. De Groot and B. J. A. Moulton, Rev. Mineral. Geochem., 2014, 78, 75–138 CrossRef CAS.
- J. E. Villinski, P. A. O'Day, T. L. Corley and M. H. Conklin, Environ. Sci. Technol., 2001, 35, 1157–1163 CrossRef CAS.
- J. Boita, M. Do Carmo Martins Alves and J. Morais, J. Synchrotron Radiat., 2014, 21, 254–258 CrossRef CAS.
- P. Ferrer, I. da Silva, J. Rubio-Zuazo, B. F. Alfonso, C. Trobajo, S. Khainakov, J. R. Garcia, S. Garcia-Granda and G. R. Castro, J. Synchrotron Radiat., 2012, 19, 93–100 CrossRef CAS.
- J. Tillier, T. Binninger, M. Garganourakis, A. Patru, E. Fabbri, T. J. Schmidt and O. Sereda, J. Electrochem. Soc., 2016, 163, H913–H920 CrossRef CAS.
- B. Lassalle-Kaiser, A. Zitolo, E. Fonda, M. Robert and E. Anxolabéhère-Mallart, ACS Energy Lett., 2017, 2, 2545–2551 CrossRef CAS.
- P. T. Kristiansen, T. C. R. Rocha, A. Knop-Gericke, J. H. Guo and L. C. Duda, Rev. Sci. Instrum., 2013, 84, 113107 CrossRef CAS.
- J. A. Van Bokhoven, A. M. J. Van Der Eerden, A. D. Smith and D. C. Koningsberger, J. Synchrotron Radiat., 1999, 6, 201–203 CrossRef CAS.
- R. Kopelent, J. A. Van Bokhoven, J. Szlachetko, J. Edebeli, C. Paun, M. Nachtegaal and O. V. Safonova, Angew. Chem., Int. Ed., 2015, 54, 8728–8731 CrossRef CAS.
- T. Huthwelker, V. Zelenay, M. Birrer, A. Krepelova, J. Raabe, G. Tzvetkov, M. G. C. Vernooij, M. Ammann, T. Huthwelker, V. Zelenay, M. Birrer, A. Krepelova, J. Raabe and G. Tzvetkov, Rev. Sci. Instrum., 2010, 81, 1–9 CrossRef.
- S. Schreck, G. Gavrila, C. Weniger and P. Wernet, Rev. Sci. Instrum., 2011, 82, 103101 CrossRef.
- C. Schwanke, L. Xi and K. M. Lange, J. Synchrotron Radiat., 2016, 23, 1390–1394 CrossRef CAS.
- M. Nagasaka, T. Hatsui, T. Horigome, Y. Hamamura and N. Kosugi, J. Electron Spectrosc. Relat. Phenom., 2010, 177, 130–134 CrossRef CAS.
- R. Qiao, Y. Xia, X. Feng, J. MacDougall, J. Pepper, K. Armitage, J. Borsos, K. G. Knauss, N. Lee, A. Allézy, B. Gilbert, A. A. MacDowell, Y.-S. Liu, P.-A. Glans, X. Sun, W. Chao and J. Guo, Rev. Sci. Instrum., 2018, 89, 013114 CrossRef.
- N. Huse, H. Wen, D. Nordlund, E. Szilagyi, D. Daranciang, T. A. Miller, A. Nilsson, R. W. Schoenlein and A. M. Lindenberg, Phys. Chem. Chem. Phys., 2009, 11, 3951–3957 RSC.
- P. Jiang, J. L. Chen, F. Borondics, P. A. Glans, M. W. West, C. L. Chang, M. Salmeron and J. Guo, Electrochem. Commun., 2010, 12, 820–822 CrossRef CAS.
- J. M. Xto, C. N. Borca, J. A. van Bokhoven and T. Huthwelker, Chem. Commun. 10.1039/C9CC03749G.
- L. Wang, I. Sondi and E. Matijević, J. Colloid Interface Sci., 1999, 218, 545–553 CrossRef CAS.
- J. W. Smith and R. J. Saykally, Chem. Rev., 2017, 117, 13909–13934 CrossRef CAS.
- J. D. Rodriguez-Blanco, S. Shaw and L. G. Benning, Nanoscale, 2011, 3, 265–271 RSC.
- D. Pontoni, J. Bolze, N. Dingenouts, T. Narayanan, M. Ballauff and V. Uni, J. Phys. Chem., 2003, 5123–5125 CrossRef CAS.
- E. M. Pouget, P. H. H. Bomans, J. A. C. M. Goos, P. M. Frederik, G. de With and N. A. J. M. Sommerdijk, Science, 2009, 323, 1455–1458 CrossRef CAS.
- A. J. Giuffre, A. C. Gagnon, J. J. De Yoreo and P. M. Dove, Geochim. Cosmochim. Acta, 2015, 165, 407–417 CrossRef CAS.
- Y. Xu and N. A. J. M. Sommerdijk, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 8469–8471 CrossRef CAS.
- M. Zeng, Y.-Y. Kim, C. Anduix-Canto, C. Frontera, D. Laundy, N. Kapur, H. K. Christenson and F. C. Meldrum, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 7670–7675 CrossRef CAS.
- Z. Zhang, Y. Xie, X. Xu, H. Pan and R. Tang, J. Cryst. Growth, 2012, 343, 62–67 CrossRef CAS.
- H. Pan and R. Tang, J. Cryst. Growth, 2012, 343, 62–67 CrossRef.
- H. F. Greer, W. Zhou and L. Guo, Mineral. Petrol., 2015, 109, 453–462 CrossRef CAS.
- W. Donald, J. Geophys. Res., 1962, 67, 4873–4874 CrossRef.
- L. Fernandez-Diaz, A. Putnis, M. Prieto and C. V Putnis, J. Sediment. Res., 1996, 66, 482–491 CAS.
- W. Sun, S. Jayaraman, W. Chen, K. A. Persson and G. Ceder, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 3199–3204 CrossRef CAS.
- F. Konrad, B. Purgstaller, F. Gallien, V. Mavromatis, P. Gane and M. Dietzel, J. Cryst. Growth, 2018, 498, 381–390 CrossRef CAS.
- C. R. Blue, A. Giuffre, S. Mergelsberg, N. Han, J. J. De Yoreo and P. M. Dove, Geochim. Cosmochim. Acta, 2017, 196, 179–196 CrossRef CAS.
- J. M. Walker, B. Marzec and F. Nudelman, Angew. Chem., Int. Ed., 2017, 56, 11740–11743 CrossRef CAS PubMed.
- L. Liu, J. Jiang and S. Yu, Cryst. Growth Des., 2014, 14, 6048–6056 CrossRef CAS.
- J. Su, F. Zhu, G. Zhang, H. Wang, L. Xie and R. Zhang, CrystEngComm, 2016, 18, 2125–2134 RSC.
- J. D. Allison, D. S. Brown and K. J. Novo-Gradac, Minteqa2/Prodefa2, a Geochemical Assessment Model for Environmental Systems: Version 3.0 User'S Manual, 1991 Search PubMed.
- J. E. Villinski, P. A. O'Day, T. L. Corley and M. H. Conklin, U.S. Geol. Surv. Toxic Subst. Hydrol. Program, Proc. Tech. Meet., Charleston, SC, 7–12 March 1999, Water Reso., pp. 217–225 Search PubMed.
- S.-Y. Chang, T. A. Kathyola, E. A. Willneff, C. J. Willis, P. Wilson, P. J. Dowding, G. Cibin, A. B. Kroner, E. J. Shotton and S. L. M. Schroeder, React. Chem. Eng., 2019, 4, 679–687 RSC.
- A. Watanabe, K. Hasegawa and Y. Abe, Sci. Rep., 2018, 8, 1–8 CrossRef CAS PubMed.
- C. J. Benmore and J. K. R. Weber, Adv. Phys.: X, 2017, 2, 717–736 CAS.
- E. Curti, J. Xto, C. N. Borca, K. Henzler, T. Huthwelker and N. I. Prasianakis, Eur. J. Mineral., 2019, 247–262 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details. See DOI: 10.1039/c9ra06084g |
| This journal is © The Royal Society of Chemistry 2019 |