DOI:
10.1039/C9RA05390E
(Paper)
RSC Adv., 2019,
9, 28112-28118
Influence of coordinating groups of organotin compounds on the Fries rearrangement of diphenyl carbonate†
Received
14th July 2019
, Accepted 29th August 2019
First published on 6th September 2019
Abstract
In this paper, the Fries rearrangement of diphenyl carbonate (DPC) catalyzed by organotin compounds with different coordination groups was studied for the first time. The electronic effect and steric hindrance of the coordinating groups were discussed with respect to the reactivity of DPC rearrangement. The results showed that both the electronic effect and steric hindrance of the coordinating groups influenced the acidity of the active tin centers and then affected the catalytic performance of organotin as a Lewis acid for the rearrangement of DPC, and the influence of the electronic effect is greater than that of steric hindrance. The catalytic activity is in the order of BuSnO(OH) > Bu2SnO > Bu2Sn(OCOC11H23)2 > BuSnCl3 > Bu3SnOSnBu3 > Bu3SnCl, and Bu2SnO showed the best catalytic activity due to its strong electron absorption effect, small steric hindrance, and good stability. Under the optimum reaction conditions, the conversion of DPC was up to 93%, and the yields of phenyl salicylate (PS) and xanthone (XA) were 62% and 28%, respectively. In addition, a reaction mechanism of DPC rearrangement catalyzed by the organotin compounds was speculated. This research can provide vigorous theoretical data support to control the byproducts produced by DPC rearrangement in the process of DPC synthesis. It also provides a new route for the preparation of PS and XA.
1 Introduction
With low toxicity and no pollution to the environment, diphenyl carbonate (DPC) is an important “green” chemical intermediate, which is commonly used to synthesize many important organic compounds and polymer materials in fields such as medicine, pesticides and plastic industries. Particularly, it is used as the main raw material for the synthesis of polycarbonate (PC) by non-phosgene processes.1,2 Among the non-phosgene processes for DPC synthesis, transesterification of dimethyl carbonate with phenol to synthesize DPC is the only non-phosgene synthetic route with industrial application prospect at present.3,4 In recent years, researchers have conducted in-depth studies on catalysts and process conditions of this synthetic approach.5–11 However, rare studies on its byproducts (except anisole12) were reported in our previous work.13 While DPC as the key raw material of non-phosgene PC, trace impurities will also seriously affect the quality of PC.14,15
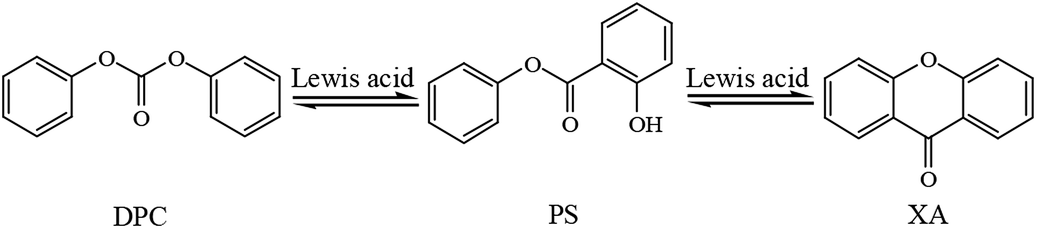
Fries rearrangement is a reaction where, when a phenolic ester is in a solvent such as nitrobenzene and nitromethane or when the phenolic ester is directly heated with the absence of solvents, the acyl group is rearranged under the catalysis of Lewis acids to produce ortho- or para-acyl phenols.16,17 Fries rearrangement has been extensively used in the synthesis of organic pharmaceuticals and gradually become a research hotspot in recent years.18–22 In the synthesis of DPC, organotin, organotitanium and other Lewis acids are usually used as catalysts, and the pilot-scale amplification reaction is carried out under high temperature and high pressure. As a phenolic ester, DPC was prone to Fries rearrangement in the condition of Lewis acid catalysts under high temperatures and pressures, which leads to the decrease of the yield and purity of DPC. As shown in Scheme 1, Fries rearrangement in DPC may produce byproducts such as phenyl salicylate (PS) and xanthone (XA). In our research group, the catalysts and process conditions in transesterification synthesis of DPC had been studied for a long time.23–27 Such byproducts as PS and XA have also been discovered in the product of the pilot-scale reaction. Based on the molecular structures of PS and XA, the transesterification catalysts and process conditions, PS and XA should be the Fries rearrangement products of DPC. The boiling points of both PS and XA are high and approximate to that of DPC, so they cannot be easily isolated during purification of DPC and are very prone to remain in DPC to affect purity of the transesterification product DPC and thus seriously affect the molecular weight and performance of PC synthesized in the next step.28–34 Therefore, in this paper, organotin were used as catalysts to study the effect of coordination groups on the reactivity of Fries rearrangement of DPC for the first time, and the possible reaction mechanism was proposed in the process. The study can provide an industrial reference for controlling the byproducts originated from DPC rearrangement in the DPC synthesis. At the same time, PS and XA have antiseptic and bactericidal effects in the intestine. They are often used as ultraviolet absorbents, plasticizers, preservatives, and stabilizers for antibacterial disinfectants and plastic products, as well as for the synthesis of bactericidal drugs and essences.35–38 So this study can also provide a method reference for the preparation of PS and XA.
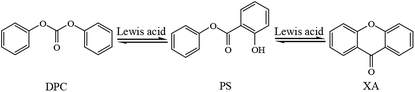 |
| | Scheme 1 Fries rearrangement of DPC to produce PS and XA. | |
2 Experimental
2.1 Instruments and reagents
A GS-0.1 intermittent high-pressure reactor (Weihai Chemical Machinery Co., Ltd); a 7820A gas chromatograph, equipped with a HP-5 column (30 m × 0.32 mm × 0.25 μm, 5% phenyl methyl siloxane) (Agilent Technologies, USA); an Ascend 400 NMR (Bruker); a Nicolet IS 10 FT-IR (Thermo Scientific); and a HP 6890/5973 GC/MS system (Agilent Technologies, USA), equipped with a HP-5 MS column (30 m × 0.32 mm × 0.25 μm) (Agilent Technologies, USA) were used in this study. PS (GC) was purchased from Alfa Aesar; XA (GC) was purchased from J&K Scientific Ltd.; DPC (GC) was purchased from Nanjing Duly Biotech Co., Ltd.; phenol (GC) was purchased from Aladdin; acetone (HPLC) was purchased from Scientific; Bu2SnO (AR) was obtained from Acros; butyl tin hydroxide [BuSnO(OH)] (AR) and tributyltin chloride (Bu3SnCl) (AR) were obtained from Merck; bis(tributyl)tin oxide (Bu3SnOSnBu3) (AR), dibutyltin dilaurate [Bu2Sn(OCOC11H23)2] (AR) and butyl tin trichloride (BuSnCl3) (AR) were obtained from Alfa Aesar. Prior to use, all solid reagents were dried in vacuum at 70 °C in a vacuum drier, and all liquid reagents were dried using a 4 Å molecular sieve.
2.2 Fries rearrangement of DPC
Fries rearrangement of DPC was carried out in a 100 mL high-pressure reactor provided with a thermometer, a nitrogen inlet, and a stirrer. DPC and organotin catalyst were added (the mass ratio of Sn and DPC is 0.05–15%, and the optimum ratio is 10%) into the reactor, and then air in the assembly was replaced with high-purity nitrogen at room temperature. Following pressurized to the required pressure (0.10–0.70 MPa), and heated to the desired temperature (140–280 °C) under stirring to start timing. After the reaction (2–14 h), the product was dissolved and diluted in acetone, qualitatively analyzed on a GC/MS system and quantitatively analyzed by external standard method on a gas chromatograph equipped with a hydrogen flame ionization detector. Analytical conditions of gas chromatography were referred to our previous work:13 the makeup nitrogen flow rate was 25 mL min−1. The air flow rate was 400 mL min−1. The hydrogen flow rate was 30 mL min−1. The column flow rate was 2.5 mL min−1. The injection volume was 0.4 μL. Samples were injected in a split mode with a split ratio of 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The injection port temperature was 280 °C. The detector temperature was 300 °C. Programmed temperature increase was performed: the initial temperature was 120 °C, and the temperature was increased at a rate of 20 °C min−1 to 280 °C and held at 280 °C for 5 min.
:1. The injection port temperature was 280 °C. The detector temperature was 300 °C. Programmed temperature increase was performed: the initial temperature was 120 °C, and the temperature was increased at a rate of 20 °C min−1 to 280 °C and held at 280 °C for 5 min.
3 Results and discussion
3.1 Catalytic performance of organotin compounds for the rearrangement of DPC
As shown in Scheme 1, DPC can generate two-step rearrangement in sequence. Fries rearrangement of DPC occurs to produce PS firstly, and then PS is rearranged to generate XA, that is, the PS is the intermediate of DPC rearrangement. Fries rearrangement reaction was usually catalyzed by Lewis acids. In this study, organotin compounds with different coordinating groups, including Bu2SnO, Bu2Sn(OCOC11H23)2, Bu3SnOSnBu3, BuSnO(OH), Bu3SnCl and BuSnCl3, were selected as catalysts for the DPC rearrangement. As shown in Fig. S1 and S2,† 1H and 13C NMR spectra of all the products revealed that DPC rearrangement did generate PS and XA. Moreover, as shown in Fig. S3,† the results of the products analysis and matching analysis of their fragment ion peaks by GC-MS also further confirmed that PS and XA produced by DPC rearrangement. Catalytic performance of organotin compounds for the DPC rearrangement reaction is presented in Table 1. The amounts of organotin compounds are calculated on the basis of tin atoms, and the mass ratio of Sn to DPC is 1:100. As shown in Table 1, with the absence of catalysts, DPC rearrangement was not observed, but a small quantity of phenol was identified. All organotin compounds can catalyze the Fries rearrangement reaction of DPC, and the conversion of DPC was all over 40%. The main products were PS and XA, and the yield of PS is higher than XA. In addition, a small quantity of phenol was also produced. The origin of phenol was analyzed by online thermal desorption-gas chromatography. No phenol was detected from DPC, indicating phenol was not a product of thermal degradation or thermal rearrangement of DPC but possibly a product of DPC hydrogenolysis, and the amount of phenol was independent of the catalyst. The catalytic activity of organotin is in the order of BuSnO(OH) > Bu2SnO > Bu2Sn(OCOC11H23)2 > BuSnCl3 > Bu3SnOSnBu3 > Bu3SnCl.
Table 1 Catalytic performance of organotin compounds for the Fries rearrangement of DPCa
| Catalysts |
DPC conversion (%) |
Yield (%) |
| PS |
XA |
Phenol |
| Reaction conditions: DPC: 15 g; mass ratio of Sn to DPC: 1%; reaction time: 8 h; temperature: 220 °C; pressure: 0.50 MPa. |
| — |
7 |
— |
— |
6 |
| BuSnO(OH) |
92 |
53 |
31 |
7 |
| Bu2SnO |
88 |
51 |
30 |
7 |
| Bu2Sn(OCOC11H23)2 |
82 |
49 |
25 |
7 |
| BuSnCl3 |
73 |
45 |
24 |
4 |
| Bu3SnOSnBu3 |
57 |
35 |
17 |
6 |
| Bu3SnCl |
41 |
26 |
12 |
3 |
When organotin compounds are used as Lewis acid to catalyze the Fries rearrangement of DPC, tin atoms play the role of active centers. The greater the steric hindrance of the groups coordinating with tin atoms is, the more coordinating chelation of tin atoms with the carbonyl oxygen atoms in DPC is disfavored, and thus the more unfavorable the rearrangement reaction is. Additionally, the electronic effect of coordinating groups will affect the acidity of tin atoms, thereby exerting influence on the catalytic performance. As an active center, each tin atom in BuSnO(OH) or Bu2SnO has three coordinating groups, among which two groups are identical (i.e. butyl group and oxygen) and the third groups are hydroxyl group and butyl group respectively in the two compounds. Since the steric hindrance of hydroxyl groups is slightly weaker than that of butyl groups while the electron-withdrawing ability of hydroxyl groups is stronger than that of butyl groups, the acidity of tin atoms in BuSnO(OH) is stronger than that in Bu2SnO; therefore, BuSnO(OH) is more catalytically active than Bu2SnO. Coordinating atoms connecting tin atoms in Bu2Sn(OCOC11H23)2 and Bu2SnO are identical, but the steric hindrance of groups coordinating with tin atoms is greater in Bu2Sn(OCOC11H23)2 than in Bu2SnO, so Bu2Sn(OCOC11H23)2 is not as active as Bu2SnO. The steric hindrance of groups coordinating with tin atoms in Bu2Sn(OCOC11H23)2 is far greater than in BuSnCl3. However, from the point of view of electronic effect, groups coordinating with tin atoms in Bu2Sn(OCOC11H23)2 are oxygen and carbon atoms, and each tin atom in Bu2Sn(OCOC11H23)2 is coordinated by two oxygen atoms and two carbon atoms, whereas each tin atom in BuSnCl3 is coordinated by three chlorine atoms and one butyl group; as the electronegativity of oxygen atoms is stronger than that of chlorine atoms, the total electron-withdrawing effect of Bu2Sn(OCOC11H23)2 is stronger than that of BuSnCl3. Therefore, the Lewis activity of tin atoms in Bu2Sn(OCOC11H23)2 is stronger than that in BuSnCl3, and Bu2Sn(OCOC11H23)2 is more catalytically active than BuSnCl3. The analytical results indicate the electronic effect of coordinating groups plays a more important role than steric hindrance in deciding the activity of organotin catalysts, which has contributed to the pronounced activity of Bu2Sn(OCOC11H23)2 so that, with it as the catalyst, the yield of the DPC rearrangement product is only slightly lower than that with BuSnO(OH) and Bu2SnO having relatively weaker steric hindrance. Regarding BuSnCl3 and Bu3SnOSnBu3, though the electronegativity of chlorine atoms coordinating with tin atoms in BuSnCl3 is weaker than that of oxygen atoms in Bu3SnOSnBu3, there are three chlorine atoms coordinating with each tin atom in BuSnCl3 but only 0.5 oxygen atom coordinating with each oxygen atom in Bu3SnOSnBu3, so the total electronegativity of atoms coordinating with tin atoms as active centers in BuSnCl3 is still greater than that in Bu3SnOSnBu3; besides, the steric hindrance of groups coordinating with tin atoms in BuSnCl3 is much weaker than that in Bu3SnOSnBu3. Therefore, the Lewis activity of tin atoms as active centers in BuSnCl3 is stronger than that in Bu3SnOSnBu3 so that it has greater catalytic activity than Bu3SnOSnBu3. Though the steric hindrance of Bu3SnCl is slightly greater than that of Bu3SnOSnBu3, the electronegativity of oxygen atoms coordinating with tin atoms in Bu3SnOSnBu3 is stronger than that of chlorine atoms coordinating with tin atoms in Bu3SnCl, and the number of tin atoms at the catalytic active centers in Bu3SnOSnBu3 is larger than that in Bu3SnCl. Therefore, the Lewis activity of tin atoms as active centers in Bu3SnOSnBu3 is stronger than that in Bu3SnCl so that it is more catalytically active than Bu3SnCl. This also verifies that the electronic effect of coordinating groups is stronger than steric hindrance with respect to the impact on the catalytic activity of organotin compounds.
It thus can be seen from the above results that the catalytic activity of organotin compounds for DPC rearrangement is affected by the steric hindrance and electronic effect of groups coordinating with tin atoms. The smaller the steric hindrance of coordinating groups is, the more favorable for the tin atom to contact with the carbonyl oxygen on DPC is. The greater the electronegativity of coordinating atoms of tin atoms is, the stronger the electron absorption effect is, the stronger the activity of tin atoms is, and the greater the catalytic activity for DPC rearrangement is. The impact of the electronic effect of coordinating groups is greater than that of steric hindrance. The results are in accordance with previously reported on organotin compounds.39 This is also the reason why the steric hindrance of Bu3SnCl is small but its catalytic activity is the lowest among the series of organotin compounds. Both BuSnO(OH) and Bu2SnO have demonstrated good catalytic performance in the rearrangement of DPC. The DPC conversion could reach over 85%, and the total yield of PS and XA could reach over 80%. However, BuSnO(OH) is unstable and easily degradable.40,41 Therefore, Bu2SnO is a better catalyst for the Fries rearrangement of DPC.
3.2 Effect of the amount of Bu2SnO on the rearrangement of DPC
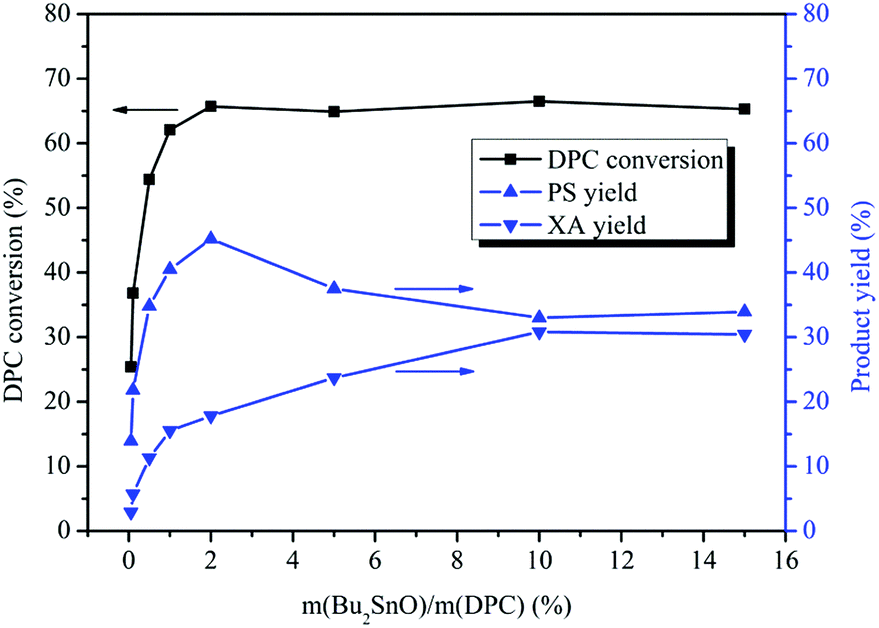
The effect of the amount of Bu2SnO on the rearrangement of DPC was investigated. The amount of the catalyst was based on the mass ratio of Bu2SnO to DPC. As shown in Fig. 1, the DPC conversion increased gradually with the increase of catalyst dosage and reached its peak (66%) when the amount of the catalyst was 2%. However, the DPC conversion did not change after 2%. The PS yield first increased and then decreased with increases in the amount of the catalyst, reaching its peak (45%) when the amount of the catalyst was 2% and beginning to decrease with further increases in the amount of the catalyst due to rearrangement of PS into XA. The XA yield showed a continually rising trend with the increase of the amount of the catalyst and reached its peak (28%) when the amount of the catalyst was 10%. However, the XA yield kept unchanged with further increases in the amount of the catalyst. This is because the generated PS will be further rearranged and converted into XA. Additionally, with increases in the amount of the catalyst, the number of acids in the catalytic system will increase, which will further promote rearrangement of PS into XA. When the amount of the catalyst is more than 10%, yields of PS and XA will keep unchanged with further increases in the amount of the catalyst as the PS rearrangement reaction to produce XA has reached equilibrium. Overall, when the amount of the catalyst is 0.05%, a small quantity of DPC can be converted into PS and XA. When the amount of the catalyst is between 0.05% and 2%, the yields of PS and XA will rise continually with increases in the amount of the catalyst; when the amount of the catalyst is between 2% and 10%, increases in the amount of the catalyst will favor further rearrangement of PS and conversion into XA. When the amount of the catalyst is 10%, the tandem rearrangement reaction of DPC will reach equilibrium. The more acidic the catalyst, the more complete the rearrangement. The result is consistent with those reported in literature.42,43
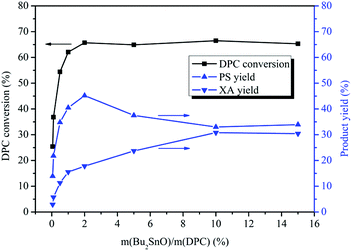 |
| | Fig. 1 Effect of the amount of Bu2SnO on the Fries rearrangement of DPC. Reaction conditions: DPC: 15 g; reaction time: 8 h; temperature: 200 °C; pressure: 0.40 MPa. | |
3.3 Effect of reaction time on the rearrangement of DPC
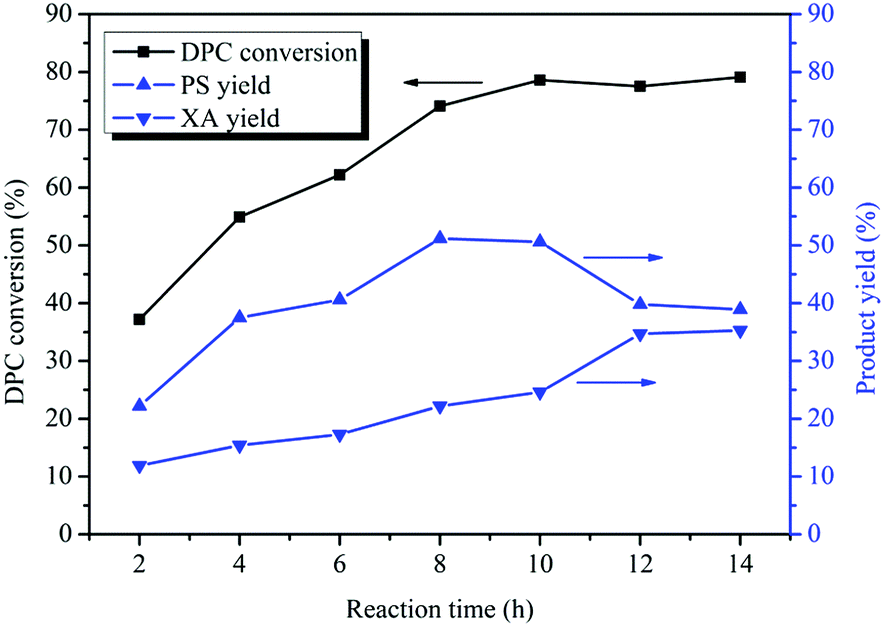
The Fries rearrangement of DPC consists of two sequential rearrangements. So the reaction time has a great influence on the distribution of rearrangement products. Fig. 2 illustrates the effect of the reaction time on Bu2SnO-catalyzed rearrangement of DPC. With the prolongation of the reaction time, the DPC conversion increased gradually. When the reaction time was 10 h, the DPC conversion reached its peak (79%). The DPC conversion did not change significantly after 10 h. The yield of PS increased gradually with the prolongation of the reaction time. When the reaction time was 2 h, the PS yield was 22%; when the reaction time was 8 h, the PS yield reached its peak (51%). The PS yield began to decrease after 10 h. The PS yield kept unchanged at 51% after 12 h. The XA yield increased gradually with the prolongation of the reaction time. When the reaction time was 2 h, the XA yield was 11% and reached its peak (35%) at 12 h. This is mainly because the first step rearrangement is synchronized with the second step rearrangement. PS converted to XA slowly before 10 h, while after 10 h, the first step was no longer occurred, and only the second step went on, so PS converted to XA quickly. After 12 h, the yield of neither PS nor XA showed significant change. Because the rearrangement reaction reached equilibrium. Between 2 h and 12 h, the longer the reaction time is, the more the tandem rearrangement of DPC is favored. In addition, the PS yield was more than XA in the whole reaction, indicating that the rearrangement in the first step was more likely to occur than that in the second step.
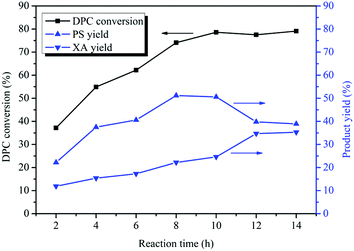 |
| | Fig. 2 Effect of the reaction time on the Fries rearrangement of DPC. Reaction conditions: DPC: 15 g; mass ratio of Bu2SnO to DPC: 2%; temperature: 200 °C; pressure: 0.40 MPa. | |
3.4 Effect of reaction temperature on the rearrangement of DPC
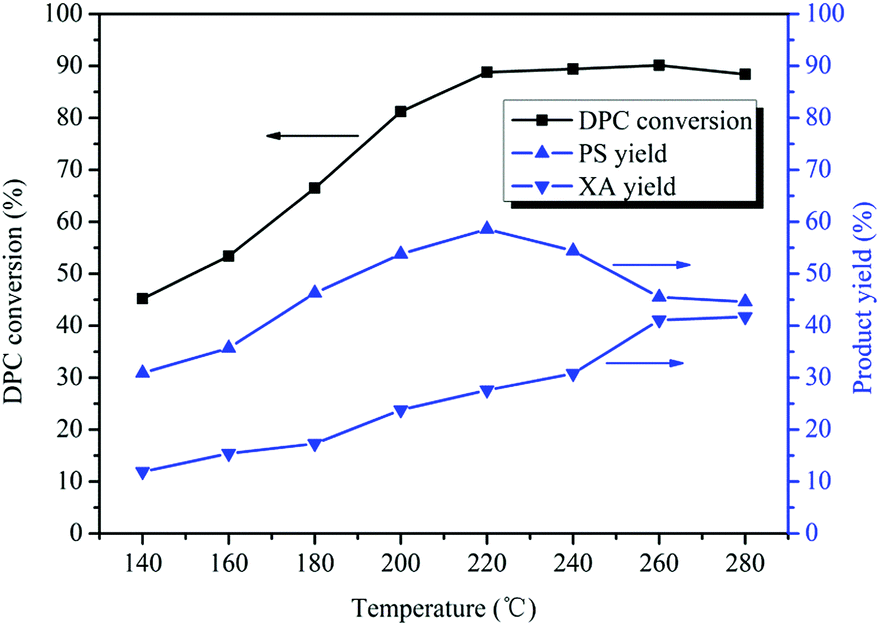
Normally, high temperatures favor Fries rearrangement reactions.16,17 Therefore, the effect of the reaction temperature on Bu2SnO-catalyzed rearrangement of DPC was investigated with temperatures ranging between 140 and 280 °C. As can be known from Fig. 3, the DPC conversion gradually increased with rises in the reaction temperature. When the reaction temperature was 140 °C, the XA yield was 11.0% and reached its peak (89%) at 220 °C. However, the DPC conversion kept unchanged with further rises in the reaction temperature. The PS yield first increased and then decreased with rises in the reaction temperature, reaching its peak (59%) when the temperature was 220 °C and gradually decreasing with further temperature rises. Fries rearrangement of DPC is an endothermic reaction. After the reaction temperature has reached 220 °C, the reaction equilibrium will move towards the endothermic direction so that the PS yield has shown some decreases. When the reaction temperature has reached 260 °C, the rearrangement reaction has reached equilibrium so the PS yield will keep unchanged. The XA yield continually increased with rises in the reaction temperature, and the increasing rate quickened after the temperature has risen to 240 °C. PS rearrangement is also an endothermic reaction, and the reaction equilibrium will move towards the endothermic direction with further temperature rises after the reaction temperature has reached 240 °C so that PS is quickly rearranged and converted into XA. The XA yield reached its peak (41%) at 260 °C. The PS rearrangement reaction has reached equilibrium so the XA yield will keep unchanged after 260 °C. Between 140 °C and 260 °C, the higher the reaction temperature is, the more the DPC rearrangement reaction is favored.
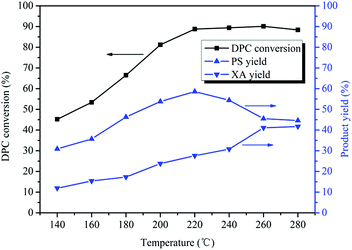 |
| | Fig. 3 Effect of the reaction temperature on the Fries rearrangement of DPC. Reaction conditions: DPC: 15 g; mass ratio of Bu2SnO to DPC: 2%; reaction time: 10 h; pressure: 0.40 MPa. | |
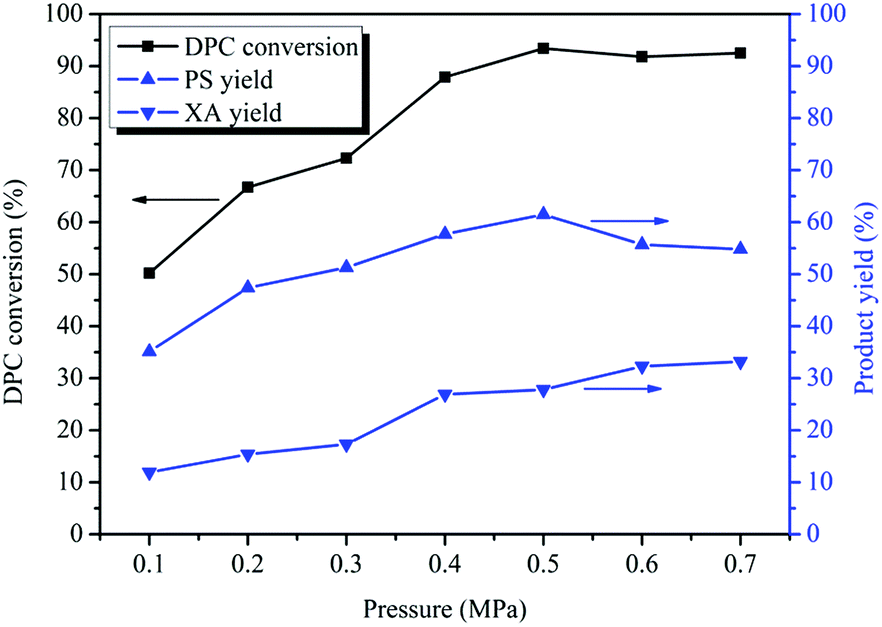
3.5 Effect of reaction pressure on the rearrangement of DPC
Fries rearrangement reaction is easy to occur under pressurized conditions,44,45 and with the process conditions of transesterification for the synthesis of DPC taken into account, the effect of the reaction pressure on Bu2SnO-catalyzed DPC rearrangement was investigated with pressures ranging between 0.10 and 0.70 MPa. As can be known from Fig. 4, the reaction pressure significantly affects the rearrangement reaction. The DPC conversion gradually increased with rises in the reaction pressure and reached its peak (93%) when the reaction pressure was 0.50 MPa; however, the DPC conversion stopped changing significantly with further rises in the reaction pressure. When the reaction pressure was 0.10 MPa, the rearrangement products PS and XA were generated, and the yields were 35% and 12%, respectively. The PS yield first increased and then decreased with rises in the reaction pressure, reaching its peak (62%) when the reaction pressure was 0.50 MPa and gradually decreasing with further pressure rises. When the reaction pressure was greater than 0.60 MPa, the PS yield stopped changing. PS rearrangement is also a reaction with spatial volume reduction, so, with further rises in the reaction pressure, the reaction will proceed towards the direction with the smaller spatial volume (i.e. XA) so that PS can be converted to XA in large quantities. Nevertheless, during the tandem rearrangement reactions, PS and XA will persistently exist in parallel, and PS will never be completely converted into XA. The XA yield continually increased with rises in the reaction pressure and reached its peak (32%) when the reaction pressure was 0.60 MPa. The PS rearrangement reaction has reached equilibrium, so the XA yield will keep unchanged when the reaction pressure was greater than 0.60 MPa. It can also be known from Fig. 4 that, within 0.6 MPa, the higher the reaction pressure is, the more the tandem rearrangement of DPC is favored.
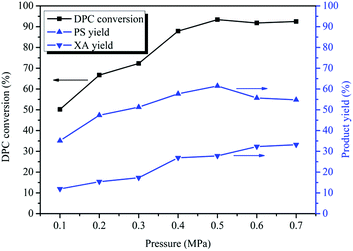 |
| | Fig. 4 Effect of the pressure on the Fries rearrangement of DPC. Reaction conditions: DPC: 15 g; mass ratio of Bu2SnO to DPC: 2%; reaction time: 10 h; temperature: 220 °C. | |
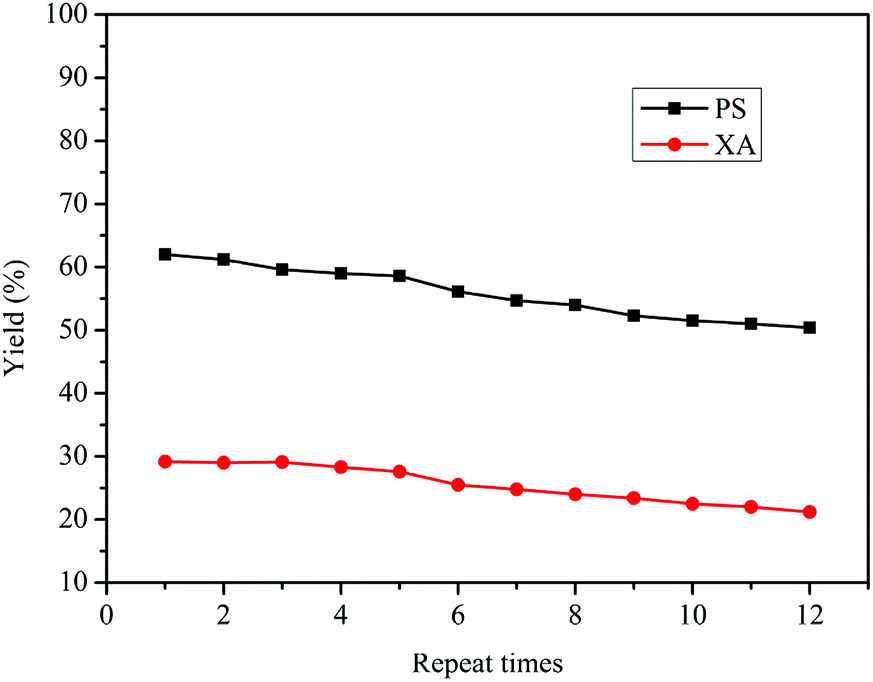
3.6 Reusability of Bu2SnO and comparison of synthetic methods
Bu2SnO is a homogeneous catalyst for the rearrangement reaction, but it can be isolated following sodium hydroxide precipitation, filtration, washing with water, liquid separation and drying from the product. The obtained Bu2SnO can be directly tested for reusability in the reaction, and the results are shown in Fig. 5. Following repeated use of Bu2SnO for 12 times, the yields of both PS and XA could be maintained above 50% and 20% with only a slight decrease, respectively, indicating tin atoms as catalytic active centers in Bu2SnO have practically not been lost or inactivated during the rearrangement reaction. The performance of the catalyst after used repeatedly for multiple times is still stable for the tandem rearrangement of DPC. As indicated in Fig. S4,† the peak position and the intensity of FT-IR for the fresh and spent Bu2SnO were all the same, which confirmed the nature of Bu2SnO unchanged during the catalyst recycling. Furthermore, we also compared this preparation method of PS and XA with those previously reported methods. As listed in Table S1,† although the yields of PS and XA are almost lower than that of other synthetic methods. The method in our work can simultaneous synthesis of PS and XA by DPC rearrangement, the process is simple, the reaction time is short, the raw material and the catalyst are low cost and easy to obtain, and the use of raw materials, catalysts, and solvents with high toxicity and corrosivity are avoided. In addition, our preparation method has few by-products, no waste acid, and waste gas in the reaction process. It is also environmentally friendly, the products are easy to separate, the catalyst can be recycled, and has good economic benefits.
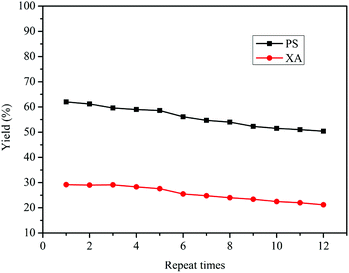 |
| | Fig. 5 Reusability of Bu2SnO for the Fries rearrangement of DPC. Reaction conditions: DPC: 15 g; mass ratio of Bu2SnO to DPC: 2%; reaction time: 10 h; temperature: 220 °C; pressure: 0.5 MPa. | |
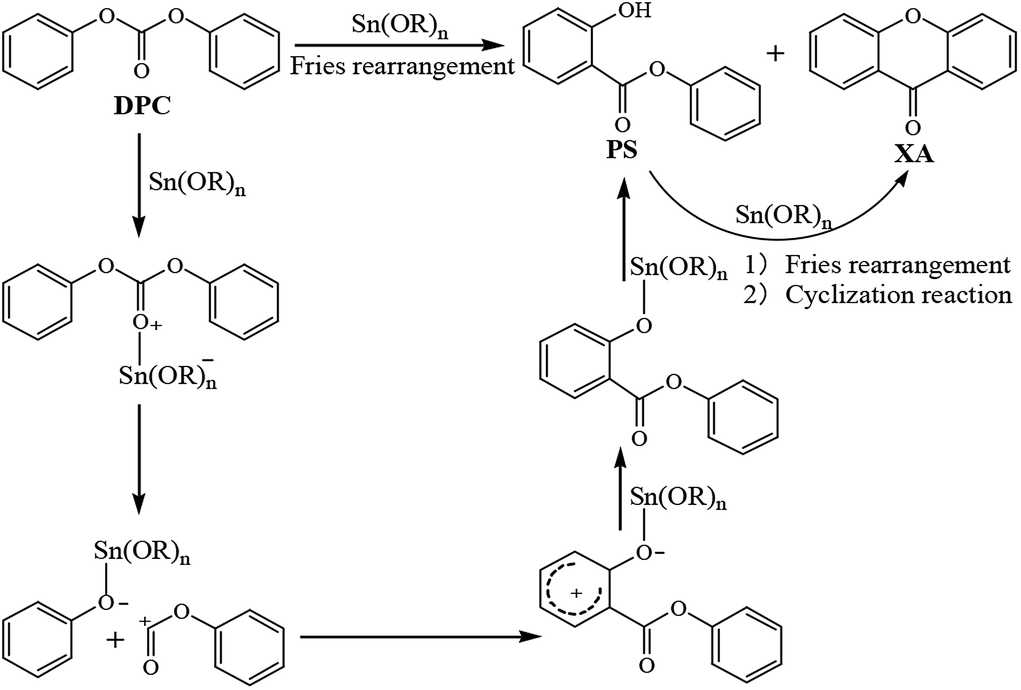
3.7 Possible mechanism of DPC rearrangement catalyzed by organotin compounds
During the Fries rearrangement of DPC, organotin compounds may react with DPC to form a class of transition metal complexes, which are soluble Lewis acid catalysts.46,47 Catalytic active centers of organotin compounds are tin atoms, and DPC is the only material involved in the rearrangement reaction, so the Fries rearrangement of DPC is an electrophilic substitution reaction. It thus can be deduced that the possible reaction mechanism of organotin-catalyzed DPC rearrangement is that shown in Scheme 2: the carbonyl oxygen atoms in DPC have lone pairs, and the electronegativity of carbonyl oxygen atoms is greater than that of carbon atoms, so the electron cloud density tends to be higher around the former than around the latter. Tin atoms with Lewis acidity in organotin compounds have vacant orbitals and are prone to chelate with carbonyl oxygen atoms in DPC to form transition metal complexes. The C–O single bond will crack to form phenolic tin compounds and acyl cations due to the weakening of the bond energy between the acyl group and the oxygen atom in the complexes. Acyl cations will react electrophilically with ortho positions with higher active sites in phenolic tin compounds to produce PS. PS can continue to undergo similar rearrangement and etherification cyclization reaction to convert to XA.
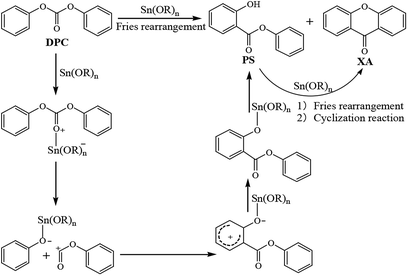 |
| | Scheme 2 Possible reaction mechanism for the Fries rearrangement of DPC. | |
Organotin compounds are Lewis acids, and tin atoms are the catalytic active centers, which form transition metal complexes by activating carbonyl oxygen atoms in DPC and their own lone pairs. For the electronic effect, the greater the electronegativity of coordinating groups connecting with tin atoms is, the stronger the Lewis acidity of tin atoms is, and the more easily carbonyl oxygen in DPC molecules and lone pairs on carbonyl oxygen atoms can be activated; the lower the electronegativity is, the less easily carbonyl oxygen and lone pairs on carbonyl oxygen atoms can be activated, and the less easily transition metal complexes can be formed, and the lower the catalytic activity of the catalysts is. For the steric hindrance effect, the larger the volumes of groups coordinating with tin atoms are, the more they will hinder tin atoms from contacting carbonyl oxygen atoms in DPC, the less easily carbonyl oxygen in DPC molecules and lone pairs on carbonyl oxygen atoms can be activated, and the lower the catalytic activity is.
4 Conclusions
Fries rearrangement of DPC catalyzed by organotin was realized for the first time. To research the effect of coordination groups on the catalytic activity of organotin revealed that the stronger the electron absorption effect of the coordination group was, the stronger the Lewis acidity of the active center tin atom was, and the better the catalytic performance of the corresponding organotin was. The small steric hindrance of the coordination group was beneficial to the activation of DPC by tin atom, and to boost the rearrangement of DPC. Among a series of organotin compounds, Bu2SnO showed the best catalytic activity because of the strong electron effect and small steric hindrance of the coordination group. Under the optimal conditions, with 2% of m(Bu2SnO)/(DPC), 10 h of reaction time, 220 °C of reaction temperature and 0.50 MPa of reaction pressure, the conversion of DPC was 93%, and the yield of PS and XA was up to 62% and 28%, respectively. The research also showed that high pressure, high temperature, increasing the amount of catalyst and prolonging the reaction time was beneficial to the rearrangement of DPC. In the process of DPC rearrangement, the two-step rearrangement products PS and XA existed at the same time, owing to PS could not be completely converted into XA. The yield of PS was always greater than XA, and the highest selectivity of XA was only 47%. Furthermore, the rearrangement reaction mechanism over organotin compounds as Lewis acid was proposed. At first, the tin atom coordinated with the oxygen over the carbonyl group of DPC to form transition state Sn complexes, which underwent nucleophilic and electrophilic substitution attack to form PS. PS proceeded with Fries rearrangement and etherification cyclization to form XA. This study can provide a theoretical and data basis for controlling the byproducts produced by DPC rearrangement in the synthesis of DPC, and also provide a new and effective route for the synthesis of PS and XA.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial support for this work from the Major Demonstration Program of Innovation Academy for Green Manufacture, Chinese Academy of Sciences (No. IAGM-2019-A10), the National High-tech R&D Program of China (863 Program, No. 2013AA031703), the National Key R&D Program of China (No. 2016YFB0301900), the Science and Technology Support Program of Sichuan Province (No. 2018JY0615), and the Science and Technology Innovation Program for Youth Team of Sichuan Province (No. 2013TD0010). The authors would like to thank Shiyanjia Lab (http:\\www.shiyanjia.com) for the FT-IR, H NMR and C NMR analysis.
References
- J. L. Gong, X. B. Ma and S. P. Wang, Appl. Catal., A, 2007, 316, 1–21 CrossRef CAS.
- M. C. Figueiredo, V. Trieu, S. Eiden, J. Heijl and M. T. M. Koper, ACS Catal., 2018, 8, 3087–3090 CrossRef CAS.
- J. Tan, Fine Spec. Chem., 2017, 25, 50–52 Search PubMed.
- Q. Liu, C. Du and L. J. Xie, Petrochem. Technol., 2017, 46, 1542–1549 Search PubMed.
- B. J. Li, R. Z. Rang, T. Chen and G. Y. Wang, Chin. J. Catal., 2012, 33, 601–604 CrossRef CAS.
- R. Z. Tang, S. L. Wang, Y. Z. Zhang, T. Chen and G. Y. Wang, Chem. J. Chin. Univ., 2014, 35, 2418–2424 CAS.
- S. L. Wang, Y. Z. Zhang and T. Chen, J. Mol. Catal. A: Chem., 2015, 398, 248–254 CrossRef CAS.
- S. L. Wang, T. Chen, G. Y. Wang, C. X. Cui, H. Y. Niu and C. G. Li, Appl. Catal., A, 2017, 540, 1–6 CrossRef CAS.
- Y. M. Qu, S. L. Wang, T. Chen and G. Y. Wang, Res. Chem. Intermed., 2017, 43, 2725–2735 CrossRef CAS.
- Y. M. Qu, H. Yang, S. L. Wang, T. Chen and G. Y. Wang, Chem. Res. Chin. Univ., 2017, 33, 804–810 CrossRef CAS.
- H. Yang, Z. L. Xiao, Y. M. Qu, T. Chen, Y. Chen and G. Y. Wang, Res. Chem. Intermed., 2018, 44, 799–812 CrossRef CAS.
- Z. Wang, Q. Q. Yu, J. M. Huang, R. X. Bai and G. X. Li, Acta Chim. Sin., 2005, 63, 1575–1580 Search PubMed.
- T. Liu, J. Hu, L. Yong, G. Zhang, Y. Zhang, T. Chen and G. Y. Wang, J. Anal. Methods Chem., 2019, 2019, 7632520 Search PubMed.
- Y. S. Eo, H. W. Rhee and S. Shin, J. Ind. Eng. Chem., 2016, 37, 42–46 CrossRef CAS.
- X. L. Sheng, Z. Q. Wang, Q. Y. Wang, S. Y. Liu and G. Y. Wang, Chin. J. Polym. Sci., 2018, 36, 1027–1035 CrossRef.
- Q. Y. Xing, W. W. Pei, R. Q. Xu and J. Pei, Basic Organic Chemistry, Higher Education Press, Beijing, 2nd edn, 2005, pp. 768–769 Search PubMed.
- M. H. Zheng, Y. Liu, J. Zhao and C. J. Wang, Prog. Chem., 2008, 27, 78–82 CAS.
- R. E. Miller, T. Rantanen, K. A. Ogilvie, U. Groth and V. Snieckus, Org. Lett., 2010, 12, 2198–2201 CrossRef CAS.
- S. Neufeind, N. Hulsken, J. M. Neudorfl, N. Schlorer and H. G. Schmalz, Chem.–Eur. J, 2011, 17, 2633–2641 CrossRef CAS.
- O. K. Rasheed, I. R. Hardcastle, J. Raftery and P. Quayle, Org. Biomol. Chem., 2015, 13, 8048–8052 RSC.
- S. K. Bankar, J. Mathew and S. S. V. Ramastry, Chem. Commun., 2016, 52, 5569–5572 RSC.
- P. A. Procopiou, D. M. Coe and G. Procopiou, Tetrahedron Lett., 2017, 58, 4766–4768 CrossRef CAS.
- S. L. Wang, R. Z. Tang, Y. Z. Zhang, T. Chen and G. Y. Wang, Chem. Eng. Sci., 2015, 138, 93–98 CrossRef CAS.
- Y. Z. Zhang, S. L. Wang, Z. L. Xiao, T. Chen and G. Y. Wang, Res. Chem. Intermed., 2016, 42, 7213–7222 CrossRef CAS.
- S. L. Wang, C. G. Li, Z. L. Xiao, T. Chen and G. Y. Wang, J. Mol. Catal. A: Chem., 2016, 420, 26–33 CrossRef CAS.
- Z. L. Xiao, H. Yang, H. Zhang, T. Chen and G. Y. Wang, Chem. Pap., 2018, 72, 2347–2352 CrossRef CAS.
- S. L. Wang, H. Y. Liu, J. J. Wang, T. Chen and G. Y. Wang, J. Alloys Compd., 2019, 777, 18–25 CrossRef CAS.
- M. Takeda, M. Mizukami, A. Hirashima, H. Ogi and H. Oki, EP 722931-A1, 1995.
- R. Sugise, S. Yashida, H. Tanaka and Y. Asada, JP 3724724-B2, 2003.
- S. Fukuoka, H. Miyaji, H. Hachiya, K. Matsuzaki, F. Shinsuke, M. Hironori, H. Hiroshi and M. Kazuhiko, WO 2006025424-A1, 2006.
- S. Fukuoka, H. Hachiya, K. Matsuzaki and H. Miyaji, WO 2006022294-A1, 2006.
- S. Fukuoka, I. Fukawa, M. Tojo, K. Oonishi, H. Hachiya, M. Aminaka, K. Hasegawa and K. Komiya, Catal. Surv. Asia, 2010, 14, 146–163 CrossRef CAS.
- K. Uchiyama and Y. Koga, JP 2016185930-A, 2016.
- X. Jing, H. R. Liu, X. Q. Dong and A. L. Geng, Chin. J. Anal. Chem., 1999, 27, 369 CAS.
- J. W. Da, R. Z. Zhang, L. Z. Li and C. W. Bao, CN 105294443-A, 2016.
- J. W. Da, R. Z. Zhang, L. Z. Li and C. W. Bao, CN 105294444-A, 2016.
- X. C. Cheng, Y. Y. Zhou, F. F. Zhang, K. Zhu, Y. Y. Liu and Y. Z. Li, Chem.–Eur. J, 2016, 22, 12655–12659 CrossRef CAS.
- A. Venkanna, P. V. K. Goud, P. V. Prasad, M. Shanker and P. V. Rao, ChemistrySelect, 2016, 1, 2271–2276 CrossRef CAS.
- S. L. Wang, Y. Z. Zhang, Y. Chen, R. Z. Tang, T. Chen and G. Y. Wang, Chem. J. Chin. Univ., 2014, 35, 2177–2181 CAS.
- F. Ribot, C. Eychenne-Baron, F. Fayon, D. Massiot and B. Bresson, Main Group Met. Chem., 2002, 25, 115–119 CAS.
- M. A. da Silva, A. S. S. dos Santos, T. V. dos Santos, M. R. Meneghetti and S. M. P. Meneghetti, Catal. Sci. Technol., 2017, 7, 5750–5757 RSC.
- F. M. Moghaddam, M. G. Dekamin and M. Ghaffarzadeh, Tetrahedron Lett., 2001, 42, 8119–8121 CrossRef.
- S. G. Yerande, D. M. Shendage, P. B. Wakchaure, G. R. Phadtare and M. Y. Bhoite, Tetrahedron Lett., 2014, 55, 2426–2429 CrossRef CAS.
- R. E. Miller, T. Rantanen, K. A. Ogilvie, U. Groth and V. Snieckus, Org. Lett., 2010, 12, 2198–2201 CrossRef CAS.
- H. Kim, K. Inoue and J. Yoshida, Angew. Chem., Int. Ed., 2017, 56, 7863–7866 CrossRef CAS.
- T. M. Serra, D. R. De Mendonca, J. P. V. Da Silva, M. R. Meneghetti and S. M. P. Meneghetti, Fuel, 2011, 90, 2203–2206 CrossRef CAS.
- Y. C. Brito, D. A. C. Ferreira, D. M. D. Fragoso, P. R. Mendes, C. M. J. De Oliveira, M. R. Meneghetti and S. M. P. Meneghetti, Appl. Catal., A, 2012, 443, 202–206 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra05390e |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a and
Gongying Wanga
*a and
Gongying Wanga