
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Simple synthesis of new imidazopyridinone, pyridopyrimidinone, and thiazolopyridinone derivatives and optimization of reaction parameters using response surface methodology†
Fahimeh Sadat
Hosseini
 and
Mohammad
Bayat
*
and
Mohammad
Bayat
*
Department of Chemistry, Faculty of Science, Imam Khomeini International University, Qazvin, Iran. E-mail: bayat_mo@yahoo.com; m.bayat@sci.ikiu.ac.ir; Tel: +98 28 33780040
First published on 25th September 2019
Abstract
The reaction between ketene animal/ketene N,S-acetals (derived from diamines/cysteamine hydrochloride and 1,1-bis(methylthio)-2-nitroethene) with aromatic aldehydes, and Meldrum's acid led to the title compounds. The reaction conditions were optimized using response surface methodology (RSM). The two independent variables (temperature and water content of aqueous ethanol), and the responses (yield of product and reaction time) were studied. The range of each parameter selected was: T = 25–100 °C and water = 0–100%. The optimal values were: T = 72 °C and water = 33%. This work offers significant advantages including use of a green solvent, experimental simplicity, absence of catalyst, a simple work-up and purification process, moderate to good yields, and preparation of potentially bioactive compounds.
Introduction
Response surface methodology (RSM) is the most popular optimization method used in recent years. RSM is a powerful mathematical and statistical technique which has been widely used for designing experiments and empirical building models with minimal experimental number. This method is exclusively used to examine the surface, or the relationship between the response and the factors affecting the response. The application of RSM to design optimization is aimed at reducing the cost of expensive analysis methods and their associated numerical noise.1–6Nitrogen and sulfur containing heterocyclic compounds broadly exist in various natural and synthetic products, and play vital roles in medicinal chemistry.7–10 Among these heterocycles, pyridine derivatives, such as imidazopyridine, pyridopyrimidine, and thiazolopyridine derivatives have received considerable attention over the past years due to their diverse biological activities and clinical applications.11,12 Imidazopyridine derivatives have shown a broad range of interesting biological activities, such as antifungal, antitumor, antiviral, antibacterial, anti-HIV,13 antipyretic, analgesic, hypnoselective, anxioselective, anti-inflammatory, anticonvulsant, antiulcer, immunomodulatory14 activities; also pyridopyrimidine derivatives15 are found to exhibit a broad spectrum of potent antibacterial, antiallergic, antimicrobial,12 anti-inflammatory, analgesic,16,17 tyrosine kinase, calcium channel antagonists,8,18 tuberculostatic, antileishmanial, antitumor,19 antifolate, antihypertensive, hepatoprotective,20 anticancer21 properties; and thiazolopyridine derivatives are associated with a wide range of biological activities such as analgesic, antioxidant,11 anticancer,22 anti-inflammatory,23 antihypertensive,24 antibacterial, and antifungal25,26 properties.
The numerous attractive features of enamines or dienamines derived from the 1,1-bis(methylthio)-2-nitroethene have made them important intermediates for the construction of a wide variety of S- and N-heterocyclic systems.27–33 Hence during recent years, they have been used for the synthesis of various fused heterocycles and drug-like compounds. For examples in 2013 Bazgir et al. described a one-pot, four-component synthesis of a novel class of functionalized imidazo[1,2-a]pyridine derivatives starting from readily available inputs including diverse diamines, 1,1-bis(methylthio)-2-nitroethene, aldehydes and activated methylene compounds in EtOH under reflux conditions (Scheme 1, Entry a).34 In 2014 Yildirim et al. reported a base-catalyzed one-pot cyclocondensation reaction of acryloyl and cinnamoyl chlorides with β-nitroenamine derivatives under mild basic conditions, which affords target thiazolopyridinone or imidazopyridinone derivatives (Scheme 1, Entry b).35 In 2017 and 2018 we developed an efficient, one-pot, multi-component synthesis of imidazopyridine, pyridopyrimidine, thiazolopyridine, and oxazolopyridine fused heterocyclic systems via reaction of various diamines, cysteamine hydrochloride, ethanolamine, 1,1-bis(methylthio)-2-nitroethene, cyanoacetohydrazide and aromatic aldehydes in ethanol at reflux (Scheme 1, Entry c).36,37 Herein we report synthesis of a new class of imidazopyridinone, pyridopyrimidinone, and thiazolopyridinone derivatives through a simple one-pot, multi-component reaction of diverse diamines or cysteamine hydrochloride, 1,1-bis(methylthio)-2-nitroethene, aromatic aldehydes, and 2,2-dimethyl-1,3-dioxane-4,6-dione (Meldrum's acid) in ethanol/water (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at 72 °C (Scheme 1, Entry d). RSM in conjunction with a central composite design was used for modeling and optimizing the synthetic process.
:1) at 72 °C (Scheme 1, Entry d). RSM in conjunction with a central composite design was used for modeling and optimizing the synthetic process.
 | ||
| Scheme 1 Use of enamines derived from 1,1-bis(methylthio)-2-nitroethene in organic synthesis. | ||
Result and discussion
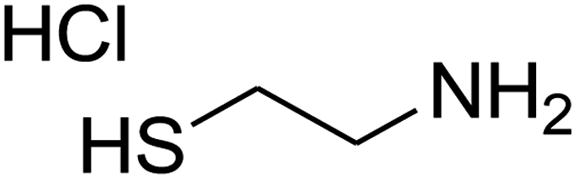
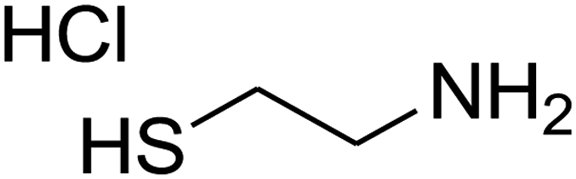
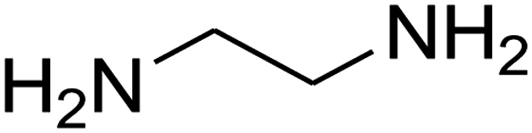
The one-pot, multi-component reaction of various diamines or cysteamine hydrochloride 1 with 1,1-bis(methylthio)-2-nitroethene 2, aromatic aldehydes 3, and 2,2-dimethyl-1,3-dioxane-4,6-dione (Meldrum's acid) 4, in EtOH/H2O (2:1) at 72 °C, gave imidazopyridinone, pyridopyrimidinone, and thiazolopyridinone derivatives 5 in moderate to good yields (Scheme 2).
 | ||
| Scheme 2 Synthetic scheme for the products 5a–o. | ||
A reaction condition is optimized using response surface methodology (RSM). A central composite design with five replicates at the center point requiring 13 treatments was used. The two independent variables (temperature (A) and water content of aqueous ethanol (B)), levels (Table 1), and the results obtained after running the experiments are represented in Table 2. The following responses were analyzed: yield of product (R1) and reaction time (R2).
| Variables | Code | Units | Levels | ||
|---|---|---|---|---|---|
| −1 | 0 | +1 | |||
| Reaction temperature | A | °C | 25 | 62.5 | 100 |
| Water content of aqueous ethanol | B | % | 0 | 50 | 100 |
| Run | A (°C) | B (%) | R 1 (%) | R 2 (min) |
|---|---|---|---|---|
| a A = temperature (°C), B = water content of aqueous ethanol (%), R1 = yield of reaction, R2 = reaction time. | ||||
| 1 | 62.50 | 50.00 | 80 | 7 |
| 2 | 35.98 | 85.36 | 50 | 5 |
| 3 | 89.02 | 14.64 | 77 | 5 |
| 4 | 62.50 | 50.00 | 83 | 6 |
| 5 | 25.00 | 50.00 | 45 | 11 |
| 6 | 62.50 | 50.00 | 84 | 5 |
| 7 | 100.00 | 50.00 | 77 | 5 |
| 8 | 62.50 | 0.00 | 90 | 5 |
| 9 | 62.50 | 50.00 | 84 | 5 |
| 10 | 89.02 | 85.36 | 62 | 6 |
| 11 | 62.50 | 100.00 | 37 | 11 |
| 12 | 62.50 | 50.00 | 82 | 5 |
| 13 | 35.98 | 14.64 | 64 | 5 |
Optimum conditions with respect to yield, purity of product and reaction time were as follow: temperature 72 °C, water content of aqueous ethanol 33%. Verification experiments, carried out at the predicted conditions showed values reasonably close to those predicted and further confirmed the adequacy of predicted models.
On the basis of the central composite design, the cubic model relationship between the experimental yield (R1) and the process variables (temperature (A) and water content of aqueous ethanol (B)) in coded units is obtained from eqn (1).
| R1 = 82.60 + 11.31A − 18.74B − 0.25AB − 10.55A2 − 9.30B2 + 11.49A2B − 5.06AB2 | (1) |
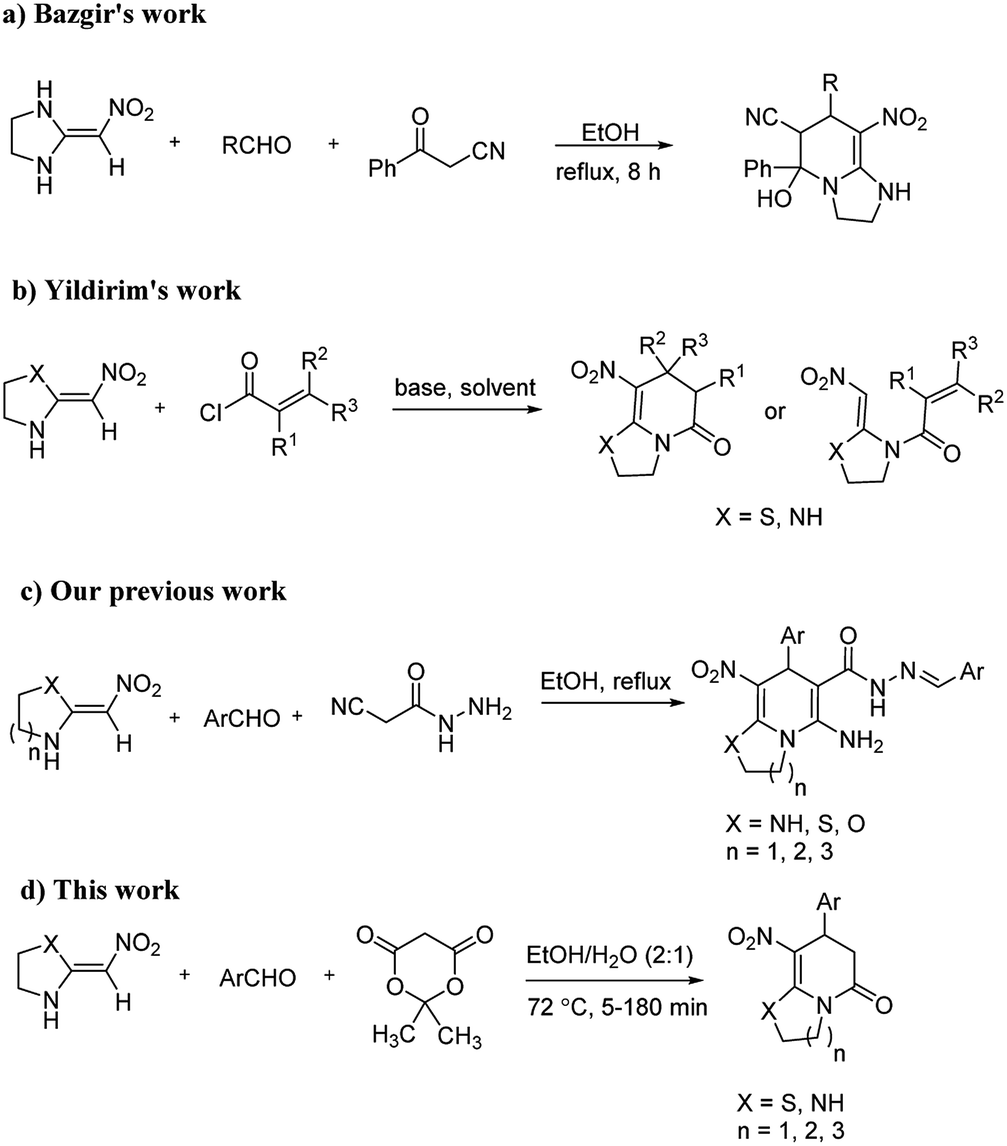
Fig. 1 illustrates the good linear correlation between the actual and predicted yield and sufficient accuracy of the forecast values. Therefore, the models can be used to predict the yield of product successfully.
 | ||
| Fig. 1 Linear correlation between the actual and predicted yield. | ||
The ANOVA for response surface cubic models are reported in Table 3. The model F-value of 189.50 implies the model is significant. The “Lack of Fit F-value” is 0.71 then it is not significant relative to the pure error. Non-significant lack of fit is good and we want the model to fit. The “Pred R-Squared” of 0.9586 is in reasonable agreement with the “Adj R-Squared” of 0.9910; i.e., the deviation is about 0.3.
| Source | Sum of square | Degree of freedom | Mean square | F value | p-Value, prob > F |
|---|---|---|---|---|---|
| Model | 3501.88 | 7 | 500.27 | 189.50 | <0.0001 significant |
| A-temperature | 512.00 | 1 | 512.00 | 193.94 | <0.0001 |
| B-percentage of water in ethanol | 1404.50 | 1 | 1404.50 | 532.01 | <0.0001 |
| AB | 0.25 | 1 | 0.25 | 0.095 | 0.7707 |
| A 2 | 774.28 | 1 | 772.28 | 293.29 | <0.0001 |
| B 2 | 601.67 | 1 | 601.67 | 227.91 | <0.0001 |
| A 2 B | 263.96 | 1 | 263.96 | 99.99 | 0.0002 |
| AB 2 | 51.28 | 1 | 51.28 | 19.43 | 0.0070 |
| A 3 | 0.000 | 0 | |||
| B 3 | 0.000 | 0 | |||
| Residual | 13.20 | 5 | 2.64 | ||
| Lack of fit | 2.0 | 1 | 2.00 | 0.71 | |
| Pure error | 11.20 | 4 | 2.80 | 0.4456 not significant | |
| Correlation total | 3515.08 | 12 |
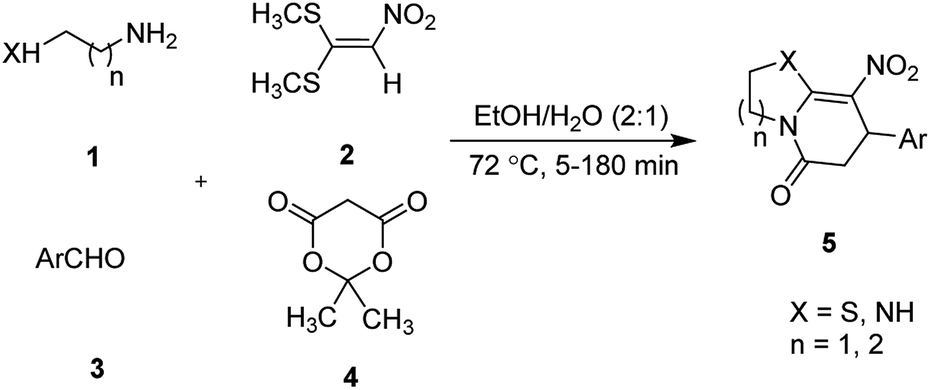
The effects of temperature and water content of aqueous ethanol on the total reaction yield are shown in Fig. 2. The total yield varied from 37% to 90%. As temperature increased, the yield increased. The changes in yield versus the water content of aqueous ethanol is minor compared to that of temperature. The highest yield (90%) was obtained at 62.5 °C and at 0% water. The lowest yield (37%) was at 62.5 °C and at 100% water.
 | ||
| Fig. 2 The effects of temperature and water content of aqueous ethanol on the total reaction yield. | ||
The effects of temperature and the water content of aqueous ethanol on the total reaction time are shown in Fig. 3. The analysis of total reaction time (R2) showed the significant Lack of fit and it is not good. So the temperature and water content of aqueous ethanol have no effect on the reaction time and the total reaction time was almost identical and short in any case.
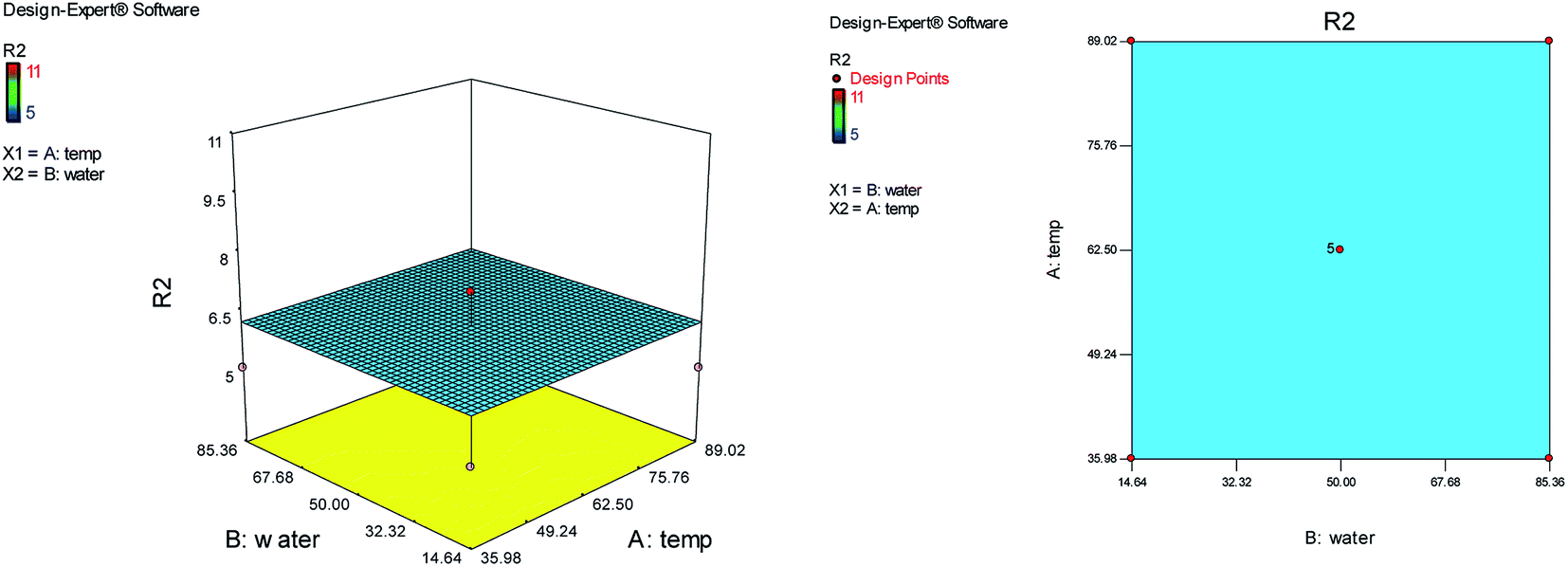 | ||
| Fig. 3 The effects of temperature and the water content of aqueous ethanol on the total reaction time. | ||
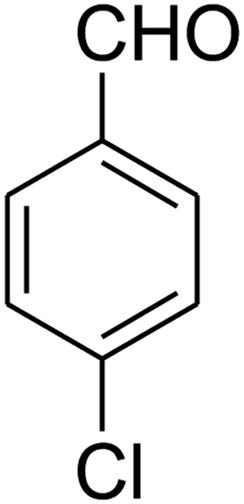
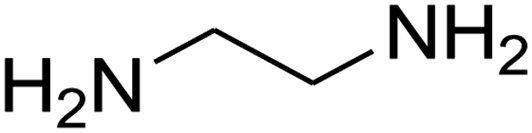
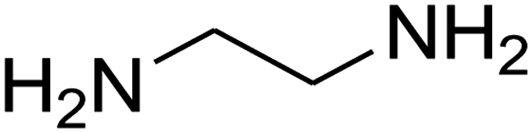
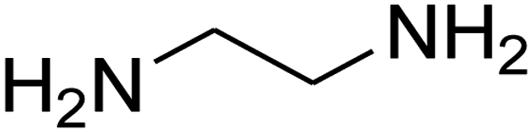
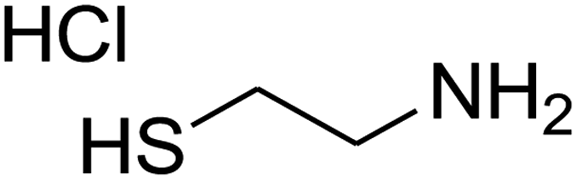
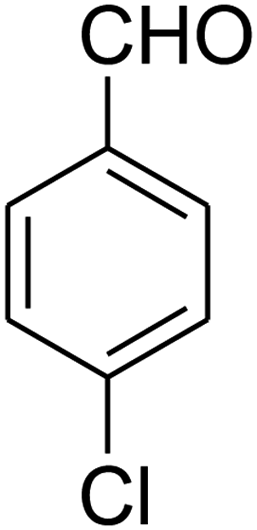
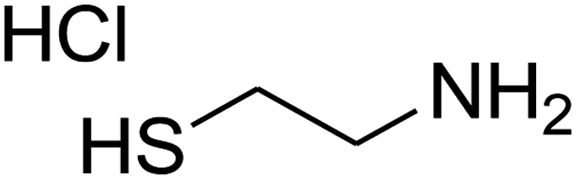
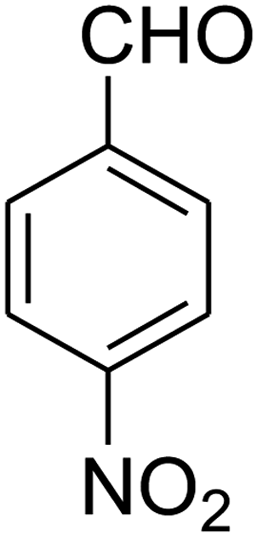
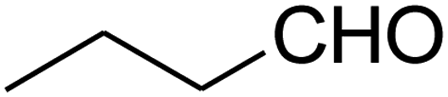
The scope of the reaction substrate was explored under optimized conditions. Several kind of aromatic aldehydes, and various diamines were tolerated. The reaction was completed after 5–180 min to afford corresponding S- and N-heterocyclic systems 5a–p, in moderate to good yields (50–89%). As shown in Table 4, reactions with diamines, usually resulted in good yields and short reaction times. Reactions with cysteamine hydrochloride, resulted in low yields and long reaction times. Also the structures of the aldehydes had obvious influence on the yield of product and reaction time. Reactions involving aromatic aldehydes usually went smoothly and resulted in good yields and short reaction times. Reaction involving heteroaromatic aldehyde (such as furfural) and aliphatic aldehyde (such as butyraldehyde), on the other hand, were normally slow and resulted in low and trace yields, respectively. When the 4-pyridinecarboxaldehyde was used the reaction did not work. The substitutions of the aromatic aldehydes had some influence on the yields and reaction times. Reaction with aromatic aldehydes carrying an electron-withdrawing group, such as chloro and nitro groups, usually resulted in good yields and short reaction times; those carrying electron-donating groups such as methoxy group, resulted in relatively poorer yields. The reaction did not work, when the reaction was performed using ethanolamine, cyclohexane-1,2-diamine and benzene-1,2-diamine.
| Entry | Aldehydea | Aminea | Product | Time (min) | Yield (%) |
|---|---|---|---|---|---|
| a Amine (1 mmol), 1,1-bis(methylthio)-2-nitroethene (1 mmol), aromatic aldehyde (1 mmol) and 2,2-dimethyl-1,3-dioxane-4,6-dione (Meldrum's acid) (1 mmol) were used. b Ref. 38. | |||||
| 1 |
|
|
|
5 | 88 |
| 2 |
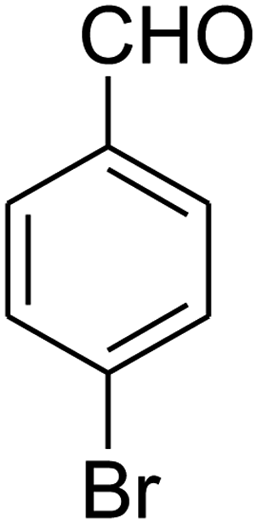
|
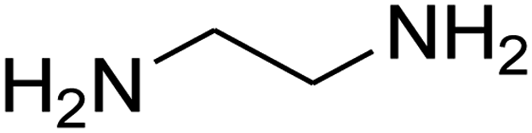
|
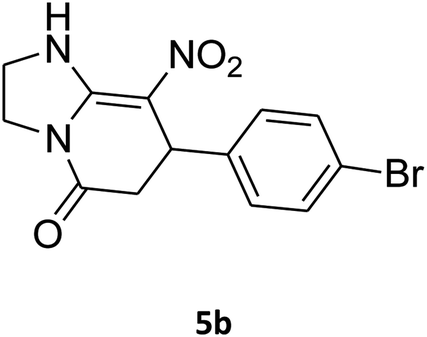
|
5 | 87 |
| 3 |
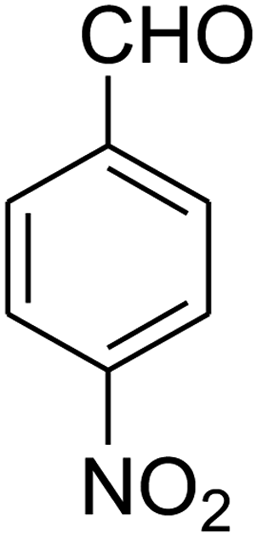
|
|
|
10 | 89 |
| 4 |
|
|
|
20 | 75 |
| 5 |
|
|
|
5 | 85 |
| 6 |
|
|
|
120 | 65 |
| 7 |
|
|
|
180 | 53 |
| 8 |
|
|
|
120 | 60 |
| 9 |
|
|
|
150 | 57 |
| 10 |
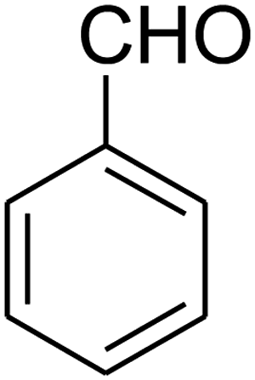
|
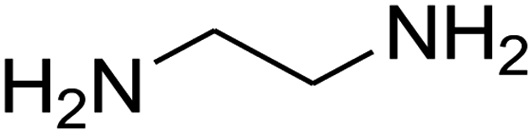
|
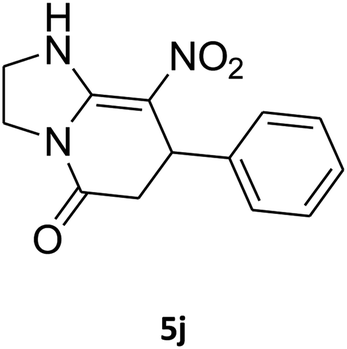
|
25 | 76 |
| 11 |
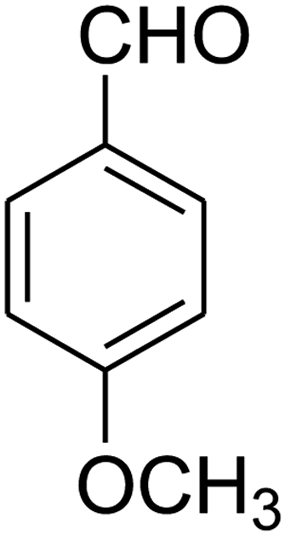
|
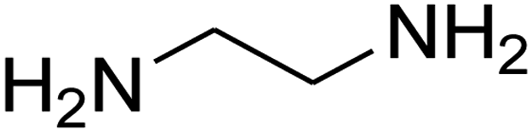
|

|
15 | 70 |
| 12 |
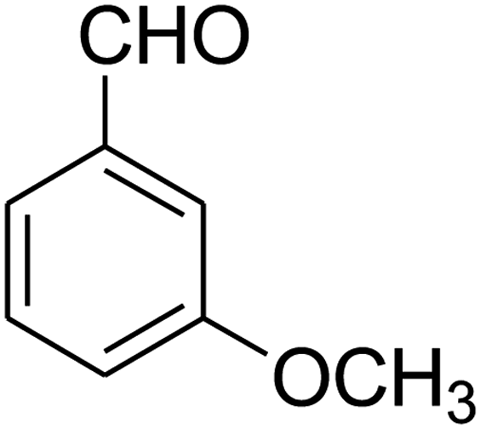
|
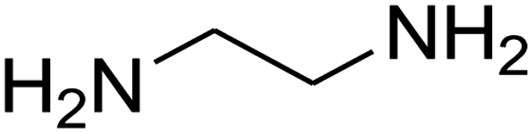
|
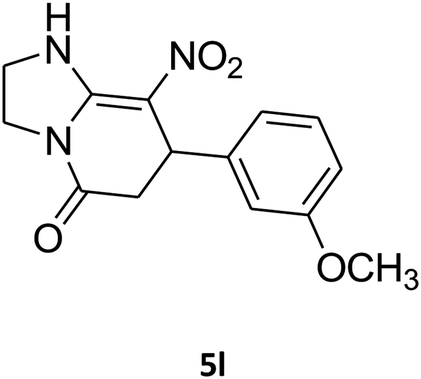
|
25 | 74 |
| 13 |
|
|
|
150 | 57 |
| 14 |
|
|
|
180 | 50 |
| 15 |
|
|
|
180 | Trace |
| 16b |
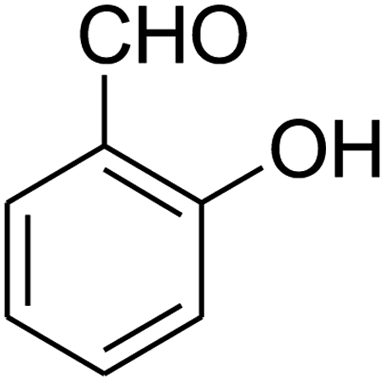
|
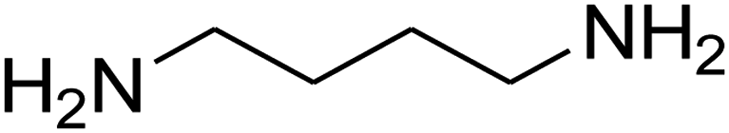
|
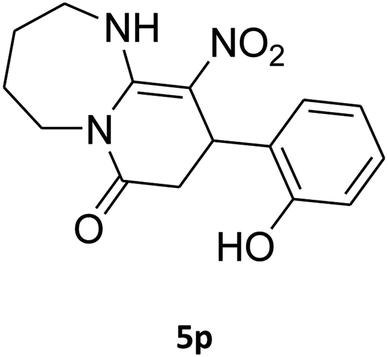
|
180 | 67 |
| 17 |

|
|
— | — | — |
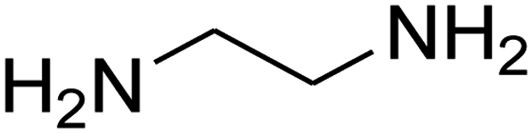
| 18 |
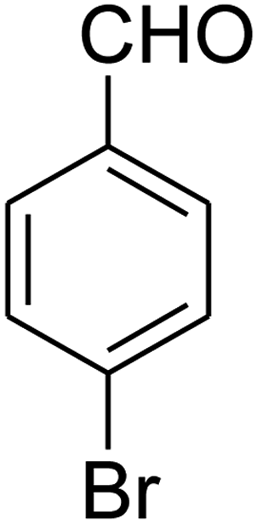
|
|
— | — | — |
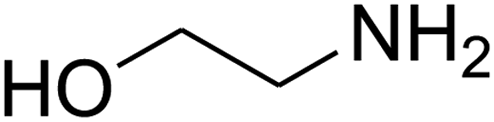
| 19 |
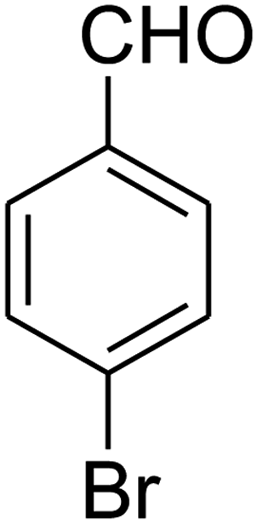
|
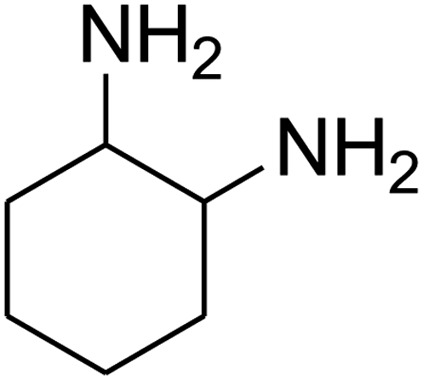
|
— | — | — |
| 20 |
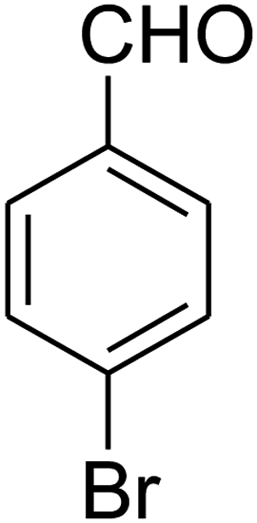
|
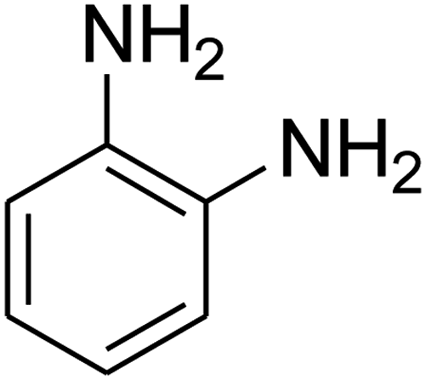
|
— | — | — |
The structures of products were fully characterized using their infrared (IR), mass spectrometry, proton hydrogen-1 nuclear magnetic resonance (1H NMR), carbon-13 nuclear magnetic resonance (13C NMR) spectra. For example, the mass spectrum of 5a displayed the molecular ion peak at m/z 293 with frequency of 53% which was in accordance with the proposed structure. The IR spectrum of this structure indicated absorption bands due to the NH stretching (3231 cm−1) as well as bands at 2922, 1704, 1623, 1486 and 1337 cm−1 due to the CH, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, CC and NO2 groups. The 1H NMR spectrum of 5a showed doublet of doublet for the CH2 group (δ 2.55, 3.21 ppm, 2JHH = 15.6 Hz, 3JHH = 7.8 Hz), multiplets for CH2NH and CH2N groups (δ 3.69–3.97 ppm), doublet for CH group (δ 4.52 ppm, 3JHH = 7.5 Hz), two doublets for the aromatic region (δ 7.16, 7.32 ppm, 3JHH = 8.4 Hz) and one singlet for NH group (δ 9.74 ppm). The 1H-decoupled 13C NMR spectrum of 5a showed 11 distinct resonances. There are three signals for CH and CH2 groups (δ 37.0, 43.0, 43.9 ppm), and signals at 106.3 and 167.6 ppm, which were assigned C–NO2 and CO groups, respectively. The 1H and 13C NMR spectra of 5b–p are similar to those of 5a except for the aryl and diamine moieties, which exhibited characteristic signals with appropriate chemical shifts (see the ESI†).
O, CC and NO2 groups. The 1H NMR spectrum of 5a showed doublet of doublet for the CH2 group (δ 2.55, 3.21 ppm, 2JHH = 15.6 Hz, 3JHH = 7.8 Hz), multiplets for CH2NH and CH2N groups (δ 3.69–3.97 ppm), doublet for CH group (δ 4.52 ppm, 3JHH = 7.5 Hz), two doublets for the aromatic region (δ 7.16, 7.32 ppm, 3JHH = 8.4 Hz) and one singlet for NH group (δ 9.74 ppm). The 1H-decoupled 13C NMR spectrum of 5a showed 11 distinct resonances. There are three signals for CH and CH2 groups (δ 37.0, 43.0, 43.9 ppm), and signals at 106.3 and 167.6 ppm, which were assigned C–NO2 and CO groups, respectively. The 1H and 13C NMR spectra of 5b–p are similar to those of 5a except for the aryl and diamine moieties, which exhibited characteristic signals with appropriate chemical shifts (see the ESI†).
An acceptable reaction mechanism for this one-pot, multi-component reaction is designated in Scheme 3. The one-pot protocol started with the reaction of various diamines or cysteamine hydrochloride 1 and 1,1-bis(methylthio)-2-nitroethene 2 to afford enamine 6. Also condensation reaction between 2,2-dimethyl-1,3-dioxane-4,6-dione (Meldrum's acid) and the aromatic aldehydes gave adduct 7, which then reacted with enamine 6 to form intermediate 8. Compound 10 was formed from 8 through imine–enamine tautomerism and cyclocondensation. Finally, decarboxylation of 10 yielded the product 5.
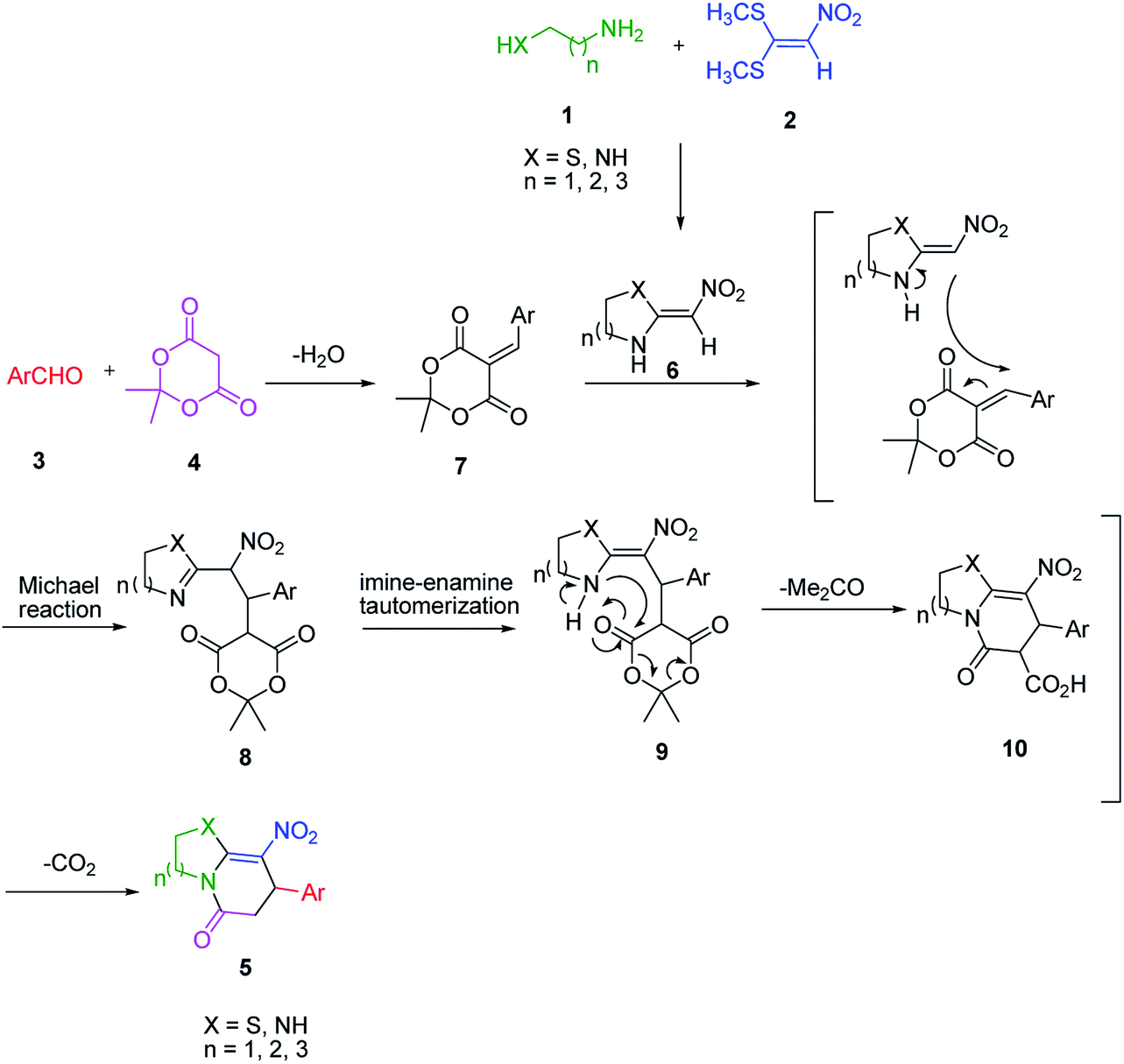 | ||
| Scheme 3 Plausible mechanism for the formation of product 5. | ||
Experimental
General
The various diamines, cysteamine hydrochloride, 1,1-bis(methylthio)-2-nitroethene, aldehydes, 2,2-dimethyl-1,3-dioxane-4,6-dione (Meldrum's acid) and solvents were purchased from Sigma-Aldrich chemical company and were used as received without further purification. Melting points were determined with an electrothermal 9100 apparatus. Infrared (IR) spectra were recorded on a Bruker Tensor 27 spectrometer. Nuclear magnetic resonance (NMR) spectra were obtained on a Bruker DRX-300 Avance instrument (300 MHz for 1H and 75.4 MHz for 13C) with DMSO as solvent. Chemical shifts are expressed in parts per million (ppm), and coupling constant (J) are reported in hertz (Hz). Elemental analyses for C, H and N were performed using a PerkinElmer 2004 series [II] CHN elemental analyzer. Mass spectra were recorded with an Agilent 5975C VL MSD with Triple-Axis Detector operating at an ionization potential of 70 eV.General procedure for the synthesis of product 5
:1) at 72 °C in a 50 mL flask was stirred for 6 h. After completion of the reaction (monitored by thin-layer chromatography, hexane/ethyl acetate 1:1), aromatic aldehyde (1 mmol), 2,2-dimethyl-1,3-dioxane-4,6-dione (Meldrum's acid) (0.144 g, 1 mmol) were added to the reaction mixture, and it was stirred under optimized condition for the time given in Table 4. Then, the reaction mixture was cooled to room temperature and filtered to give the crude product. The solid was washed with ethanol to give product in good yields.
:1), and Et3N (140 μL, 1 mmol) at 72 °C in a 50 mL flask was stirred for 5 h. After completion of the reaction (monitored by thin-layer chromatography, hexane/ethyl acetate 1:1), aromatic aldehyde (1 mmol), 2,2-dimethyl-1,3-dioxane-4,6-dione (Meldrum's acid) (0.144 g, 1 mmol) were added to the reaction mixture, and it was stirred under optimized condition for the time given in Table 4. The reaction mixture was monitored by thin layer chromatography (hexane/ethyl acetate 1:1); after completion of the reaction, the reaction mixture was cooled to room temperature and the product (as colorless crystals) was filtered. The solid was washed with ethanol to give product in moderate yields.
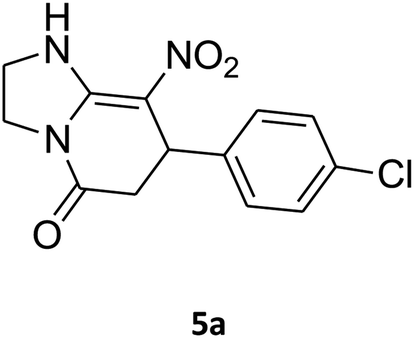
7-(4-Chlorophenyl)-2,3,6,7-tetrahydro-8-nitroimidazo[1,2-a]pyridin-5(1H)-one (5a). White solid; yield: 0.257 g (88%); mp 299–301 °C. 1H NMR (300 MHz, DMSO-d6): δ 2.55 (d, 2JHH = 15.6 Hz, 1H, CH2), 3.21 (dd, 2JHH = 15.6 Hz, 3JHH = 7.8 Hz, 1H, CH2), 3.69–3.97 (m, 4H, CH2NH and CH2N), 4.52 (d, 3JHH = 7.5 Hz, 1H, CH), 7.16 (d, 3JHH = 8.4 Hz, 2H, Ar), 7.32 (d, 3JHH = 8.4 Hz, 2H, Ar), 9.74 (s, 1H, NH). 13C NMR (75.4 MHz, DMSO-d6): δ 37.0, 43.0, 43.9, 106.3, 128.8, 129.0, 131.9, 141.9, 153.0, 167.6. IR (KBr) (νmax/cm−1): 3231 (NH), 2922 (CH), 1704 (C
O), 1623 (CC), 1486 and 1337 (NO2), 1214 (C–N). MS (EI, 70 eV): m/z (%) = 293 (M+, 53), 276 (42), 240 (100), 218 (23), 182 (59), 151 (35), 136 (45), 70 (18), 42 (14). Anal. calc. for C13H12ClN3O3 (293.71): C, 53.16; H, 4.12; N, 14.31. Found: C, 53.01; H, 4.51; N, 14.03.
7-(4-Bromophenyl)-2,3,6,7-tetrahydro-8-nitroimidazo[1,2-a]pyridin-5(1H)-one (5b). White solid; yield: 0.294 g (87%); mp 305–307 °C. 1H NMR (300 MHz, DMSO-d6): δ 2.55 (d, 2JHH = 16.6 Hz, 1H, CH2), 3.21 (dd, 2JHH = 16.6 Hz, 3JHH = 7.8 Hz, 1H, CH2), 3.69–3.97 (m, 4H, CH2NH and CH2N), 4.51 (d, 3JHH = 7.8 Hz, 1H, CH), 7.10 (d, 3JHH = 7.2 Hz, 2H, Ar), 7.45 (d, 3JHH = 7.2 Hz, 2H, Ar), 9.73 (s, 1H, NH). 13C NMR (75.4 MHz, DMSO-d6): δ 37.1, 43.1, 44.0, 106.3, 120.3, 129.1, 131.9, 142.3, 153.0, 167.6. IR (KBr) (νmax/cm−1): 3224 (NH), 2921 (CH), 1699 (C
O), 1623 (CC), 1486 and 1337 (NO2), 1215 (C–N). MS (EI, 70 eV): m/z (%) = 337 (M+-1, 25), 320 (22), 291 (58), 240 (100), 212 (12), 182 (51), 136 (29), 116 (33), 70 (49), 42 (22). Anal. calc. for C13H12BrN3O3 (338.16): C, 46.17; H, 3.58; N, 12.43. Found: C, 46.52; H, 3.21; N, 12.12.
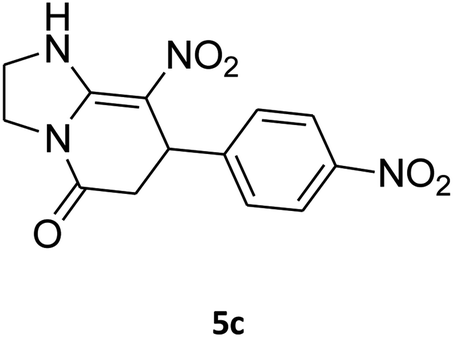
2,3,6,7-Tetrahydro-8-nitro-7-(4-nitrophenyl)imidazo[1,2-a]pyridin-5(1H)-one (5c). Yellow solid; yield: 0.270 g (89%); mp 264–266 °C. 1H NMR (300 MHz, DMSO-d6): δ 2.59 (d, 2JHH = 15.6 Hz, 1H, CH2), 3.26 (dd, 2JHH = 15.6 Hz, 3JHH = 8.1 Hz, 1H, CH2), 3.71–3.99 (m, 4H, CH2NH and CH2N), 4.68 (d, 3JHH = 7.5 Hz, 1H, CH), 7.44 (d, 3JHH = 8.7 Hz, 2H, Ar), 8.12 (d, 3JHH = 8.7 Hz, 2H, Ar), 9.79 (s, 1H, NH). 13C NMR (75.4 MHz, DMSO-d6): δ 37.6, 43.1, 44.0, 105.8, 124.3, 128.3, 146.9, 150.8, 153.0, 167.2. IR (KBr) (νmax/cm−1): 3227 (NH), 2923 (CH), 1712 (C
O), 1628 (CC), 1520 and 1345 (NO2), 1219 (C–N). Anal. calc. for C13H12N4O5 (304.26): C, 51.32; H, 3.98; N, 18.41. Found: C, 51.56; H, 3.64; N, 18.16.
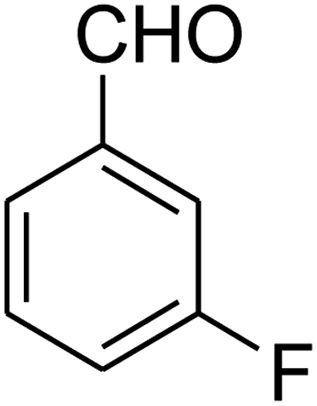
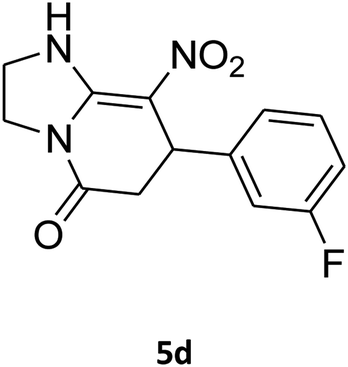
7-(3-Fluorophenyl)-2,3,6,7-tetrahydro-8-nitroimidazo[1,2-a]pyridin-5(1H)-one (5d). White solid; yield: 0.207 g (75%); mp 253–255 °C. 1H NMR (300 MHz, DMSO-d6): δ 2.59 (d, 2JHH = 16.6 Hz, 1H, CH2), 3.20 (m, 2JHH = 16.6 Hz, 3JHH = 8.1 Hz, 1H, CH2), 3.70–3.94 (m, 4H, CH2NH and CH2N), 4.55 (d, 3JHH = 7.5 Hz, 1H, CH), 6.96–7.06 (m, 3H, Ar), 7.28–7.35 (m, 1H, Ar), 9.75 (s, 1H, NH). 13C NMR (75.4 MHz, DMSO-d6): δ 37.4, 43.1, 44.0, 106.2, 113.8 (d, 2JCF = 21 Hz), 114.2 (d, 2JCF = 21 Hz), 122.8, 131.1, 145.9, 153.1, 162.8 (d, 1JCF = 244 Hz), 167.6. Anal. calc. for C13H12FN3O3 (277.25): C, 56.32; H, 4.36; N, 15.16. Found: C, 56.70; H, 4.06, N, 15.38.
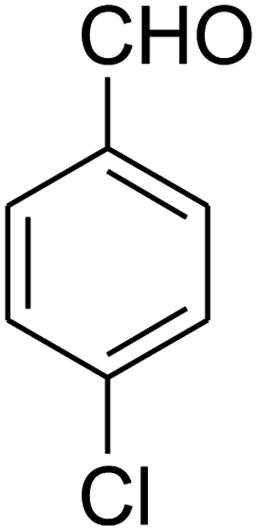
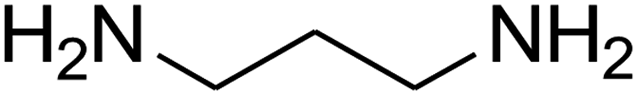
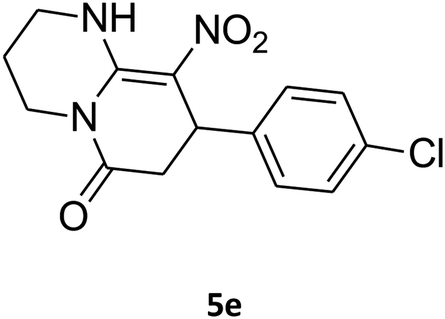
8-(4-Chlorophenyl)-1,2,3,4,7,8-hexahydro-9-nitropyrido[1,2-a]pyrimidin-6-one (5e). White solid; yield: 0.260 g (85%); mp 282–284 °C. 1H NMR (300 MHz, DMSO-d6): δ 1.89–1.98 (m, 2H, CH2), 2.65 (d, 2JHH = 15.0 Hz, 1H, CH2), 3.21 (dd, 2JHH = 15.0 Hz, 3JHH = 7.5 Hz, 1H, CH2), 3.41–3.89 (m, 4H, CH2NH and CH2N), 4.59 (d, 3JHH = 6.6 Hz, 1H, CH), 7.15 (d, 3JHH = 8.4 Hz, 2H, Ar), 7.31 (d, 3JHH = 7.2 Hz, 2H, Ar), 11.55 (s, 1H, NH). 13C NMR (75.4 MHz, DMSO-d6): δ 19.5, 36.0, 38.6, 108.6, 128.8, 129.0, 131.9, 140.9, 152.9, 168.6. IR (KBr) (νmax/cm−1): 3089 (NH), 2927 (CH), 1710 (C
O), 1618 (CC), 1502 and 1330 (NO2), 1232 (C–N). MS (EI, 70 eV): m/z (%) = 307 (M+, 23), 290 (17), 261 (100), 196 (20), 178 (13), 151 (37), 115 (19), 84 (31), 56 (29). Anal. calc. for C14H14ClN3O3 (307.73): C, 54.64; H, 4.59; N, 13.65. Found: C, 54.26; H, 4.90; N, 13.49.
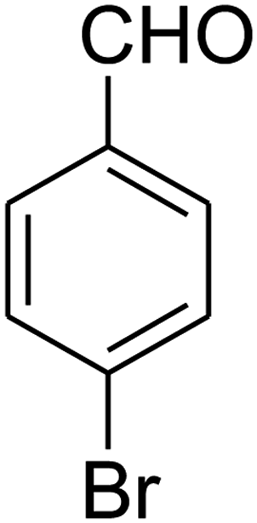
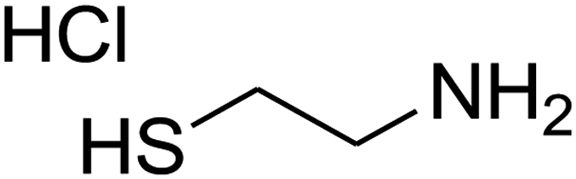
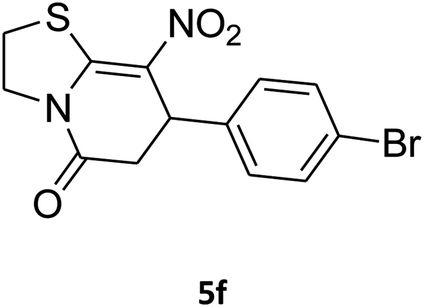
7-(4-Bromophenyl)-2,3,6,7-tetrahydro-8-nitrothiazolo[3,2-a]pyridin-5-one (5f). Yellow solid; yield: 0.230 g (65%); mp 204–206 °C. 1H NMR (300 MHz, DMSO-d6): δ 2.60 (d, 2JHH = 16.5 Hz, 1H, CH2), 3.17–3.39 (m, 3H, CH2 and CH2S), 3.96–4.06 (m, 1H, CH2N), 4.32–4.39 (m, 1H, CH2N), 4.64 (d, 3JHH = 8.4 Hz, 1H, CH), 7.14 (d, 3JHH = 8.4 Hz, 2H, Ar), 7.36 (d, 3JHH = 8.4 Hz, 2H, Ar). 13C NMR (75.4 MHz, DMSO-d6): δ 29.1, 37.7, 49.4, 120.9, 124.8, 129.0, 132.3, 140.3, 159.9, 166.6. IR (KBr) (νmax/cm−1): 2918 (CH), 1699 (C
O), 1573 (CC), 1420 and 1350 (NO2), 1223 (C–N). MS (EI, 70 eV): m/z (%) = 355 (M+, 58), 339 (100), 309 (40), 281 (17), 213 (16), 182 (22), 140 (18), 102 (21), 60 (15). Anal. calc. for C13H11BrN2O3S (355.21): C, 43.96; H, 3.12; N, 7.89. Found: C, 43.56; H, 3.33; N, 8.26.
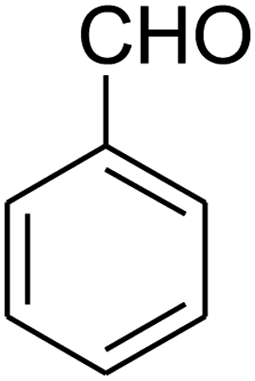
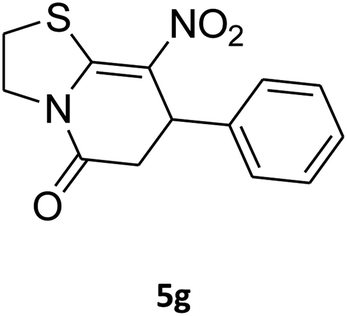
2,3,6,7-Tetrahydro-8-nitro-7-phenylthiazolo[3,2-a]pyridin-5-one (5g). Yellow solid; yield: 0.146 g (53%); mp 175–177 °C. 1H NMR (300 MHz, DMSO-d6): δ 2.66 (d, 2JHH = 16.2 Hz, 1H, CH2), 3.25–3.40 (m, 3H, CH2 and CH2S), 3.95–4.05 (m, 1H, CH2N), 4.33–4.41 (m, 1H, CH2N), 4.63 (d, 3JHH = 7.5 Hz, 1H, CH), 7.09–7.32 (m, 5H, Ar). 13C NMR (75.4 MHz, DMSO-d6): δ 29.0, 38.3, 49.4, 125.3, 126.7, 127.8, 129.5, 140.9, 159.6, 166.8.
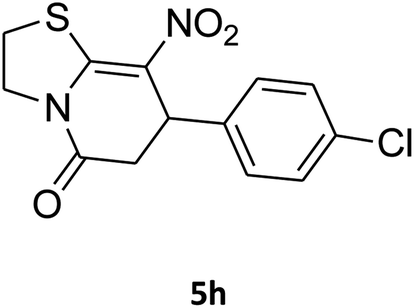
7-(4-Chlorophenyl)-2,3,6,7-tetrahydro-8-nitrothiazolo[3,2-a]pyridin-5-one (5h). Yellow solid; yield: 0.186 g (60%); mp 128–130 °C. 1H NMR (300 MHz, DMSO-d6): δ 2.65 (d, 2JHH = 16.2 Hz, 1H, CH2), 3.18–3.40 (m, 3H, CH2 and CH2S), 3.96–4.06 (m, 1H, CH2N), 4.32–4.40 (m, 1H, CH2N), 4.65 (d, 3JHH = 7.5 Hz, 1H, CH), 7.14 (d, 3JHH = 8.7 Hz, 2H, Ar), 7.35 (d, 3JHH = 8.4 Hz, 2H, Ar). 13C NMR (75.4 MHz, DMSO-d6): δ 29.1, 37.7, 49.5, 124.9, 128.7, 129.4, 132.4, 139.9, 159.9, 166.6. IR (KBr) (νmax/cm−1): 2918 (CH), 1703 (C
O), 1573 (CC), 1507 and 1350 (NO2), 1246 (C–N).
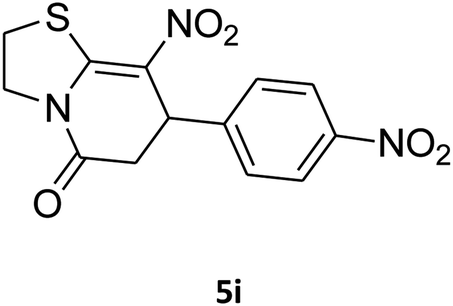
2,3,6,7-Tetrahydro-8-nitro-7-(4-nitrophenyl)thiazolo[3,2-a]pyridin-5-one (5i). Yellow solid; yield: 0.182 g (57%); mp 145–147 °C. 1H NMR (300 MHz, DMSO-d6): δ 2.70 (d, 2JHH = 16.8 Hz, 2H, CH2), 3.07–3.46 (m, 3H, CH2 and CH2S), 3.99–4.09 (m, 1H, CH2N), 4.33–4.41 (m, 1H, CH2N), 4.81 (d, 3JHH = 8.1 Hz, 1H, CH), 7.42 (d, 3JHH = 8.7 Hz, 2H, Ar), 8.15 (d, 3JHH = 8.7 Hz, 2H, Ar). 13C NMR (75.4 MHz, DMSO-d6): δ 29.1, 38.2, 49.5, 124.2, 124.6, 128.3, 147.2, 148.7, 160.4, 166.3. IR (KBr) (νmax/cm−1): 2919 (CH), 1709 (C
O), 1573 (CC), 1522 and 1357 (NO2), 1226 (C–N). MS (EI, 70 eV): m/z (%) = 321 (M+, 36), 304 (100), 274 (45), 246 (12), 199 (8), 140 (6), 60 (11). Anal. calc. for C13H11N3O5S (321.04): C, 48.59; H, 3.45; N, 13.08. Found: C, 48.79; H, 3.82; N, 13.50.
2,3,6,7-Tetrahydro-8-nitro-7-phenylimidazo[1,2-a]pyridin-5(1H)-one (5j). White solid; yield: 0.196 g (76%); mp 266–268 °C. 1H NMR (300 MHz, DMSO-d6): δ 2.55 (d, 2JHH = 16.2 Hz, 1H, CH2), 3.22 (dd, 2JHH = 16.2 Hz, 3JHH = 7.5 Hz, 1H, CH2), 3.73–3.96 (m, 4H, CH2NH and CH2N), 4.51 (d, 3JHH = 7.2 Hz, 1H, CH), 7.02–7.29 (m, 5H, Ar), 9.73 (s, 1H, NH). 13C NMR (75.4 MHz, DMSO-d6): δ 37.6, 43.0, 43.9, 106.6, 126.8, 127.3, 129.1, 142.9, 153.1, 167.8.
2,3,6,7-Tetrahydro-7-(4-methoxyphenyl)-8-nitroimidazo[1,2-a]pyridin-5(1H)-one (5k). White solid; yield: 0.202 g (70%); mp 307–309 °C. 1H NMR (300 MHz, DMSO-d6): δ 2.55 (d, 2JHH = 16.5 Hz, 1H, CH2), 3.14 (dd, 2JHH = 16.5 Hz, 3JHH = 7.8 Hz, 1H, CH2), 3.68 (s, 1H, OCH3), 3.72–3.97 (m, 4H, CH2NH and CH2N), 4.32 (d, 3JHH = 7.5 Hz, 1H, CH), 6.81 (d, 3JHH = 7.5 Hz, 2H, Ar), 7.03 (d, 3JHH = 7.8 Hz, 2H, Ar), 9.69 (s, 1H, NH). 13C NMR (75.4 MHz, DMSO-d6): δ 36.7, 43.0, 43.9, 55.4, 107.0, 114.4, 127.8, 134.7, 153.0, 158.6, 167.9.
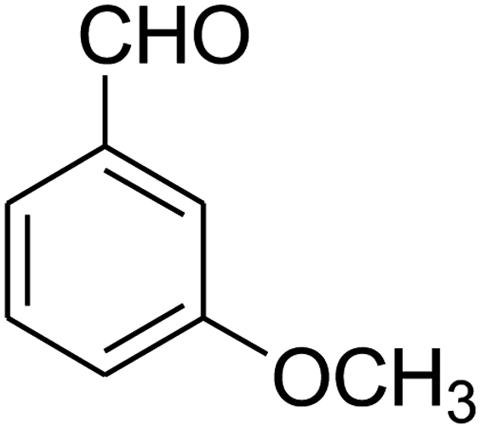
2,3,6,7-Tetrahydro-7-(3-methoxyphenyl)-8-nitroimidazo[1,2-a]pyridin-5(1H)-one (5l). White solid; yield: 0.213 g (74%); mp 213–215 °C. 1H NMR (300 MHz, DMSO-d6): δ 2.56 (d, 2JHH = 16.5 Hz, 1H, CH2), 3.14 (dd, 2JHH = 16.5 Hz, 3JHH = 8.1 Hz, 1H, CH2), 3.69 (s, 1H, OCH3), 3.79–3.97 (m, 4H, CH2NH and CH2N), 4.49 (d, 3JHH = 7.5 Hz, 1H, CH), 6.66 (s, 2H, Ar), 6.77 (d, 3JHH = 7.2 Hz, 1H, Ar), 7.18 (t, 3JHH = 8.1 Hz, 1H, Ar), 9.72 (s, 1H, NH). 13C NMR (75.4 MHz, DMSO-d6): δ 37.5, 43.0, 43.9, 55.3, 106.5, 112.2, 113.1, 118.6, 130.2, 144.5, 153.1, 159.9, 167.8.
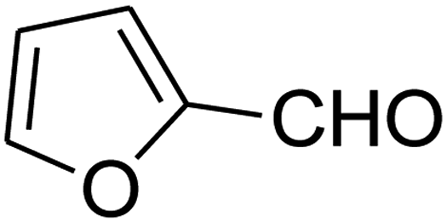
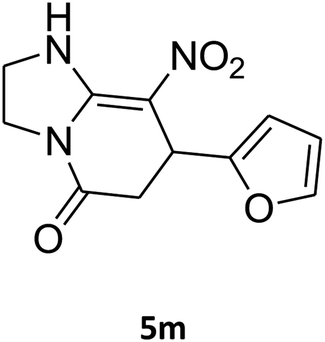
7-(Furan-2-yl)-2,3,6,7-tetrahydro-8-nitroimidazo[1,2-a]pyridin-5(1H)-one (5m). White solid; yield: 0.141 g (57%); mp 241–243 °C. 1H NMR (300 MHz, DMSO-d6): δ 2.67 (d, 2JHH = 16.5 Hz, 1H, CH2), 3.10 (dd, 2JHH = 16.5 Hz, 3JHH = 7.5 Hz, 1H, CH2), 3.70–3.97 (m, 4H, CH2NH and CH2N), 4.61 (d, 3JHH = 6.9 Hz, 1H, CH), 6.04 (s, 1H, Ar), 6.31 (s, 1H, Ar), 7.50 (s, 1H, Ar), 9.63 (s, 1H, NH). 13C NMR (75.4 MHz, DMSO-d6): δ 32.0, 36.6, 43.0, 43.8, 104.9, 105.7, 110.8, 142.6, 152.7, 154.7, 167.7.
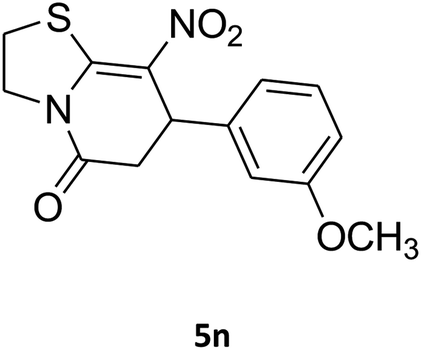
2,3,6,7-Tetrahydro-7-(3-methoxyphenyl)-8-nitrothiazolo[3,2-a]pyridin-5-one (5n). Yellow solid; yield: 0.153 g (50%); mp 130–132 °C. 1H NMR (300 MHz, DMSO-d6): δ 2.70 (d, 2JHH = 16.5 Hz, 1H, CH2), 3.27–3.40 (m, 3H, CH2 and CH2S), 3.70 (s, 1H, OCH3), 3.93–4.03 (m, 1H, CH2N), 4.33–4.41 (m, 1H, CH2N), 4.61 (d, 3JHH = 7.2 Hz, 1H, CH), 6.61–6.83 (m, 3H, Ar), 7.21 (t, 3JHH = 7.8 Hz, 1H, Ar). 13C NMR (75.4 MHz, DMSO-d6): δ 29.0, 38.2, 49.4, 55.4, 112.8, 112.9, 118.3, 125.1, 130.6, 142.5, 159.6, 160.1, 166.8.
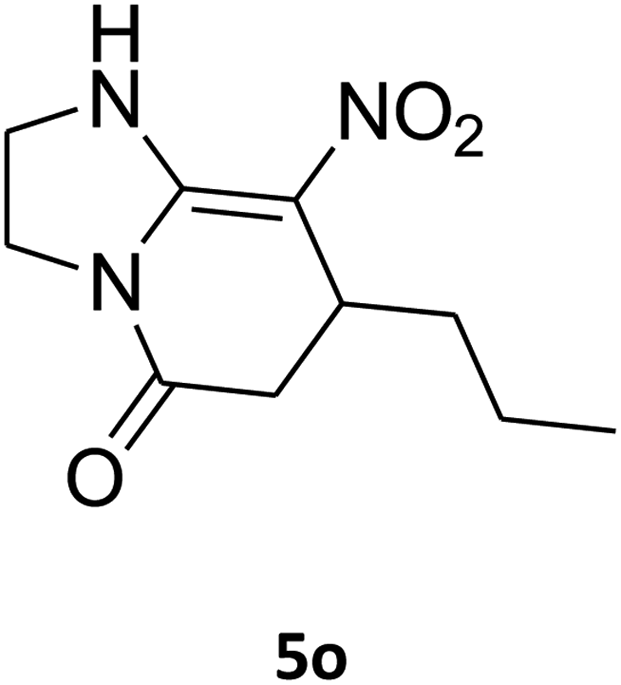
2,3,6,7-Tetrahydro-8-nitro-7-propylimidazo[1,2-a]pyridin-5(1H)-one (5o). White solid; mp 174–176 °C. 1H NMR (300 MHz, DMSO-d6): δ 0.83 (t, 3H, CH3), 1.15–1.45 (m, 4H, 2CH2), 2.42 (d, 2JHH = 16.8 Hz, 1H, CH2), 2.77 (dd, 2JHH = 16.8 Hz, 3JHH = 7.2 Hz, 1H, CH2), 3.62–3.93 (m, 5H, CH and CH2NH and CH2N), 9.54 (s, 1H, NH).
Conclusion
We have reported a simple and clean synthesis of imidazopyridinone, pyridopyrimidinone, and thiazolopyridinone derivatives. We were able to optimize the reaction conditions using RSM in conjunction with a central composite design. Further experimentation revealed the optimal values of the water content of aqueous ethanol (33%), and reaction temperature (72 °C). Also good yield of product in mild conditions show that the experimental results are in good agreement with the predicted values, and the model successfully can be used to predict the synthesis of such heterocyclic compounds.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We gratefully acknowledge the financial support of this research from Imam Khomeini International University. We thank the Iran National Science Foundation (No. 97021367) for financial support.Notes and references
- D. Bas and I. H. Boyaci, J. Food Eng., 2007, 78, 836–845 CrossRef CAS.
- H. Tang, Q. Xiao, H. Xu and Y. Zhang, Org. Process Res. Dev., 2013, 17, 632–640 CrossRef CAS.
- J. Palaniraja, S. M. Roopan and G. M. Rayalu, RSC Adv., 2016, 6, 24610–24616 RSC.
- B. Maherani, M. Barzegar and M. A. Sahari, J. Food Sci. Technol., 2007, 44, 250–254 CAS.
- V. K. Garlapati and L. Roy, J. Young Pharm., 2017, 9, 417–421 CrossRef CAS.
- A. Ryad, K. Lakhdar, K. S. Majda, A. Samia, A. Mark, A. D. Corinne and G. Eric, Int. J. Mol. Sci., 2010, 11, 4726–4740 CrossRef.
- I. R. Parrey and A. A. Hashmi, Catal. Sustainable Energy, 2016, 3, 1–6 CAS.
- L. Yurttas, Y. Ozkay, H. K. Gencer and U. Acar, J. Chem., 2015, 2015, 1–7 CrossRef.
- Y. Cui and P. E. Floreancig, Org. Lett., 2012, 14, 1720–1723 CrossRef CAS.
- A. Lozynskyi, B. Zimenkovsky, L. Radko, S. Stypula-Trebas, O. Roman, A. K. Gzella and R. Lesyk, Chem. Pap., 2018, 72, 669–681 CrossRef CAS.
- G. M. Ziarani, N. H. Nasab, M. Rahimifard and A. A. Soorki, J. Saudi Chem. Soc., 2015, 19, 676–681 CrossRef.
- S. M. Roopan, S. M. Patil and J. Palaniraja, Res. Chem. Intermed., 2016, 42, 2749–2790 CrossRef.
- L. R. Wen, Z. R. Li, M. Li and H. Cao, Green Chem., 2012, 14, 707–716 RSC.
- S. H. Li, Y. H. Shen, N. Gao and J. T. Li, Eur. J. Chem., 2010, 7, 779–784 CAS.
- I. Yavari, M. Nematpour and E. Sodagar, Monatsh. Chem., 2015, 146, 2135–2138 CrossRef CAS.
- M. Mamaghani and R. Hossein Nia, J. Heterocycl. Chem., 2017, 54, 1700–1722 CrossRef CAS.
- S. Abdolmohammadi and M. Afsharpour, Chin. Chem. Lett., 2012, 23, 257–260 CrossRef CAS.
- K. C. Majumdar, S. Ponra and D. Ghosh, Synthesis, 2011, 1132–1136 CrossRef CAS.
- A. M. Rad and M. Mokhtary, Int. Nano Lett., 2015, 5, 109–123 CrossRef CAS.
- A. H. Shamroukh, A. E. Rashad and F. M. E. Abdelmegeid, J. Chem. Pharm. Res., 2016, 8, 734–772 CAS.
- M. Boominathan, M. Nagaraj, C. Maheshwaran, S. Muthusubramanian and N. Bhuvanesh, J. Heterocycl. Chem., 2014, 51, 244–248 CrossRef CAS.
- G. A. El-Hiti, Monatsh. Chem., 2003, 134, 837–841 CrossRef CAS.
- M. E. Azab, G. A. M. El-Hag Ali and A. H. F. Abd El-Wahab, Acta Pharm., 2003, 53, 213–221 CAS.
- H. L. Chen and H. Y. Guo, J. Chem. Res., 2012, 36, 162–165 CrossRef CAS.
- F. Shi, C. Li, M. Xia, K. Miao, Y. Zhao, S. Tu, W. Zheng, G. Zhang and N. Ma, Bioorg. Med. Chem. Lett., 2009, 19, 5565–5568 CrossRef CAS.
- R. A. Mekheimer, N. M. Hilmy, A. A. Hameed, S. Dacrory and K. U. Sadek, Synth. Commun., 2011, 41, 2511–2516 CrossRef CAS.
- K. M. Wang, S. J. Yan and J. Lin, Eur. J. Org. Chem., 2014, 1129–1145 CrossRef CAS.
- P. H. Yang, Res. Chem. Intermed., 2016, 42, 5617–5637 CrossRef CAS.
- L. Pan, X. Bi and Q. Liu, Chem. Soc. Rev., 2013, 42, 1251–1286 RSC.
- I. Yavari, N. Zahedi, L. Baoosi and S. Skoulika, Mol. Diversity, 2018, 22, 11–19 CrossRef CAS.
- L. Zhang, J. Dong, X. Xu and Q. Liu, Chem. Rev., 2016, 116, 287 CrossRef CAS.
- D. P. Sangi, J. L. Monteiro, K. L. Vanzolini, Q. B. Cass, M. W. Paixao and A. G. Correa, J. Braz. Chem. Soc., 2014, 25, 887–889 CAS.
- D. Elumalai, R. Gnanasekaran, S. Leelakrishnan, G. Nachimuthu, T. Kannan, T. P. Paramasivam and K. Jayabal, ChemistrySelect, 2018, 3, 2070–2079 CrossRef CAS.
- S. K. Movahed, M. Dabiri and A. Bazgir, Helv. Chim. Acta, 2013, 96, 525–532 CrossRef.
- M. Yildirim, D. Celikel, N. Evis, D. W. Knight and B. M. Kariuki, Tetrahedron, 2014, 70, 5674–5681 CrossRef CAS.
- M. Bayat and F. S. Hosseini, Tetrahedron Lett., 2017, 58, 1616–1621 CrossRef CAS.
- M. Bayat and F. S. Hosseini, J. Sulfur Chem., 2018, 39, 279–293 CrossRef CAS.
- M. Bayat and M. Rezaei, J. Iran. Chem. Soc., 2018, 15, 769–777 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra06054e |
| This journal is © The Royal Society of Chemistry 2019 |