
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConverse transitions between the micelles and the vesicles of pyrrolidone-based AIE amphiphilic copolymers in polar and apolar solvents†
Xiaolong He,
Beibei Wang,
Xuefeng Li * and
Jinfeng Dong*
* and
Jinfeng Dong*
Engineering Research Center of Organosilicon Compounds & Materials, Ministry of Education, College of Chemistry and Molecular Sciences, Wuhan University, Wuhan 430072, P. R. China. E-mail: lixuefeng@whu.edu.cn; jfdong@whu.edu.cn
First published on 6th September 2019
Abstract
Herein, a new family of aggregation-induced emission (AIE) amphiphilic copolymers, named poly(N-(2-methacryloyloxyethyl)pyrrolidone)-b-poly(lauryl methacrylate-co-1-ethenyl-4-(1,2,2-triphenylethenyl)benzene), PNMPx-b-P(LMAy-co-TPEz), was developed by the reversible addition–fragmentation chain transfer (RAFT) polymerization method. The polymerization degree x of the NMP segment was kept constant at 35, whereas that of the LMA segment ranged from 9 to 55 with the polymerization degree ratio y/z of the LMA and TPE segments being around 9. As a result, the PNMPx-b-P(LMAy-co-TPEz) copolymer gradually transformed from being water soluble to oil soluble with an increase in the length of the P(LMAy-co-TPEz) segment. Moreover, these copolymers could form self-organized normal and reverse assemblies in both water and n-dodecane. Various morphologies, including spherical micelles, worm-like micelles and vesicles, were confirmed by the transmission electron microscopy (TEM) observation. Specifically, the micelle-to-vesicle transition via worm-like micelles occurred in the aqueous solution upon increasing the length of the P(LMAy-co-TPEz) segment, whereas the reverse transition occurred in n-dodecane. Because of the presence of the AIE-active TPE segment, both the aqueous and the n-dodecane solutions of PNMPx-b-P(LMAy-co-TPEz) were highly luminescent, and their fluorescence quantum yields significantly depended on the polarity of the solvent and the morphology of the assemblies. Due to the strong luminescence properties of PNMPx-b-P(LMAy-co-TPEz) assemblies, these AIE-active amphiphilic copolymers acted as excellent bioimaging probes with high efficiency.
1 Introduction
Currently, studies on the application of luminescent probe molecules as cell imaging reagents, especially in biological systems, are attracting significant interest.1–6 The development of highly efficient and biocompatible luminescent probes has been the key project.7 However, the commonly presented aggregation-caused quenching (ACQ) phenomena have significantly limited and impeded the potential of the classic fluorescent molecules.8 Alternatively, luminescent molecules processing anti-ACQ and showing aggregation-induced emission (AIE) can overcome the abovementioned shortcomings well.9–11 Since the first report on the AIE phenomenon by the Tang's group in 2001,12 the AIE effect has attracted significant attention; on the one hand, a series of AIE molecules has been developed based on the general luminescence mechanism that the AIE effect increases upon the restriction of intramolecular motion (RIM) including the restriction of both intramolecular rotation (RIR) and intramolecular vibration (RIV).13–18 Moreover, the AIE phenomena have been applied in wide fields beyond the original imaging function. Note that AIE molecules with some particular amphiphilicity have always been in demand, which provide the primary driving force during the self-assembly processes. In other words, AIE molecules could be considered as amphiphiles containing some particular AIE-active moieties. As a result, numerous AIE molecules with interesting functions have been developed in recent years.19–24The stability of amphiphilic copolymer assemblies is often more excellent than that of classic surfactants.25 Accordingly, amphiphilic copolymers show promising potential in various application fields of biotechnology,26 the development of functional materials,27 drug-controlled release systems,28 catalysis,29 and so forth. Based on the classic synthetic methods such as the reversible addition–fragmentation chain transfer (RAFT) polymerization, atom transfer radical polymerization (ATRP), and nitroxide-mediated radical polymerization (NMR), numerous novel amphiphilic copolymers have been developed and their corresponding self-assembly behaviors have also been reported in the past few decades.30–37 Recent studies on the polymerization-induced self-assembly (PISA) of amphiphilic copolymers have attracted significant attention because the development of block copolymers and the preparation of aggregates with various morphologies have been simultaneously achieved, thereby further enriching the amphiphilic copolymers.38–42
Our group has reported some pyrrolidone-based amphiphilic diblock copolymers synthesized by the RAFT method for the first time; these copolymers not only show rich self-assembly behaviors in both polar and apolar solvents but also display some interesting stimuli-responses. For example, the micellization of poly(methacrylate acid)-b-poly(N-(2-methacryloyloxyethyl)pyrrolidone) (PMAA-b-PNMP) in the aqueous solution is sensitive to both pH and temperature that significantly benefits the development of gold nanoparticles with controlled size;43 alternatively, their corresponding esterified products, i.e. poly(N-(2-methacryloyloxyethyl)-pyrrolidone)-b-poly(methyl methacrylate) (PNMP-b-PMMA), form organogels in isopropanol with thermally reversible transitions between 3D micellar networks and spherical micelles.44 In a recent study, Armes and coworkers have reported the interesting assembly behaviors of pyrrolidone-based amphiphilic diblock copolymers in n-dodecane.45 Recently, we have reported the detailed self-assembly behaviors of poly(N-(2-methacryloyloxyethyl)-pyrrolidone)-b-poly(lauryl methacrylate) (PNMP-b-PLMA) in n-dodecane,46 where aggregates with rich morphologies, including spherical micelles, worm-like nanoparticles, and vesicles, can be formed by slightly changing the molar ratio of the PNMP and the PLMA segments. In addition, we have noticed interesting self-assembly behaviors of them in water. This suggests the promising self-assembly of PNMP-b-PLMA in both water and n-dodecane, and similar behaviors have been rarely reported previously for amphiphiles in two distinct solvents simultaneously. The introduction of an additional AIE-active segment into these particular amphiphilic copolymers might significantly extend the application potential of these coploymers.
In this study, a new family of the AIE amphiphilic copolymers PNMPx-b-P(LMAy-co-TPEz), named poly(N-(2-methacryloyloxyethyl)pyrrolidone)-b-poly(lauryl methacrylate-co-1-ethenyl-4-(1,2,2-triphenylethenyl)benzene), was developed by the RAFT method. Their self-assembly behaviors in both water and n-dodecane were comprehensively studied by the dynamic light scattering (DLS), confocal laser scanning microscopy (CLSM) and transmission electron microscopy (TEM) techniques. Moreover, the structure–property relationship of PNMPx-b-P(LMAy-co-TPEz) was established. Because of the presence of the TPE segment, these AIE-active amphiphilic copolymers could act as luminescent probes and were applied in bioimaging using HeLa cells as substrates.
2 Experimental
2.1 Materials
2-Cyano-2-propyl dithiobenzoate (CPDB) was synthesized according to a previously reported procedure.47 2,2′-Azobisisobutyronitrile (AIBN, 99%, Shanghai HATECH Co. Ltd.) was recrystallized from methanol. Methacryloyl chloride (98%, Shanghai HATECH Co. Ltd.) was distilled under reduced pressure prior to use. Lauryl methacrylate (LMA, 99%, TCI) was passed through a column of silica gel to remove the inhibitor. All other chemicals were used as received.2.2 Synthesis of the PNMP-b-P(LMA-co-TPE) diblock copolymer
The synthetic route of the target diblock copolymer is shown in Scheme 1, and the procedure is similar to our previously reported procedure with slight modification.46 | ||
| Scheme 1 Synthetic route of PNMPx-b-P(LMAy-co-TPEz). | ||
The detailed synthetic procedure is described as follows by taking PNMP35 as an example. A 250 mL round-bottom flask was charged with NMP (50.0 g, 254 mmol), 2-cyano-2-propyl dithiobenzoate (CPDB, 1.52 g, 6.88 mmol), 2,2′-azobisisobutyronitrile (AIBN, 226 mg, 1.38 mmol, and the molar ratio of CPDB/AIBN = 5.0), and 52.5 mL chloroform. The sealed reaction vessel was purged with nitrogen for 30 min and placed in a preheated oil bath at 60 °C for 24 h. The resulting PNMP was purified by precipitation in excess ethyl ether. The degree of polymerization of the synthesized macro-CTA was calculated to be 35 using 1H NMR spectroscopy, in which the integrated aromatic proton signals of CPDB at 7–8 ppm to the two oxymethylene PNMP protons at 3.7–4.2 were employed.
The synthetic procedure of the PNMP35-b-P(LMA38-co-TPE4.7) copolymer is typically described as follows. LMA (3.60 g, 14.2 mmol), TPE (0.51 g, 1.42 mmol), the AIBN initiator (11.5 mg, 0.07 mmol), and PNMP35 macro-CTA (2.03 g, 0.28 mmol, and the molar ratio of macro-CTA/initiator = 4.0) were dissolved in 23.5 mL chloroform. The reaction mixture was sealed in a 50 mL round-bottom flask and purged with nitrogen gas for 30 min. The deoxygenated solution was then placed at 60 °C for 24 h. The resultant products were sequentially purified by dialysis against methanol and chloroform. The final sample was concentrated and dried in a vacuum oven at 25 °C for 12 h.
2.3 Preparation of PNMP-b-P(LMA-co-TPE) diblock copolymer assemblies
Self-assemblies in water were prepared by directly dissolving the copolymers (1 wt%) in water in an ice-water bath to achieve a 1 wt% aqueous solution. Similarly, self-assembles in alkane were prepared by directly dissolving the copolymers (1 wt%) in n-dodecane at 100 °C and then slowly cooling down the mixture to room temperature as previously reported.46 To fully integrate PNMP50-b-PLMA10 with the AIE copolymers, these two substances were dissolved in the mixed solvent of methanol and THF, and then, dialysis was carried out to replace the organic solvent with water.2.4 Characterization methods
2.5 Cytotoxicity measurements
The MTT (3-[4,5-dimethyl-2-thiazolyl]-2,5-diphenyl-2-H-tetrazolium bromide) assay was employed to evaluate the cytotoxicity of the complex against HeLa cells. At the density of 1 × 103 cells per well, the HeLa cells were placed in 96-well plates in 100 μL of DMEM (Dulbecco's modified Eagle medium). After 24 h, the previous medium was discarded, 200 μL of culture medium containing self-assemblies was added, and incubation was continued for 24 h in a cell incubator under the 5% CO2 and 37 °C condition. Then, 20 μL of MTT solution (5 mg mL−1) was put in a cell plate and continued to culture for 4 h; moreover, 150 μL of dimethyl sulfoxide was added to replace the culture medium. Finally, the absorbance was tested by a microplate reader (BioTek ELx800) at 490 nm.2.6 Cell-imaging experiments
At the density of 2 × 103 cells per well, HeLa cells were put in a cell culture dish with the DMEM including 10% fetal bovine serum (FBS) and 1% antibiotics and incubated in a cell incubator under the 5% CO2 and 37 °C condition. After culturing the cells for 24 h, the aggregates were cultured with the HeLa cells for 3 h and then washed three times with phosphate buffer saline (PBS). Images were obtained by CLSM at 400× magnification.3 Results and discussion
3.1 Self-assembly of PNMPx-b-P(LMAy-co-TPEz)
From the viewpoint of molecular structure, the PNMPx-b-P(LMAy-co-TPEz) homopolymers are a typical category of amphiphiles, in which the PNMP and PLMA segments are the hydrophilic and hydrophobic parts, respectively, as mentioned previously.46 Although the water-insoluble TPE moiety has only slight affinity to n-dodecane, the copolymers still exhibit excellent solubility in both water and n-dodecane by rationally adjusting the composition. In this study, five amphiphilic PNMPx-b-P(LMAy-co-TPEz) copolymers were developed, and the detailed information is provided in Table 1. The polymerization degree of PNMP for all copolymers was kept constant at 35, indicating the same hydrophilicity. Therefore, their hydrophobicity should mainly depend on the polymerization degree of P(LMAy-co-TPEz), in which the ratio of y/z was kept nearly constant at about 9. Accordingly, the solubility of PNMP35-b-P(LMAy-co-TPEz) in water was gradually weakened upon increasing the length of the P(LMAy-co-TPEz) segment. In contrast, the corresponding affinity to n-dodecane gradually increased from the copolymer No. 1 to No. 5.| Entry | Composition | PNMP | LMA | TPE | Mn (g mol−1) | Mw/Mn | |
|---|---|---|---|---|---|---|---|
| Wta | Conv. | Conv. | 1H NMR | GPC | GPC | ||
| a Weight fraction of the PNMP block in the block copolymer. | |||||||
| No. 1 | PNMP35-b-P(LMA9-co-TPE0.9) | 71% | 81% | 99% | 9700 | 6300 | 1.10 |
| No. 2 | PNMP35-b-P(LMA18-co-TPE1.9) | 56% | 86% | 99% | 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 400 |
8200 | 1.21 |
| No. 3 | PNMP35-b-P(LMA24-co-TPE2.7) | 50% | 76% | 99% | 13700 |
10200 |
1.13 |
| No. 4 | PNMP35-b-P(LMA38-co-TPE4.7) | 37% | 80% | 99% | 18500 |
13100 |
1.09 |
| No. 5 | PNMP35-b-P(LMA55-co-TPE6.3) | 30% | 83% | 99% | 23200 |
14500 |
1.07 |
 | ||
| Fig. 1 Optical and fluorescence images (a) of 1 wt% PNMP35-b-P(LMAy-co-TPEz) aqueous solution and the corresponding size distribution (b) of them. CLSM images of 1 wt% PNMP35-b-P(LMA18-co-TPE1.9) (c), PNMP35-b-P(LMA24-co-TPE2.7) (d) and PNMP35-b-P(LMA55-co-TPE6.3) (e) aqueous solution. Bars represent 25 μm. | ||
To have a better understanding of the micro-structural characteristics of the assemblies, TEM measurements were also performed. Generally, a spherical micelle-to-vesicle transition via worm-like micelles occurred in the 1 wt% PNMP35-b-P(LMAy-co-TPEz) aqueous solution upon increasing the length of the hydrophobic P(LMAy-co-TPEz) segment. Specifically, nearly spherical micelles of about 30–40 nm were formed in the PNMP35-b-P(LMA9-co-TPE0.9) aqueous solution, and similar aggregates were also observed in the PNMP35-b-P(LMA18-co-TPE1.9) (ESI Fig. S5a†) system. Instead, worm-like micelles and vesicles became the dominant morphologies for PNMP35-b-P(LMA24-co-TPE2.7) and PNMP35-b-P(LMA55-co-TPE6.3), as shown in Fig. 2b and c, respectively. Moreover, a binary mixture of worm-like micelles and vesicles was observed in PNMP35-b-P(LMA38-co-TPE4.7) (ESI Fig. S5b†). Thus, TEM observation clearly confirms the morphological transition from spherical micelles to vesicles, and the increased hydrophobic chain is the major reason for the occurrence of this transition.37
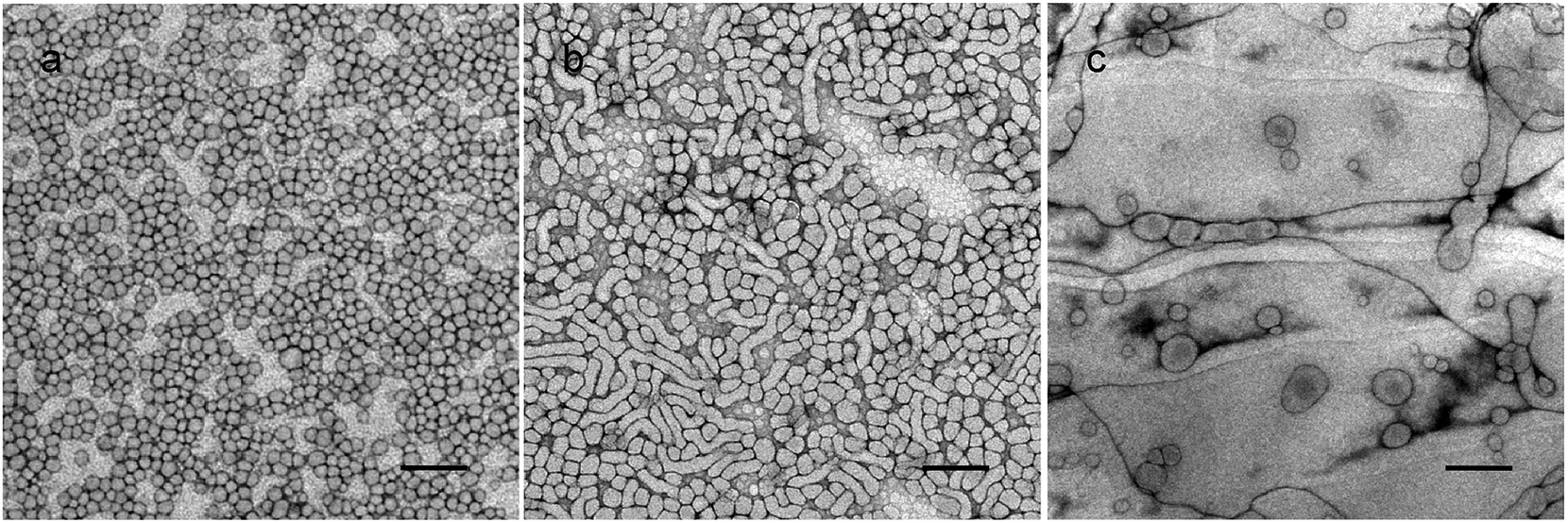 | ||
| Fig. 2 TEM images of 1 wt% PNMP35-b-P(LMA9-co-TPE0.9) (a), PNMP35-b-P(LMA24-co-TPE2.7) (b) and PNMP35-b-P(LMA55-co-TPE6.3) (c) aqueous solutions. Bars represent 200 nm. | ||
 | ||
| Fig. 3 Optical and fluorescence images (a) of 1 wt% PNMP35-b-P(LMAy-co-TPEz) in n-dodecane and the corresponding size distribution (b). CLSM images of 1 wt% PNMP35-b-P(LMA9-co-TPE0.9) (c), PNMP35-b-P(LMA18-co-TPE1.9) (d) and PNMP35-b-P(LMA55-co-TPE6.3) (e) in n-dodecane. Bars represent 10 μm. | ||
Fig. 3c–e show the typical CLSM images of PNMP35-b-P(LMA9-co-TPE0.9), PNMP35-b-P(LMA18-co-TPE1.9) and PNMP35-b-P(LMA55-co-TPE6.3) in n-dodecane, respectively, and distinguishable assemblies with different morphologies have been observed. Particularly, those of PNMP35-b-P(LMA18-co-TPE1.9) and PNMP35-b-P(LMA55-co-TPE6.3) seemed to be spherical dots dominantly, whereas that of PNMP35-b-P(LMA24-co-TPE2.7) seemed to be rod-like particles. Their corresponding TEM images (Fig. 4) showed that vesicles were formed in the PNMP35-b-P(LMA9-co-TPE0.9) n-dodecane dispersion. Alternatively, worm-like micelles and spherical micelles were formed in the PNMP35-b-P(LMA18-co-TPE1.9) and PNMP35-b-P(LMA55-co-TPE6.3) systems, respectively. In addition, the aggregate morphologies of PNMP35-b-P(LMA24-co-TPE2.7) and PNMP35-b-P(LMA38-co-TPE4.7) were confirmed to be worm-like micelles and spherical micelles, respectively, by the TEM observation (ESI Fig. S6†). Thus, it could be concluded safely that a vesicle-to-spherical micelle transition via worm-like micelles occurred upon increasing the length of the solvophilic P(LMA-co-TPE) segment.46
 | ||
| Fig. 4 TEM images of 1 wt% PNMP35-b-P(LMA9-co-TPE0.9) (a), PNMP35-b-P(LMA18-co-TPE1.9) (b), and PNMP35-b-P(LMA55-co-TPE6.3) (c) in n-dodecane. Bars represent 200 nm. | ||
3.2 Effect of self-assembly structure on AIE
One of the major specialties of PNMP35-b-P(LMAy-co-TPEz) copolymers is the introduced TPE moieties, which endow them with aggregation-induced emission in either water (Fig. 1a) or n-dodecane (Fig. 3a), as observed from the fluorescence images. To clarify the AIE effect, the representative fluorescence spectra of 1 wt% PNMP35-b-P(LMA18-co-TPE1.9) in water, n-dodecane and chloroform were examined, as shown in Fig. 5. Nearly no fluorescence characteristics could be observed from the chloroform solution. However, apparent fluorescence was observed in both the water and n-dodecane solutions. It was also noticed that the fluorescence intensity in n-dodecane was significantly stronger. This clearly suggested the aggregation-induced emission of PNMP35-b-P(LMA18-co-TPE1.9) in the selected solvents. Since chloroform is a good solvent for both the PNMP and the P(LMA-co-TPE) segments, PNMP35-b-P(LMA18-co-TPE1.9) is mainly present in its unimolecular form and shows a slight fluorescence. Instead, self-assemblies with different morphologies were formed in both water and n-dodecane, resulting in strong fluorescence. | ||
| Fig. 5 (a) Fluorescence emission spectra of PNMP35-b-P(LMA18-co-TPE1.9) in n-dodecane, water and chloroform; the inset images represent the corresponding fluorescence images. (b) Fluorescence quantum yield of PNMP35-b-P(LMAy-co-TPEz) in water and n-dodecane with the constant concentration of 20 μg mL−1 for the TPE moiety. | ||
Since there is a slight difference between the fluorescence responses of PNMP35-b-P(LMAy-co-TPEz) in water and n-dodecane, the fluorescence quantum yields (QY) of both solutions have been measured, as shown in Fig. 5b. Clearly, the value of QY significantly depended on the nature of the solvent for a specific copolymer, and significantly different variation tendency was observed for PNMP35-b-P(LMAy-co-TPEz) homologues in water and n-dodecane. In the aqueous solutions, all quantum yields of NMP35-b-P(LMA9-co-TPE0.9), PNMP35-b-P(LMA18-co-TPE1.9) and PNMP35-b-P(LMA24-co-TPE2.7) were small, whereas those of PNMP35-b-P(LMA38-co-TPE4.7) and PNMP35-b-P(LMA55-co-TPE6.3) were significantly enhanced. Generally, the higher the polymerization degree of TPE, the larger the QY. Alternatively, the effect of polymerization degree on QY became more complex due to which the quantum yields of PNMP35-b-P(LMAy-co-TPEz) in n-dodecane fluctuated in the region between about 3% and 9%. In addition, those with the averaged polymerization degree of TPE below 4.7 showed a larger QY in n-dodecane when compared with the case of water, whereas that of NMP35-b-P(LMA55-co-TPE6.3) became smaller.
In addition to the effect of copolymer chain configuration on the QY of PNMP35-b-P(LMAy-co-TPEz), QY should be mainly affected by the microstructural characteristics of the assemblies because of the constant concentration of the TPE moiety. Specifically, the morphology of the assemblies and the location of the TEP moieties in the assemblies were the major cause of the QY variation. In n-dodecane, the P(LMA-co-TPE) segment is the solvophilic part, which is solvated by n-dodecane regardless of the morphology of the assemblies. Accordingly, the effect of the assembly morphology on QY should be weakened dramatically because the micro-environment of the TPE moieties is kept nearly constant. However, the formation of large assemblies still favors the enhancement of QY. For example, large assemblies as vesicles were formed in PNMP35-b-P(LMA9-co-TPE0.9), whereas its QY remained weak. The extremely small polymerization degree of the TPE moieties was the major cause, where the aggregation of the TPE moieties within a molecule by π–π interactions through the variation of copolymer configuration became difficult because of the solvation of the P(LMA-co-TPE) segment. Alternatively, a relatively larger QY was obtained in PNMP35-b-P(LMA18-co-TPE1.9) by slightly increasing the polymerization degree of the TPE moieties. This was because the formation of long and entangled worm-like micelles benefitted the aggregation of the TPE moieties within molecules and assemblies. Although a further increase in the polymerization degree of the TPE moieties was favorable, the formation of small assemblies, such as spherical micelles, became a disadvantage for the enhancement of QY, i.e., PNMP35-b-P(LMA55-co-TPE6.3) had smaller QY when compared with PNMP35-b-P(LMA18-co-TPE1.9).
In the aqueous solution, the P(LMA-co-TPE) segment was always located in the hydrophobic core of the various assemblies regardless of the micelle or vesicle form. In other words, both the increased polymerization degree of TPE moieties and the formed assemblies of PNMP35-b-P(LMAy-co-TPEz) should benefit the enhancement of QY. On the one hand, the formation of assemblies favors the aggregation of the TPE moieties and thereby increases the QY for a specific copolymer, as shown in Fig. 5a. The larger the assemblies, the higher the QY. Moreover, the increased polymerization degree of the TPE moieties benefitted the self-aggregation of copolymer chains within molecules, which was also favorable for the enhancement of QY. For copolymers, including PNMP35-b-P(LMA9-co-TPE0.9), PNMP35-b-P(LMA18-co-TPE1.9) and PNMP35-b-P(LMA24-co-TPE2.7), with a relatively smaller polymerization degree of the TPE moieties, the QY values remained low because mainly small assemblies, such as spherical and rod-like micelles, were formed. In addition, a slight increase in the polymerization degree of the TPE moieties enhanced the QY. However, the QY values of PNMP35-b-P(LMA38-co-TPE4.7) and PNMP35-b-P(LMA55-co-TPE6.3) were increased dramatically due to the increased polymerization degree of the TPE moieties and the formation of large assemblies such as worm-like micelles and vesicles synergistically.
3.3 Cell imaging
As is well-known, one of the major advantages of pyrrolidone-containing chemicals is their excellent biocompatibility; therefore, they are widely applied in biological and drug delivery systems.48 Due to the apparent fluorescence characteristics of the PNMP35-b-P(LMAy-co-TPEz) copolymers based on the AIE, these copolymers might show promising potential in cell imaging. To weaken the biotoxicity of the TPE moiety in PNMP35-b-P(LMAy-co-TPEz) and match the requirement of bioimaging, the biocompatible (Fig. 6a) and micelle-forming block copolymer PNMP50-b-PLMA10 (ESI Fig. S7†) was introduced. Fig. 6 shows the cell viability of the PNMP35-b-P(LMAy-co-TPEz)/PNMP50-b-PLMA10 binary systems with HeLa cells as the objective substrate. Obviously, the biotoxicity (red columns in Fig. 6a) was enhanced upon increasing the content of PNMP35-b-P(LMA55-co-TPE6.3) at the constant polymer concentration of 100 μg mL−1, whereas over 85% cell viability was still retained when the content of PNMP35-b-P(LMA55-co-TPE6.3) was increased to 40 wt%. Moreover, the cell viability varied slightly around 90% upon increasing the total copolymer concentration from 10, 25, 50, 75 to 100 μg mL−1 with the constant PNMP35-b-P(LMA55-co-TPE6.3) content of 40 wt%, as shown by the blue to red columns. In addition, the cell viability was highly dependent on the AIE copolymers, and the increased polymerization degree of the TPE moieties was disadvantageous to the biocompatibility of these copolymers under the same conditions as shown in Fig. 6b. Generally, the larger the TPE concentration, the higher the biotoxicity. | ||
| Fig. 6 Cell viability of the PNMP35-b-P(LMAy-co-TPEz)/PNMP50-b-PLMA10 binary system with HeLa cells. (a) The effect of PNMP35-b-P(LMA55-co-TPE6.3) content on the cell viability with a constant concentration of 100 μg mL−1, and the effect of polymer concentration on the cell viability at the constant content of 40 wt% of PNMP35-b-P(LMA55-co-TPE6.3). (b) The effect of copolymer structure on the cell viability with the constant concentration of 100 μg mL−1 and constant content of 40 wt% of PNMP35-b-P(LMAy-co-TPEz). | ||
Moreover, three AIE copolymers i.e. PNMP35-b-P(LMA24-co-TPE2.7), PNMP35-b-P(LMA38-co-TPE4.7) and PNMP35-b-P(LMA55-co-TPE6.3) were employed in cell imaging, and micelles were commonly formed in the corresponding PNMP35-b-P(LMAy-co-TPEz)/PNMP50-b-PLMA10 binary aqueous solutions with the constant PNMP35-b-P(LMAy-co-TPEz) content of 40 wt% (ESI Fig. S8†). To ensure sufficient fluorescence and biocompatibility, the total copolymer concentration of 50 μg mL−1 was used. Fig. 7 shows the corresponding CLSM images of the HeLa cells in the presence of different AIE copolymers. Obviously, each cell could be distinguished clearly. However, the cells treated by PNMP35-b-P(LMA24-co-TPE2.7) and PNMP35-b-P(LMA38-co-TPE4.7) had much higher resolution than those treated by PNMP35-b-P(LMA55-co-TPE6.3). The relatively higher biotoxicity of PNMP35-b-P(LMA55-co-TPE6.3) (Fig. 6b) might be the major cause. Moreover, the microstructural characteristics of the cells shown in Fig. 7a and b were clearly presented. This evidently confirms that these AIE copolymers as probes clearly meet the requirements of bioimaging.
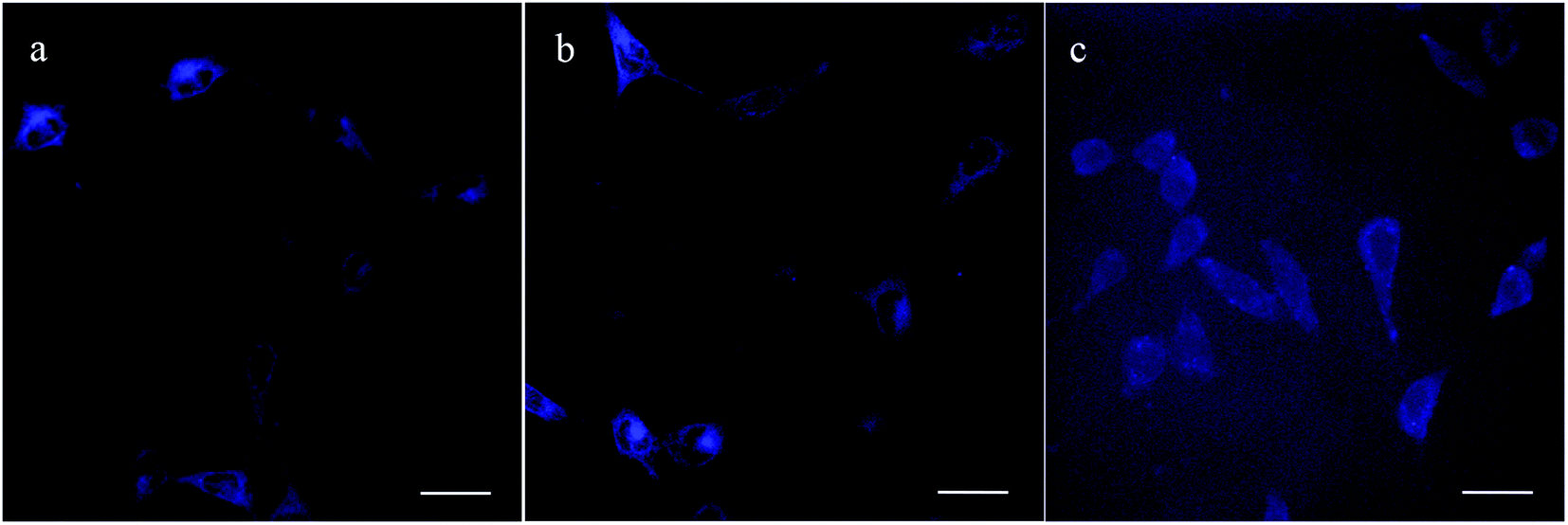 | ||
| Fig. 7 CLSM images of the HeLa cells treated with PNMP35-b-P(LMA24-co-TPE2.7) (a), PNMP35-b-P(LMA38-co-TPE4.7) (b), and PNMP35-b-P(LMA55-co-TPE6.3) (c). Bars represent 25 μm. | ||
4 Conclusion
In summary, in this study, a series of AIE amphiphilic copolymers, namely poly(N-(2-methacryloyloxyethyl)pyrrolidone)-b-poly(lauryl methacrylate-co-1-ethenyl-4-(1,2,2-triphenylethenyl)benzene), PNMPx-b-P(LMAy-co-TPEz), was developed by the RAFT polymerization method. Their corresponding self-assembly behaviors in both water and n-dodecane were studied systematically by employing the DLS, CLSM and TEM techniques. Accordingly, their structure–property relationships were illustrated well. Generally, an increase in the length of the P(LMAy-co-TPEz) segment with the ratio of y/z being around 9 enhanced the hydrophobicity of copolymers and thereby resulted in spherical micelle-to-vesicle transition via worm-like micelles in the aqueous solution. Instead, the reversed morphological transition from vesicles to spherical micelles via worm-like micelles occurred in n-dodecane because of the increased solvophilicity.46 It was also noticed that the polarity of the solvent and the morphology of the assemblies significantly affected the apparent fluorescence quantum yields of PNMP35-b-P(LMAy-co-TPEz), especially in the case of the aqueous solution. Although the introduced TPE moieties showed particular biotoxicity, the AIE-active amphiphilic copolymers could be potentially employed as excellent and highly efficient bioimaging probes under suitable conditions.7Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC 21573164 and 21773174). The authors thank Prof. Yi Liu and Fenglei Jiang of Wuhan University for the measurements of cell imaging.References
- B. Liu, X. He, C. Xu, L. Xu, S. Ai, S. Cheng and R. Zhuo, Biomacromolecules, 2018, 19, 2957–2968 CrossRef CAS.
- C. Liu, F. Xiong, H. Jia, X. Wang, H. Cheng, Y. Sun, X. Zhang, R. Zhuo and J. Feng, Biomacromolecules, 2013, 14, 358–366 CrossRef CAS.
- M. Gong, J. Wu, B. Chen, R. Zhuo and S. Cheng, Langmuir, 2015, 31, 5115–5122 CrossRef CAS.
- J. Li, Y. Cheng, C. Zhang, H. Cheng, J. Feng, R. Zhuo, X. Zeng and X. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 5287–5295 CrossRef CAS.
- Y. Li, H. Cheng, Z. Zhang, C. Wang, J. Zhu, Y. Liang, K. Zhang, S. Cheng, X. Zhang and R. Zhuo, ACS Nano, 2008, 2, 125–133 CrossRef CAS.
- G. Luo, X. Xu, J. Zhang, J. Yang, Y. Gong, Q. Lei, H. Jia, C. Li, R. Zhuo and X. Zhang, ACS Appl. Mater. Interfaces, 2012, 4, 5317–5324 CrossRef CAS.
- Y. Zhao, Y. Wu, S. Chen, H. Deng and X. Zhu, Macromolecules, 2018, 51, 5234–5244 CrossRef CAS.
- M. Huang, R. Yu, K. Xu, S. Ye, S. Kuang, X. Zhu and Y. Wan, Chem. Sci., 2016, 7, 4485–4491 RSC.
- T. Jadhav, B. Dhokale, Y. Patil and R. Misra, RSC Adv., 2015, 5, 68187–68191 RSC.
- T. Jadhav, B. Dhokale, S. M. Mobin and R. Misra, J. Mater. Chem. C, 2015, 3, 9981–9988 RSC.
- L. Viglianti, N. L. C. Leung, N. Xie, X. Gu, H. H. Y. Sung, Q. Miao, I. D. Williams, E. Licandro and B. Tang, Chem. Sci., 2017, 8, 2629–2639 RSC.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Tang, Chem. Commun., 2001, 1740–1741 RSC.
- J. Chen, C. C. W. Law, J. W. Y. Lam, Y. Dong, S. M. F. Lo, I. D. Williams, D. Zhu and B. Tang, Chem. Mater., 2003, 15, 1535–1546 CrossRef CAS.
- X. Fan, J. Sun, F. Wang, Z. Chu, P. Wang, Y. Dong, R. Hu, B. Tang and D. Zou, Chem. Commun., 2008, 2989–2991 RSC.
- S. Li, Q. Wang, Y. Qian, S. Wang, Y. Li and G. Yang, J. Phys. Chem. A, 2007, 111, 11793–11800 CrossRef CAS.
- Y. Hong, J. W. Y. Lam and B. Tang, Chem. Commun., 2009, 4332–4353 RSC.
- J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Tang, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS.
- Q. Peng, Y. Yi, Z. Shuai and J. Shao, J. Am. Chem. Soc., 2007, 129, 9333–9339 CrossRef CAS.
- T. Wu, J. Huang and Y. Yan, Chem.–Asian J., 2019, 14, 730–750 CrossRef CAS.
- M. Huo, Q. Ye, H. Che, X. Wang, Y. Wei and J. Yuan, Macromolecules, 2017, 50, 1126–1133 CrossRef CAS.
- Z. Huang, X. Zhang, X. Zhang, S. Wang, B. Yang, K. Wang, J. Yuan, L. Tao and Y. Wei, RSC Adv., 2015, 5, 89472–89477 RSC.
- Y. Zhao, Y. Wu, G. Yan and K. Zhang, RSC Adv., 2014, 4, 51194–51200 RSC.
- Q. Zhou, C. Fan, C. Li, Y. Wang, Z. Chen, Q. Yu and M. Zhu, Mater. Horiz., 2018, 5, 474–479 RSC.
- N. Zhang, H. Chen, Y. Fan, L. Zhou, S. Trépout, J. Guo and M. Li, ACS Nano, 2018, 12, 4025–4035 CrossRef CAS.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- K. Haraguchi, K. Kubota, T. Takada and S. Mahara, Biomacromolecules, 2014, 15, 1992–2003 CrossRef CAS.
- A. Bousquet, E. Ibarboure, E. Papon, C. Labrugère and J. Rodríguez-Hernández, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1952–1961 CrossRef CAS.
- Z. Liu, Y. Huang, X. Zhang, X. Tu, M. Wang, L. Ma, B. Wang, J. He, P. Ni and H. Wei, Macromolecules, 2018, 51, 7672–7679 CrossRef CAS.
- Z. Wang, M. C. M. Oers, F. P. J. T. Rutjes and J. C. M. Hest, Angew. Chem., 2012, 124, 10904–10908 CrossRef.
- P. Gu, C. Lu, F. Ye, J. Ge, Q. Xu, Z. Hu, N. Li and J. Lu, Chem. Commun., 2012, 48, 10234–10236 RSC.
- X. Shen, Y. Shi, B. Peng, K. Li, J. Xiang, G. Zhang, Z. Liu, Y. Chen and D. Zhang, Macromol. Biosci., 2012, 12, 1583–1590 CrossRef CAS.
- X. Zhang, X. Zhang, B. Yang, M. Liu, W. Liu, Y. Chen and Y. Wei, Polym. Chem., 2014, 5, 356–360 RSC.
- X. Zhang, X. Zhang, B. Yang, J. Hui, M. Liu, Z. Chi, S. Liu, J. Xu and Y. Wei, Polym. Chem., 2014, 5, 683–688 RSC.
- X. Zhao, X. Fan, X. Chen, C. Chai and Q. Zhou, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4656–4667 CrossRef CAS.
- Y. Miura and M. Okada, Polymer, 2004, 45, 6539–6546 CrossRef CAS.
- J. Zhong, H. Luo, Q. Tang, Z. Lei and Z. Tong, Macromol. Chem. Phys., 2019, 220, 1800554 CrossRef.
- H. Luo, Q. Tang, J. Zhong, Z. Lei, J. Zhou and Z. Tong, Macromol. Chem. Phys., 2019, 220, 1800508 CrossRef.
- M. Semsarilar, N. J. W. Penfold, E. R. Jones and S. P. Armes, Polym. Chem., 2015, 6, 1751–1757 RSC.
- D. Zehm, L. P. D. Ratcliffe and S. P. Armes, Macromolecules, 2013, 46, 128–139 CrossRef CAS.
- V. Ladmiral, A. Charlot, M. Semsarilar and S. P. Armes, Polym. Chem., 2015, 6, 1805–1816 RSC.
- Y. Pei, K. Jarrett, L. G. Garces, M. Saunders, J. Croue, P. J. Roth, C. E. Buckley and A. B. Lowe, RSC Adv., 2016, 6, 28130–28139 RSC.
- M. J. Derry, L. A. Fielding and S. P. Armes, Polym. Chem., 2015, 6, 3054–3062 RSC.
- J. Zhang, S. Cheng, X. Li and J. Dong, Acta Phys.-Chim. Sin., 2016, 32, 2018–2026 CAS.
- S. Cheng, Y. Xue, Y. Lu, X. Li and J. Dong, ACS Omega, 2017, 2, 105–112 CrossRef CAS.
- V. J. Cunningham, S. P. Armes and O. M. Musa, Polym. Chem., 2016, 7, 1882–1891 RSC.
- X. He, X. Li and J. Dong, Colloids Surf., A, 2019, 577, 493–499 CrossRef CAS.
- M. Benaglia, E. Rizzardo, A. Alberti and M. Guerra, Macromolecules, 2005, 38, 3129–3140 CrossRef CAS.
- J. Sun, Y. Peng, Y. Chen, Y. Liu, J. Deng, L. Lu and Y. Cai, Macromolecules, 2010, 43, 4041–4049 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra05997k |
| This journal is © The Royal Society of Chemistry 2019 |