
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEvaluation of anti-inflammatory, antinociceptive and biological activities of Cenostigma macrophyllum standardized extracts and determination and quantification of the main metabolites
Bruno Oliveira Moreira a,
Anaildes Lago de Carvalhob,
Clayton Queiroz Alvesc,
Lorena Lôbo Brito Morbecka,
Mariluze Peixoto Cruza,
Regiane Yatsudaa,
Juceni Pereira Davidd and
Jorge Mauricio David*d
a,
Anaildes Lago de Carvalhob,
Clayton Queiroz Alvesc,
Lorena Lôbo Brito Morbecka,
Mariluze Peixoto Cruza,
Regiane Yatsudaa,
Juceni Pereira Davidd and
Jorge Mauricio David*d
aInstituto Multidisciplinar em Saúde, Universidade Federal da Bahia, 45029-094, Vitória da Conquista, BA, Brazil
bDepartamento de Ciências Naturais, Universidade Estadual do Sudoeste da Bahia, 45083-900, Vitória da Conquista, BA, Brazil
cDepartamento de Ciências Exatas, Universidade Estadual de Feira de Santana, 44036-336, Feira de Santana, BA, Brazil
dInstituto de Química, Universidade Federal da Bahia, 40170-115, Salvador, BA, Brazil. E-mail: jmdavid@ufba.br
First published on 13th December 2019
Abstract
The stem barks and leaves of Cenostigma macrophyllum are used in Brazilian folk medicines in the treatment of stomach and intestinal diseases. However, there are no reports of chromatographic methods used to evaluate the bioactives of its standardized extracts and for biological evaluation. An analytical method was developed and validated for simultaneous determination and quantification of the bioactive phenolics gallic acid, methyl gallate, ellagic acid and, the biflavonoids agathisflavone and amentoflavone in the leaves and stem bark of C. macrophyllum. HPLC operating conditions were optimized and the parameters such as selectivity, linearity, precision, accuracy, LOD, LOQ and, robustness of the method were also evaluated. Robustness was evaluated using a multivariate optimization technique. Linear relationships within the range of investigated concentrations were observed with their correlation coefficients greater than 0.9991. The method was validated for repeatability (RSD ≤ 2.88%), intermediate precision (RSD ≤ 3.38%) with recovery between 84.12 and 106.64% and the RSD less than 3.40% and proved to be robust. Besides, antioxidant, acetylcholinesterase inhibition, anti-inflammatory and antinociceptive activities of the standardized hydromethanolic extracts of leaves and stem bark of this species were evaluated. The method was successfully applied in the quantification of the gallic acid, methyl gallate, ellagic acid, agathisflavone and amentoflavone of standardized extracts. The results showed the present method developed was simple, sensitive, reproducible, accurate and precise. The standardized hydromethanolic extracts of leaves and stem bark of C. macrophyllum showed antioxidant activity (EC50 69.09 and 83.06 μg mL−1), acetylcholinesterase inhibition (52.23 and 83.36%) and they were able to inhibit the formalin-induced nociception and also reduced the edema formations at 100 mg kg−1 doses. The anti-inflammatory potentials were evaluated by the decrease of the Cg-induced neutrophils migrations at the same doses.
1. Introduction
Cenostigma macrophyllum Tul. is a tree belonging to the family Leguminoseae. This species is popularly known as “canela-de-velho”, “caneleiro” and “catingueira” and can be found in the semi-arid region of Brazilian northeastern.1,2 The decoction or infusion of stem barks, leaves and flowers of this species are used in folk medicine for the treatment of stomach and intestinal diseases.3 Cenostigma macrophyllum of the “Caatinga” Brazilian biome was classified until recently as another specie (C. gardnerianum Tul.) but they were considered as synonyms after the morphological variation analysis. The study was carried out in populations of its habitats that would be consistent with C. gardnerianum, from the old system, however the taxonomic polymorphism in the chemical characteristics demonstrated proposes a reevaluation of the taxonomic equation of this species.4,5Previous studies with extracts and fractions of the leaves, stem barks and seeds of a variety of this plant (C. macrophyllum Tul. var. acuminata Teles Freitas) revealed different biological activities. The hydroalcoholic extracts presented antioxidant activity6 and effect against gastric ulcers induced by ethanol or HCl/ethanol, ischemia-reperfusion and cold restraint stress with lesion inhibitions at 100 or 200 mg kg−1. In other studies dealing with antinociceptive actions of the ethanol, ethyl acetate extract and emulsion of hexane extract obtained from stem bark in streptozotocin (STZ)-induced diabetic rats showed a pain threshold higher than those in the STZ-vehicle group indicating antinociceptive activity but in this study the chemical composition of the extracts were presumed based on simple thin layer chromatography analysis and no evidences about the phenolics content.8–10
Phytochemical studies with C. macrophyllum have led to the isolation and identification of flavonoids, biflavonoids, benzoic acid derivatives, peptide derivatives, triterpenoids, steroids, vitamin A and E, fatty acids, coumarin and volatile esters.3,11,12 However, there are no reports in the literature on the development of a validated chromatographic method for the quantification of active principles present in this species. Thus, in the present study, a HPLC-DAD method was developed to simultaneously detect and quantify five bioactive compounds (Fig. 1) in C. macrophyllum standardized extracts. The method was applied in the analysis of leaves and stem bark of the species. These is the first study of antioxidant activity, acetylcholinesterase inhibition, anti-inflammatory and antinociceptive activities of the standardized with chemical markers of hydromethanolic extracts of leaves and stem bark of C. macrophyllum were evaluated.
 | ||
| Fig. 1 Chemical structures of the five bioactive compounds investigated in C. macrophyllum. | ||
2. Experimental
2.1 Instrumentation, materials, reagents and standards
Chromatographic determinations were performed using a Dionex UltiMate 3000 HPLC equipment equipped with a quaternary pump with on-line vacuum degasser of four channels, autosampler, column temperature control and diode array detector (DAD). Chromatographic analysis was carried out on a XBridge, BEH C18 column (100 × 3.0 mm i.d., 2.5 μm, Waters) and Acclaim RSLC 120 C18 column (100 × 2.1 mm i.d., 5 μm, Thermo Fisher Scientific). Analytical grade MeOH and DCM (Qhemis) and deionized water were used for extraction procedures, while HPLC grade acetonitrile, MeOH, acetic acid and formic acid (TEDIA or J. T. Baker) were used in chromatographic analyses. Water was purified using a NANOpure Diamond system (Barnstead®, Dubuque, Iowa, USA). The sample solution was filtered through a 0.45 μm Millipore membrane before sample injection. Three reference standards, including gallic acid (1), ellagic acid (3) and amentoflavone (5) were purchased from the Sigma-Aldrich (St. Louis, Missouri, USA). Methyl gallate (2) and agathisflavone (4) were isolated and purified from of the leaves of Schinopsis brasiliensis (Anacardiaceae)13 and C. macrophyllum,10 respectively. These compounds were identified by spectroscopic techniques and the purity were determined injecting 20 μL of the appropriate solution (at a concentration of about 0.1 mg mL−1) into an analytical HPLC-DAD under the conditions described below. All standards showed at least 97% of purity determined by calculating the HPLC peak area percentage.2.2 Plant material
Leaves and stem barks of Cenostigma macrophyllum were collected in the Petrônio Portella campus of the Federal University of Piauí, Teresina, Brazil (5°03′34.3′′S 42°48′03.6′′W) and identified by Prof. Luciano P. de Queiróz. A voucher specimen (HUEFS 78.424) was deposited at the Herbarium of the State University of Feira de Santana (Bahia, Brazil).2.3 Extract preparations
The botanical material was dried at room temperature and then powdered. Part of ground powder (10.0 g of the leaves and 10.0 g of the stem bark) were macerated at 40 °C in MeOH (200 mL) separately, by three consecutive extractions, under stirring, for about 48 h, and the filtrate obtained was concentrated under reduced pressure, yielding the methanolic extracts of the leaves (2.14 g) and the stem bark (1.81 g). The methanolic extracts were dissolved in H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH (4:6, v/v) and partitioned between DCM, yielding the DCM and hydromethanol soluble fractions. The hydrometanolic portions of the leaves (MLCM) and the stem bark of C. macrophyllum (MBCM) were subjected to HPLC analysis.
:MeOH (4:6, v/v) and partitioned between DCM, yielding the DCM and hydromethanol soluble fractions. The hydrometanolic portions of the leaves (MLCM) and the stem bark of C. macrophyllum (MBCM) were subjected to HPLC analysis.
2.4 Chromatographic conditions
Several attempts of optimization of the chromatographic conditions were carried out, modifying solvents, the gradient of elution, flow rate, analytical columns, and column temperature. After optimization of the method, the chromatographic separation was performed using an XBridge, BEH C18 column (100 × 3.0 mm i.d., 2.5 μm, Waters). The separations were obtained by gradient elution using solvent A (0.2% aqueous formic acid) and solvent B (acetonitrile). The following multi-step linear gradient profile was applied: 0.1% B (4 min), 0.1–8% B (16 min), 8–10% B (20 min), 10–30% B (15 min), 30–100% B (4 min) and 100% B (1 min) in a 60 min. total time analysis. Sample injection volumes were 10 μL, column temperature of 30 °C and a 0.6 mL min−1 mobile phase flow rate were set. The column was conditioned at 0.1% B for five minutes between each analysis. UV spectra were recorded between λ = 190 and 400 nm and the chromatographic profiles were recorded at λ = 254, 265, 290 and 330 nm, but the detection wavelengths were set at λ = 265 nm.2.5 Preparation of standard solutions
Five stock solutions of gallic acid (1), methyl gallate (2), ellagic acid (3), agathisflavone (4) and amentoflavone (5) in MeOH were separately prepared at concentrations of 400 mg L−1, 400 mg L−1, 200 mg L−1, 200 mg L−1 and 24 mg L−1, respectively. Serial dilutions of each stock solution afforded the working solutions for generating calibration curves and for method validation.2.6 Method validation
Selectivity, linearity, precision, accuracy, limit of detection (LOD), limit of quantification (LOQ) (Table 1) and robustness were determinated according to the requirements of the Brazilian Government proposed by National Agency of Sanitary Vigilance (ANVISA),14 International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use – ICH,15 and suggestions proposed by Ribani et al.16 The selectivity of the method was evaluated by the spectral homogeneity of the chromatographic peaks, by comparing the retention times of the standards with the chromatographic peaks of the analyzed compounds, by the overlapping of the chromatograms and the UV spectra obtained from the standards and extract. The analysis of the spectral homogeneity of the peaks was performed by observing the spectra recorded in the ascending, apical and descending regions of the peaks, being considered pure when they presented exact overlap (Fig. 2). The standard stock solutions were diluted with methanol to provide solutions with a range of five different concentrations of the standards were analyzed by three replicate injections (Table 1). Calibration graphs generated by plotting peak areas vs. concentrations were analyzed by linear regression. Linearity was accessed by analyzing regression parameters, by residual plot analysis and by analysis of variance (ANOVA, with p = 95%).| Compound | RTa (min) | Regression equationb | R2c | Linear range (μg mL−1) | LOD (μg mL−1) | LOQ (μg mL−1) | RSD (%) |
|---|---|---|---|---|---|---|---|
| a RT = retention times in minutes.b In the regression equation y = ax + b, x refers to the concentration (μg mL−1) and y is the peak area.c R2 is the correlation coefficient of the equation.d LOD is the limit of detection. LOQ is the limit of quantification. | |||||||
| Gallic acid (1) | 2.94 | y = 0.3892x − 0.1168 | 0.9992 | 2.0–4.0 | 0.21 | 0.65 | 2.98 |
| Methyl gallate (2) | 14.04 | y = 0.6421x + 0.0389 | 0.9991 | 2.0–8.0 | 0.39 | 1.17 | 3.31 |
| Ellagic acid (3) | 35.56 | y = 1.7063x + 0.00224 | 0.9993 | 7.5–17.5 | 0.56 | 1.70 | 3.54 |
| Agathisflavone (4) | 58.49 | y = 1.0105x + 1.0377 | 0.9996 | 3.0–16.0 | 0.56 | 1.70 | 2.38 |
| Amentoflavone (5) | 58.63 | y = 2.8362x + 0.4478 | 0.9993 | 1.0–9.0 | 0.44 | 1.34 | 3.03 |
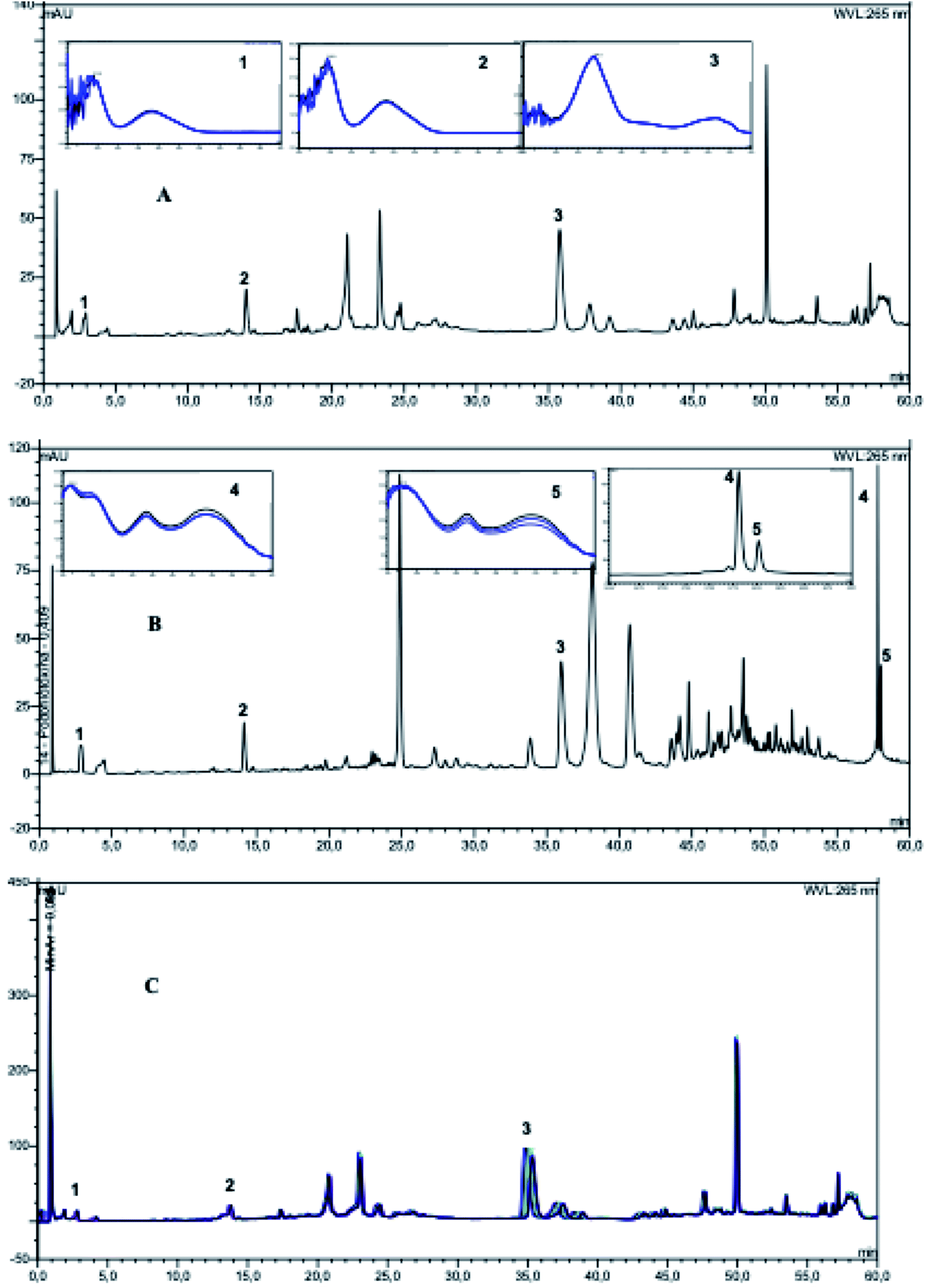 | ||
| Fig. 2 HPLC chromatograms of extracts MBCM (A) and MLCM (B) recorded at 265 nm and their UV spectra obtained by the DAD detector in the ascending, apical and descending regions of the peaks. (C) HPLC chromatograms (λ = 265 nm) of repetitive injections (n = 10) of a solution of MBCM extract (2.0 mg mL−1). | ||
The repeatability (intraday precision) and the intermediate precision (interday precision) of the method were determined in three different concentrations. The repeatability was performed on a single day with the same equipment and analyst, the analysis was performed in quadruplicate of the standard solutions in different levels of concentration: low, medium and high (Table 2). Intermediate precision was assessed by performing triplicate analyzes on four non-consecutive days of the standard solutions at the same concentrations used for intra-day precision. The relative standard deviation (RSD) was taken as a measure of precision.
| Compound | Concentration (μg mL−1) | Precision | |
|---|---|---|---|
| Repeatability (intraday) | Intermediate precision (interday) | ||
| 1 – gallic acid | 2.0 | 1.85 | 3.38 |
| 3.0 | 1.57 | 3.46 | |
| 4.0 | 1.63 | 2.80 | |
| 2 – methyl gallate | 2.0 | 2.53 | 1.23 |
| 5.0 | 1.53 | 2.04 | |
| 8.0 | 1.81 | 1.77 | |
| 3 – ellagic acid | 7.5 | 1.89 | 2.30 |
| 12.5 | 0.76 | 1.62 | |
| 17.5 | 1.59 | 1.04 | |
| 4 – agathisflavone | 3.0 | 2.88 | 1.79 |
| 9.0 | 2.04 | 1.58 | |
| 16.0 | 0.50 | 0.87 | |
| 5 – amentoflavone | 1.0 | 1.99 | 1.40 |
| 5.0 | 1.51 | 0.86 | |
| 9.0 | 1.66 | 0.77 | |
Accuracy was determined in recovery experiments using both MLCM and MBCM extracts of. The MBCM extract was fortified with gallic acid (1), methyl gallate (2) and ellagic acid (3) and the MLCM extract was fortified with agathisflavone (4) and amentoflavone (5) once the biflavonoids did not occur in barks of C. macrophyllum. All fortifications were performed in triplicate and at three different concentration levels (low, medium and high) for each standard (Table 3).
| Compound | Initial concentration in the extractb (μg mL−1) | Amount added (μg mL−1) | Concentration after addition (μg mL−1) | Recovery (%) | RSD (%) |
|---|---|---|---|---|---|
| a The analyzes were performed in triplicate, for each level of concentration and the results expressed as mean.b Recoveries of compounds 1–3 were determined in MBCM extracts, while the recoveries of compounds 4 and 5 were measured in MBCM extracts. | |||||
| 1 – gallic acid | 3.49 | 2.0 | 4.62 | 84.12 | 0.55 |
| 3.0 | 5.69 | 87.75 | 1.80 | ||
| 4.0 | 6.91 | 92.31 | 1.90 | ||
| 2 – methyl gallate | 5.15 | 2.0 | 7.58 | 106.08 | 1.44 |
| 5.0 | 10.82 | 106.64 | 0.84 | ||
| 8.0 | 13.40 | 101.92 | 0.78 | ||
| 3 – ellagic acid | 13.52 | 7.5 | 20.60 | 97.98 | 1.47 |
| 12.5 | 24.90 | 95.70 | 0.17 | ||
| 17.5 | 31.57 | 101.77 | 0.24 | ||
| 4 – agathisflavone | 11.49 | 3.0 | 14.79 | 102.07 | 0.50 |
| 9.0 | 17.59 | 85.87 | 0.94 | ||
| 16.0 | 27.35 | 99.48 | 3.62 | ||
| 5 – amentoflavone | 4.12 | 1.0 | 4.94 | 96.51 | 2.78 |
| 5.0 | 8.71 | 95.53 | 0.22 | ||
| 9.0 | 11.64 | 88.74 | 0.48 | ||
The linear regression equation of the calibration curve was used to determine the LOD and LOQ (Table 1). The LOD and LOQ were calculated based on the standard deviation (SD) of the y-intercept and the slope (s) as 3.3 SD/s and 10 SD/s, respectively. To evaluate the behavior of the analytical system, when small changes were made in the optimal operating conditions, the robustness test was performed. A standard solution of 12.5 μg mL−1 ellagic acid was used in all experiments. This analysis was performed using the previously optimized chromatographic conditions. In the evaluation of the robustness, the following parameters were used: flow (0.55, 0.60 and 0.65 mL min−1), percentage of formic acid in water (0.1%, 0.2% and 0.3%) and column temperature (25, 30 and 35 °C). The robustness evaluation was performed through a full two-level factorial design and the results interpreted from the analysis of variance (ANOVA). The central point was determined in triplicate to allow an estimation of the error and to verify if the modifications cause curvature in the model and the other experiments were performed in duplicate. All experiments were performed randomly. Table 4 shows the experimental matrix. The results were treated in the STATISTICA 8.0 program with a 95% confidence level.
| Experiment | Flow rate (mL min−1) | % aqueous formic acid | Column temperature (°C) | Response |
|---|---|---|---|---|
| Peak area | ||||
| a Central point. | ||||
| 1 | + (0.65) | + (0.3) | + (35) | 24.1565 |
| 26.0040 | ||||
| 2 | + (0.65) | + (0.3) | − (25) | 25.2578 |
| 25.3579 | ||||
| 3 | + (0.65) | − (0.1) | + (35) | 16.2218 |
| 15.0967 | ||||
| 4 | + (0.65) | − (0.1) | − (25) | 15.0967 |
| 15.2856 | ||||
| 5 | − (0.55) | + (0.3) | + (35) | 30.8812 |
| 28.5862 | ||||
| 6 | − (0.55) | + (0.3) | − (25) | 28.6600 |
| 28.0132 | ||||
| 7 | − (0.55) | − (0.1) | + (35) | 20.4211 |
| 20.1353 | ||||
| 8 | − (0.55) | − (0.1) | − (25) | 15.3884 |
| 15.9604 | ||||
| CPa | 0 (0.60) | 0 (0.2) | 0 (30) | 26.9623 |
| CPa | 0 (0.60) | 0 (0.2) | 0 (30) | 26.0129 |
| CPa | 0 (0.60) | 0 (0.2) | 0 (30) | 25.9894 |
2.7 Biological activity
The percentage of free radical scavenging (% FRS) was calculated as:
| % FRS = [SA(CA − SA)/AC] × 100 |
The efficient concentration, amount of antioxidant needed to reduce the initial concentration of DPPH by 50% (EC50), was calculated by linear regression, where the abscissa axis represented sample (μg mL−1) or positive control concentrations and ordinate the percentage of free radical sequestration (% FRS). When necessary, dilutions were carried out at the concentrations of the samples and the standard to enable the calculation of the EC50.
| AA = (DRC − DRA)/DRC × 100. |
| % I = [(AChE − AChI)]/AChE. |
2.7.4.1 Animals. All experiments were performed with male Balb/C mice (bodyweights between 20–25 g) obtained and maintained at our Animal Facility at the Multidisciplinary Health Institute of the Federal University of Bahia. The animals were maintained in a temperature-controlled room (25 ± 2 °C) with controlled humidity (50–70%) and a 12 h light/dark cycle. The animals were kept in polypropylene boxes containing wood shavings at the base of the box with free access to food (Labina®, Purina) and filtered water. The mice were homogeneously distributed among the groups. All animals were allowed to acclimatize to the air-conditioned laboratory for at least two hours before the tests, which were performed during the light cycle phase.
2.7.4.2 Acetic acid-induced writhing test. The antinociceptive effect was evaluated in mice using the acid acetic writhing test according to the procedures previously described.21 Animals were treated subcutaneously (s.c.) with the MLCM and MBCM extracts (100 or 200 mg kg−1) 30 min prior to intraperitoneal (i.p.) injection of 1.0% acetic acid (0.1 mL 10 g−1, Sigma, St. Louis, MO, USA). The negative control received only vehicle (negative control group, s.c.). The mice were placed in separate boxes and, after the administration of the acetic acid, the number of writhes and stretching movements (contraction of the abdominal musculature and extension of the hind limbs) was counted at 5 min intervals for a period of 30 min. Antinociceptive activity was expressed as the writhing scores over 30 min.
2.7.4.3 Nociception induced by intraplantar injection of formalin. Formalin-induced hypernociception behavior was assessed as described previously.20 Mice were allowed to acclimate for 15 min in a plastic box and pretreated 30 min prior to the injection with MLCM and MBCM extract (100 mg kg−1, s.c.), vehicle (s.c.), or morphine (5 mg kg−1, s.c.). Then, the mice were injected subcutaneously with 20 μL of 1.5% formaldehyde (St. Louis, MO, USA) in 0.9% saline into the plantar right hindpaws. The mice were observed for 30 min after the formalin injection, and the pain behavior was determined by the number of flinches, biting and licking of the injected paw counted during the observation period time. The acute phase (phase 1) was defined as 0–15 min after injection, and the persistent tonic phase (phase 2) was defined as 15–30 min after injection.
2.7.4.4 Paw edema induced by intraplantar injection of formalin. After the end of the experiment of nociception induced by intraplantar injection of formalin, both posterior paws were removed and weighed using an analytical balance. The contralateral paw was used as a control. The weight of the paw that did not receive the formalin injection was subtracted from the weight of the injected paw to determine edema formation. The results were express in grams (g). Percentage of antiedematogenic effect was also calculated according to the following formula:
| % AN = [(control group average of the edema − test group average of edema)/(control group average of the edema)] × 100. |
2.7.4.5 Evaluation of neutrophil migration. To investigate neutrophil migration to the peritoneal cavity, the MLCM and MBCM extracts (100 mg kg−1), were administered subcutaneously 30 min prior to the administration of the inflammatory stimulus: an intraperitoneal injection of Carrageenan (Cg – Sigma, St. Louis, MO, USA) at a dose of 500 μg per cavity.20 The negative control group received the vehicle. The mice were sacrificed 4 h after Cg administration, and the peritoneal cavity cells were harvested by washing the cavity with 3 mL of phosphate-buffered saline (PBS) containing 1 mM EDTA. The recovered volume was similar in all experimental groups and equated to approximately 95% of the injected volume. Total counts were performed in a Newbauer chamber. Differential cell counts (100 cells in total) were performed on a cytocentrifuge (PRESVAC CT12, Curitiba, Brazil) using slides stained with Panoptic. The results are presented as the number of neutrophils per cavity.
2.8 Statement to confirm the experimental protocols
Animal care and research protocols were in accordance with the guidelines and regulations adopted by the Brazilian College of Animal Experimentation (COBEA) and were approved by the Ethical Committee for Animal Research of the University of Uberaba, Brazil (protocol # 0107/2009). The number of animals used was the minimum number necessary to show consistent effects of the drug treatments.3. Results and discussion
For the optimization of the chromatographic separation conditions the influence of the following factors in the chromatographic peak elutions were evaluated: different stationary and mobile phases, changes in the elution program, mobile phase flow, volume injection, wavelength and, column temperature. Two columns, XBridge, BEH C18 column (100 × 3.0 mm i.d., 2.5 μm) and Acclaim RSLC 120 C18 column (100 × 2.1 mm i.d., 5 μm), were tested for their separation efficiencies. The results showed that the best separation was achieved on an XBridge, BEH C18 column.The choice of the best wavelength for the analysis of the phenolics was performed by monitoring the wavelength range of 190 to 400 nm and the UV absorption spectrum of each peak was registered. The wavelength selected for the analyzes was 265 nm, in which good sensitivity was obtained for most of the substances present in the extracts. The mobile phase composed of acetonitrile–water presented the best separation resolution. The presence of acid modifiers in the mobile phase suppressed the tailing effect of the peaks and an improvement in the separation was observed (formic acid 0.2% (v/v) and flow-rate of 0.6 mL min−1). The separation was better when the column temperature was kept at 30 °C rather than other temperatures. Fig. 2 shows the HPLC-DAD chromatograms of MLCM and MBCM extracts and the chromatograms were used to considerate the obtained extracts as standardized.
The identification compounds present in the extracts were carried out by comparison of their retention times and UV spectra with the standards (in the ascending, apical and descending regions of the peaks), confirming the selectivity of the method. Before starting the linearity study, the reproducibility of sample preparation based on the evaluation of retention time and peak area was evaluated for the chromatographic peaks referring to compounds 1–3 present in the MBCM extract (Fig. 2C).21 The results demonstrated the reproducibility of the sample preparation, since the RSD was less than 2.38% for triplicate analyses. The MBCM extract was analyzed in replicate (n = 10) to evaluate the precision of the injection, which was measured by sequential repetitive injections of the same sample. The analysis of the results of the injection precision showed that the preparation of the samples presented good reproducibility in the optimized method (RSD < 5%, for the peaks referring to 1, 2 and 3).
The linearity was determined by the external standard method. Table 1 describes the retention times, regression equation, the correlation coefficient for triplicate analyses, range, LOD, LOQ and, RSD (%). The five compounds analyzed showed satisfactory linearity with the correlation coefficient (R2) greater than 0.999 for all compounds. The optimized method presented high sensitivity for all the compounds, which indicated an excellent capacity to respond to distinctly small differences in concentrations. The residual dispersion of all analytes followed a random distribution pattern, which indicates adequacy of the linear models applied to the obtained responses, which confirms the good linear fit of the analytical curve. The analysis of variance (ANOVA) allowed evaluating the linearity of the method and the validity of the regression. One way to verify the validity of the regression is by comparing the values of calculated F and tabulated F value. If it is verified that calculated F > tabulated F (at least 10 times greater), there is enough statistical evidence to believe in the existence of a linear relationship between the variables y and x. The values of calculated F were 880.04, 1810.27, 4723.36, 7621.22 and 4738.07 for the data obtained in the construction of the analytical curves of the standards 1–5 respectively, which are much higher than the tabulated F (F1,13 = 4.67) at a confidence level of 95%. Therefore, the linearity of the method is accepted, and the regression can be considered significant.
Good sensitivity was demonstrated with LODs and LOQs in the range of 0.21–0.56 μg mL−1 and 0.65–1.70 μg mL−1, respectively for the gallic acid derivatives (1–3) and in the range of 0.44–0.56 μg mL−1 and 1.34–1.70 μg mL−1, respectively for the bioflavonoids 4 and 5, respectively. The values were comparable to those reported in previous analysis for compounds 1, 2, 3 and 4.22–27
The results obtained for repeatability and intermediate precision are also presented in Table 2. For both tests, the RSD was less than 5% for triplicate analyses at the three different concentrations, as recommended by ICH protocols and ANVISA.14,15 Accuracy was determined in recovery experiments and the results are presented in Table 3. The method proposed in the present study presented a good recovery for all the compounds in the three concentration levels, in the range of 84.12 to 106.64% and with RSD less than 5%, all values being within acceptable limits.
There are few reports of the use of multivariate optimization techniques to determine robustness in validation studies.28,29 The results for robustness evaluation are presented in the Pareto chart (Fig. 3) and demonstrate that the modifications performed in the optimal conditions of the chromatographic method did not present significant effects for the variables flow, column temperature and percentage of formic acid in water. Thus, the chromatographic method presented robustness and therefore the validation parameters indicate that the conditions used in the quantitative determination were satisfactory.
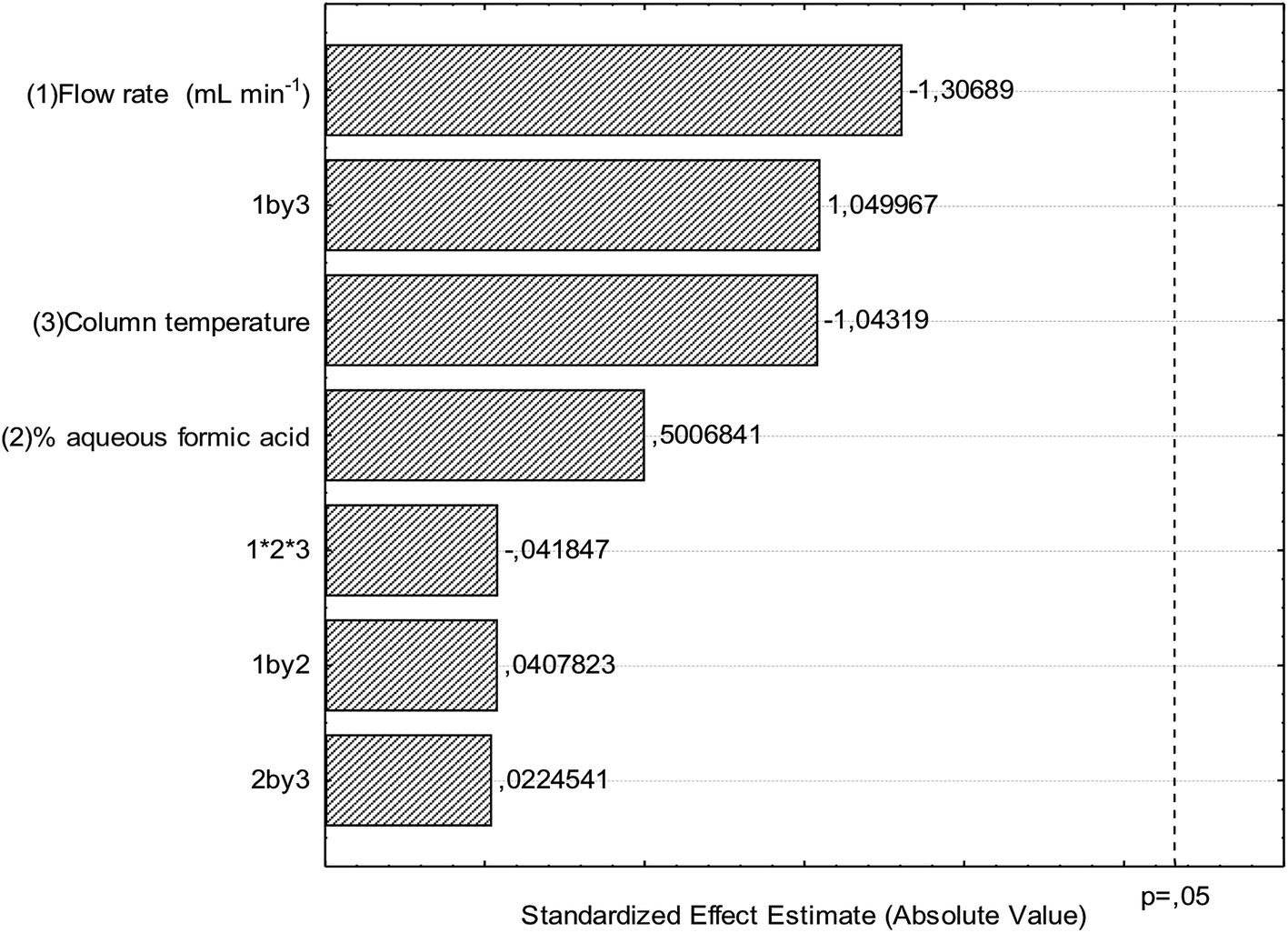 | ||
| Fig. 3 Pareto chart for robustness evaluation using peak area as independent variable. | ||
The presented chromatographic method was successfully applied in the simultaneous determination of 1–5 in the extracts MBCM and MLCM (Table 5). Ellagic acid (3) is the compound present in major concentrations in both extracts and the bioflavonoids agathisflavone (4) and amentoflavone (5) were detected only on leaves which is in accordance with the literature, since 4 and 5 were isolated only from the leaves of C. macrophyllum,7,11 whereas ellagic acid was isolated from both leaves and stem bark3,7 and, this is the first report of gallic acid and methyl gallate in the stem bark of this species. In the previous work11 from stem bark from a specimen collected in different habitat was isolated bergenin as the main compound of this species. However, it is not detected in both extracts of the specimen studied. Other flavonoid derivates also previously isolated from leaves C. macrophyllum were not also detected in the present species collected in different bioma. As pointed previously C. macrophyllum species of the Caatinga biome was classified until recently as another species, C. gardnerianum Tul. and later it was considered a synonym of C. macrophyllum.
| Compound | MBCM | MLCM |
|---|---|---|
| a The results are presented as the means ± SD (standard deviation, n = 3). Values followed by the same letter did not show significant differences, p < 0.05. Statistical significance was calculated by ANOVA followed by Bonferroni's test. ND – not detected. | ||
| 1 | 3.49 ± 0.09a | 2.23 ± 0.03b |
| 2 | 5.15 ± 0.12c | 3.52 ± 0.20a |
| 3 | 13.52 ± 0.10d | 12.34 ± 0.36e |
| 4 | ND | 7.01 ± 0.12f |
| 5 | ND | 2.23 ± 0.08b |
The substances quantified in this work present different biological activities30–35 and even if they are not directly responsible for the use of the species in traditional medicine, it is expected with this analytical methodology to contribute to a future application in the quality control of future phytotherapics of this medicinal plant.
The analysis of the HPLC fingerprints of the MLCM and MBCM extracts allows the realization of biological and pharmacological assays in order to investigate the folk medicinal employment of this plant. So, antioxidant, inhibition of AChE, antinociceptive and anti-inflammatory activities of the standardized extracts of MBCM and MLCM were evaluated.
Table 6 shows the EC50 values of AA of the extracts and the control obtained by the two known methods. According to literature36,37 the AA by the DPPH method can be classified as good if the EC50 of the extract is up to three times the EC50 of the standard. Following this criterion, the two extracts presented a good antioxidant activity. Regarding the statistical analysis of the results, it was verified that the extract MLCM showed a higher free radical scavenging activity with concerning MBCM extract (p < 0.05). This result can be justified by the presence of the flavonoids agathisflavone and amentoflavone in this extract. The results of the assay using the β-carotene/linolenic acid system indicated that the MLCM extract presented a higher AA when compared to the MBCM extract in the two analyzed times, statistically significant differences were verified (p < 0.05), which corroborates with the results obtained in the evaluation of the AA by the DPPH method. There was a decrease in activity over time for both extracts and standards, and this result was significant for the extracts and not significant for the standards (p < 0.05). The AA of the standards was statistically higher when compared to the extracts. These results encouraged the evaluation of the antinociceptive and anti-inflammatory activities of both extracts.
| Extracts/positive control | DPPH | β-Carotene | Inhibition of AChE | ||
|---|---|---|---|---|---|
| EC50a (μg mL−1) | 30 min | 60 min | 30 min | 60 min | |
| EC50a (μg mL−1) | EC50a (μg mL−1) | % Ia | % Ia | ||
| a The results are presented as the means ± SD (standard deviation, n = 3). — Quercetin as standard for AA and Eserine as a standard for inhibition of AChE. Values followed by the same letter did not show significant differences, p < 0.05. Statistical significance was calculated by ANOVA followed by Bonferroni's test. | |||||
| MBCM | 83.06 ± 2.09a | 17.54 ± 0.10d | 24.44 ± 1.13e | 83.36 ± 1.07f | 86.91 ± 1.76f,g |
| MLCM | 69.09 ± 2.38b | 13.00 ± 0.89d | 16.61 ± 0.82d | 52.23 ± 0.99h | 68.77 ± 1.20i |
| Quercetin | 36.56 ± 0.93c | 21.93 ± 1.27d,e | 23.59 ± 1.20e | — | — |
| Eserine | — | — | — | 89.67 ± 1.54g | 92.97 ± 1.98g |
To evaluate the peripheral antinociceptive effect extracts the acetic acid-induced writhing test was performed with 100 and 200 mg kg−1 doses of MBCM and MLCM (Fig. 4). Both 200 mg kg−1 doses resulted in a significant reduction (P < 0.05) in the number of writhing episodes compared to the group treated with the vehicle (negative control group). But, at the doses of 100 mg kg−1 only the MBCM extract was able to significantly reduce (P < 0.05) the number of writhing episodes compared to the group treated with the vehicle.
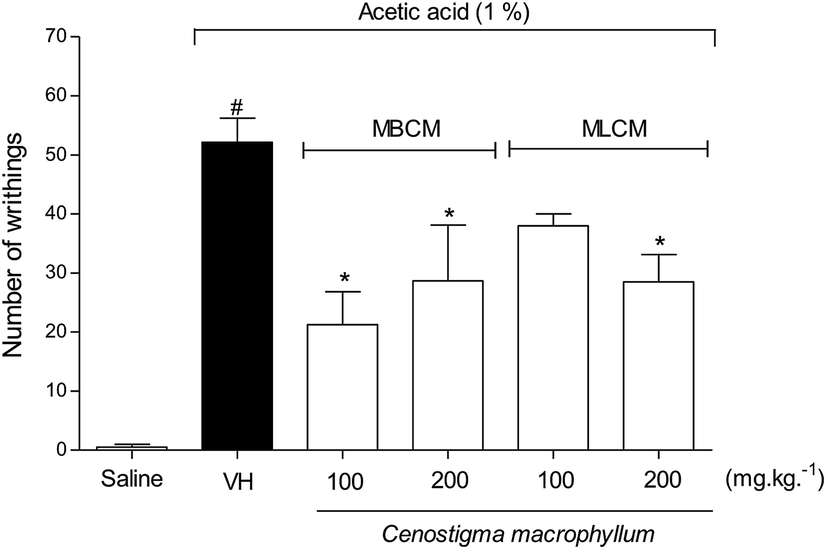 | ||
| Fig. 4 Effect of pretreatment of the mice with 100 and 200 mg kg−1 of the MBCM and MLCM extracts acetic acid-induced (1.0%) writhing test. VH is the vehicle group (control group). The MBCM and MLCM extracts was tested at doses of 100 and 200 mg kg−1 (s.c.). The results are presented as the means ± SD of writhing in mice (n = 6). Statistical significance was calculated by ANOVA followed by Bonferroni's test. *P < 0.05 compared to the vehicle-negative control treated group. #P > 0.05 compared to the vehicle-negative control treated group. | ||
Formalin model was used to confirm the antinociceptive effect of extracts. In this model, the antinociceptive and also a possible anti-inflammatory effect were evaluated (Fig. 5). The administration of MBCM and MLCM extracts at 100 mg kg−1 dose 30 min before formalin nociception were able to inhibit the formalin-induced nociception produced during the neurogenic phase in the animal paw (P < 0.05) (Fig. 5, 0–5 min). Morphine showed good activity in reduction of flinches compared to negative control (P < 0.05) and the MBCM extract did not present statistical difference in relation to the positive control (morphine, P < 0.05). Flinches in the inflammatory phase (Fig. 5, 5–30 min) were significantly decreased by the two extracts compared to the vehicle-treated group (P < 0.05). The MLCM extract did not present a statistical difference in relation to the positive control (morphine, P < 0.05) in the inflammatory phase.
 | ||
| Fig. 5 Effects of MBCM and MLCM extracts on the response to nociception induced by intraplantar formalin injection in mice during the neurogenic phase (0–5 min) and inflammatory phase (10–30 min). Morphine (5 mg kg−1, s.c.) was the positive control group (+). VH is the vehicle group (negative control). The MBCM and MLCM extract was tested at dose of 100 mg kg−1 (s.c.). The results are presented as the mean ± SD (n = 8) of the number of flinches in the injected paw for a period of 30 min. Statistical significance was calculated by ANOVA followed by Bonferroni's test. *P < 0.05 compared to the vehicle (negative control treated group). #P > 0.05 compared to the morphine (positive control group). | ||
The treatment of mice with the MBCM and MLCM extracts showed a significant reduction in the edema formation induced by formalin injection in the paws when compared to the vehicle group (P < 0.05), indicating the anti-inflammatory activity of the extracts (Fig. 6).
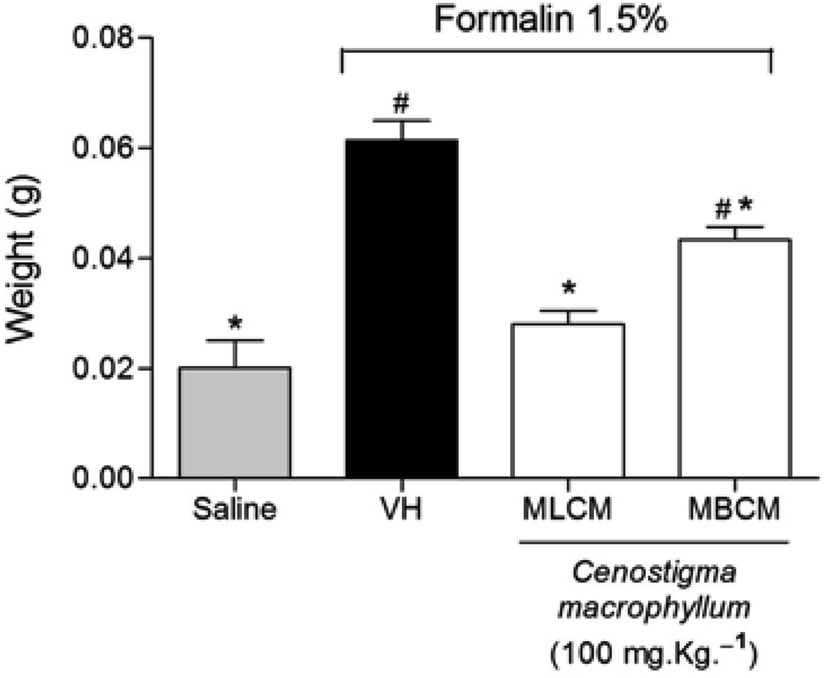 | ||
| Fig. 6 Effect of MBCM and MLCM extracts on the induction of paw edema in mice pretreated with an intraplantar injection of 1.5% formalin. VH is the vehicle group (negative control). The MBCM and MLCM extracts were tested at dose of 100 mg kg−1 (s.c.). The results are presented as the mean ± SD (n = 8). Statistical significance was calculated by ANOVA followed by Bonferroni's test. *P < 0.05 compared to the vehicle negative control treated group. #P < 0.05 when the vehicle group was compared to the saline group. | ||
The anti-inflammatory potentials of the MBCM and MLCM extracts were also evaluated using the inhibition of Cg-induced neutrophil migration to the peritoneal cavity (Fig. 7). Pre-treatment with 100 mg kg−1 (s.c.) with both extracts promoted a decrease in neutrophil migration (P < 0.05) to the peritoneal cavity. Further tests are needed to elucidate the mechanism of action of the extract.
 | ||
| Fig. 7 Effect of the MBCM and MLCM extracts on neutrophil migration into the peritoneal cavity of mice pretreated subcutaneously 30 min prior to Cg injection (500 μg per cavity) to induce peritonitis. VH is the vehicle group (negative control). The MBCM and MLCM extracts was tested at dose 100 mg kg−1 (s.c.). The results are presented as the mean ± SD (n = 6). Statistical significance was calculated by ANOVA followed by Bonferroni's test. *P < 0.05 compared to the vehicle negative control treated group. | ||
Previously the antinociceptive activity reported in the literature for bergenin isolated from barks of C. macrophyllum,11 a specimen collected in the Caatinga of Bahia. In the present study, this compound is not responsible for the activity presented by the extract MBCM, since bergenin was not found in this extract. Piaulino et al.9 investigated the antinociceptive actions of the ethanol extract and ethyl acetate fraction from C. macrophyllum stem bark in streptozotocin-induced diabetic rats and the involvement of opioid and nitrergic mechanisms. The authors showed that analysis by thin layer chromatography using authentic standard and suggested the presence of ellagic acid and valoneic acid dilactone. Thus, the results on antinociceptive activity presented in the present study confirm the results reported in the literature, since the MBCM extract ellagic acid was detected as the main compound. Besides that, the qualitative analysis of the HPLC fingerprints of the MBCM extract showed the presence of ellagic acid derivatives. Besides, the compound 1 that shows recognized antinociceptive activity38 was also quantified in the present extracts. The MLCM extract also showed antinociceptive activity, in addition to compounds 1 and 3 (the main compounds), this activity is related to the presence of biflavonoids 4 and 5. The anti-inflammatory activity presented by both extracts is due to the presence of compounds 1, 3, 4 and 5, which have recognized activity.34,36,39,40
The AChE inhibitory activity of standardized extracts was also evaluated. MBCM extract presented higher AChE inhibitory activity than the MLCM (Table 6) in the two analyzed periods of time and, statistically significant differences (p < 0.05) between the extracts were observed. The MBCM activity in 60 min test did not show statistically significant differences in relation to the standard (p < 0.05). This result shows excellent activity of this extract. Previously, from leaves of C. macrophyllum were isolated two peptides inhibitors of AChE12 but the compounds present in the extracts did not show AChE inhibition.
4. Conclusions
An HPLC-PAD method was developed to evaluate standardized C. macrophyllum extracts by chromatographic fingerprints and it also permitted to validate a simultaneous analysis gallic acid, methyl gallate, ellagic acid, amentoflavone and agathisflavone present in the leaves and stem bark of this plant species. The selectivity, correlation coefficients, LODs, LOQs, repeatability, intermediate precision, and accuracy were in good agreement with the requirements for the developed method. The robustness evaluation was performed through a full two-level factorial design and the results showed that the method was robust. The analytical method was successfully applied to determine the five compounds in extracts being ellagic acid as the major component. The standardized extracts presented expressive antioxidant, AChE inhibitory, antinociceptive and anti-inflammatory activities.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank CNPq-Conselho Nacional de Desenvolvimento Científico e Tecnológico (# 406427/2018-6) for grants and scholarship.References
- M. F. Silva, L. A. G. Souza and L. M. Carreira, Nomes Populares das Leguminosas do Brasil, EDUA/IMPA/FAPEAM, Manaus, 2004 Search PubMed.
- L. P. Queiroz, Distribuição das espécies de leguminosae na caatinga, in Vegetação e flora da caatinga, Associação Plantas do Nordeste – APNE/Centro Nordestino de informações sobre Plantas – CNIP, ed. EVSB Sampaio, et al., Recife, 2002, pp. 141–154 Search PubMed.
- H. R. Silva, C. C. M. Silva, L. B. C. Neto, J. A. D. Lopes, A. M. G. L. Citó and M. H. Chaves, Constituintes químicos das cascas do caule de Cenostigma macrophyllum: ocorrência de colesterol, Quim. Nova, 2007, 30, 1877–1881, DOI:10.1590/S0100-40422007000800015.
- G. P. Lewis, Legumes of Bahia, Royal Botanic Gardens Kew, England, 1987, p. 369 Search PubMed.
- M. C. Warwick and G. P. Lewis, Revision of Plathymenia (Leguminosae–Mimosoideae), Edinb. J. Bot., 2003, 60, 111–119, DOI:10.1017/S0960428603000106.
- C. M. M. Sousa, H. R. Silva, G. M. Vieira-Jr, M. C. C. Ayres, C. L. S. Costa, D. S. Araújo, L. C. D. Cavalcante, E. D. S. Barros, P. B. M. Araújo, M. S. Brandão and M. H. Chaves, Fenóis totais e atividade antioxidante de cinco plantas medicinais, Quim. Nova, 2007, 30, 351–355, DOI:10.1590/S0100-40422007000200021.
- A. F. S. C. Viana, H. B. Fernandes, F. V. Silva, I. S. Oliveira, F. F. B. P. Freitas, F. D. F. Machado, C. L. S. Costa, D. D. R. Arcanjo, M. H. Chaves, F. A. Oliveira and R. C. M. Oliveira, Gastro protective activity of Cenostigma macrophyllum Tul. var. acuminata Teles Freire leaves on experimental ulcer models, J. Ethnopharmacol., 2013, 150, 316–323, DOI:10.1016/j.jep.2013.08.047.
- M. F. Cavalcanti, H. B. Fernandes, S. S. Pereira, C. A. Piauilino, C. L. S. Costa, M. H. Chaves, J. O. Ibiapina, R. B. Marques, F. A. Oliveira and F. R. C. Almeida, Antinociceptive and anti-inflammatory effects of the hydroalcoholic fraction from leaves of Cenostigma macrophyllum Tul. var. acuminata Teles Freire (Leguminosae) in rodents, Orthopedics and Rheumatology Open Access Journal, 2017, 8(2), 555733, DOI:10.19080/OROAJ.2017.08.555733.
- C. A. Piaulino, F. C. B. Carvalho, B. C. Almeida, M. H. Chaves, F. R. C. Almeida and S. M. R. C. Brito, The stem bark extracts of Cenostigma macrophyllum attenuates tactile allodynia in streptozotocin-induced diabetic rats, Pharm. Biol., 2013, 51, 1243–1248, DOI:10.3109/13880209.2013.786096.
- N. P. M. F. Coelho, V. C. Nogueira, M. A. G. Cardoso, L. S. Lopes, P. P. Nascimento, E. S. Rocha, C. L. P. Silva and E. Â. L. Arisawa, Cenostigma macrophyllum Tul. on the healing of skin wounds in rats with Diabetes mellitus, Acta Bras. Cir., 2013, 28, 594–600, DOI:10.1590/S0102-86502013000800007.
- C. Q. Alves, J. M. David, J. P. David, C. F. Villareal, M. B. P. Soares, L. P. Queiroz and R. M. Aguiar, Flavonoids and other bioactive phenolics isolated from Cenostigma macrophyllum (Leguminosae), Quim. Nova, 2012, 35, 1137–1140, DOI:10.1590/S0100-40422012000600013.
- C. Q. Alves, L. S. Lima, J. M. David, M. V. B. Lima, J. P. David, F. W. M. Lima, K. C. M. C. Pedroza and L. P. Queiroz, In vitro acetylcholinesterase activity of peptide derivatives isolated from two species of Leguminosae, Pharm. Biol., 2013, 51, 936–939, DOI:10.3109/13880209.2013.770536.
- M. P. Cardoso, L. S. Lima, J. P. David, B. O. Moreira, E. O. Santos, J. M. David and C. Q. Alves, A New Biflavonoid from Schinopsis brasiliensis (Anacardiaceae), J. Braz. Chem. Soc., 2015, 26, 1527–1531, DOI:10.5935/0103-5053.20150101.
- Brasil, Resolution RE N 899, of May 29, 2003, Guide for validation of analytical and bioanalytical methods, Official Diary of the Union, Brazil, 2003, http://portal.anvisa.gov.br/legislacao#/visualizar/353660 Search PubMed.
- ICH, topic Q2 (R1) validation of analytical procedures: text and methodology, Int. Conf. Harmon., 2005, http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q2_R1/Step4/Q2_R1__Guideline.pdf Search PubMed.
- M. Ribani, C. B. G. Bottoli, C. H. Collins, I. C. S. F. Jardim and L. F. C. Melo, Validation for chromatographic and electrophoretic methods, Quim. Nova, 2004, 27, 771–780, DOI:10.1590/S0100-40422004000500017.
- A. W. L. Andrade, K. C. Machado, K. C. Machado, D. D. R. Figueiredo, J. M. David, M. T. Slam, S. J. Uddin, J. A. Shilpi and J. P. Costa, In vitro antioxidant properties of the biflavonoid agathisflavone, Chem. Cent. J., 2004, 12, 75–84, DOI:10.1186/s13065-018-0443-0.
- J. P. David, M. Meira, J. M. David, H. N. Brandão, A. Branco, M. F. Agra, M. R. V. Barbosa, L. P. Queiroz and A. M. Giulietti, Radical scavenging, antioxidant and cytotoxic activity of Brazilian Caatinga plants, Fitoterapia, 2007, 78, 215–218, DOI:10.1016/j.fitote.2006.11.015.
- M. R. Ahn, S. Kumazawa, T. Hamasaka, K. S. Bang and T. Nakayama, Antioxidant activity and constituents of propolis collected in various areas of Korea, J. Agric. Food Chem., 2004, 52, 7286–7292, DOI:10.1016/j.foodchem.2006.03.045.
- M. P. Cruz, C. M. F. Andrade, K. O. Silva, E. P. Souza, R. Yatsuda, L. M. Marques, J. P. David, J. M. David, M. H. Napimoga and J. T. C. Napimoga, Antinoceptive and anti-inflammatory activities of the ethanolic extract, fractions and flavones isolated from Mimosa tenuiflora (Willd.) Poir (Leguminosae), PLoS One, 2016, 11, e0150839, DOI:10.1371/journal.pone.0150839.
- X. H. Fan, Y. Cheng, Z. L. Ye, R. C. Lin and Z. Z. Qian, Multiple chromatographic fingerprinting and its application to the quality control of herbal medicines, Anal. Chim. Acta, 2006, 555, 217–224, DOI:10.1016/j.aca.2005.09.037.
- C. C. Wyrepkowski, D. L. M. G. Costa, A. P. Sinhorin, W. Vilegas, R. A. Grandis, F. A. Resende, E. A. Varanda and L. C. Santos, Characterization and quantification of the compounds of the ethanolic extract from Caesalpinia ferrea stem bark and evaluation of their mutagenic activity, Molecules, 2014, 19, 16039–16057, DOI:10.3390/molecules191016039.
- Y. Shen, H. Yang, G. Xia, J. Wang, B. Cai and X. Jia, Isolation of gallic acid and methyl gallate from folium Toonea sinensis and validated method for their quantitation using LC-based technologies, Acta Chromatogr., 2013, 25, 687–701, DOI:10.1556/AChrom.25.2013.4.7.
- J. Li, X. He, M. Li, W. Zhao, L. Liu and X. Kong, Chemical Fingerprint and Quantitative Analysis for Quality Control of Polyphenols Extracted from Pomegranate Peel by HPLC, Food Chem., 2015, 176, 7–11, DOI:10.1016/j.foodchem.2014.12.040.
- T. Dhanani, S. Shah and S. Kumar, A validated high-performance liquid chromatography method for determination of tannin-related marker constituents gallic acid, corilagin, chebulagic acid, ellagic acid and chebulinic acid in four Terminalia species from India, J. Chromatogr. Sci., 2014, 53, 625–632, DOI:10.1093/chromsci/bmu096.
- U. M. Acunã, K. Dastmalchi, M. J. Basile and E. J. Kennelly, Quantitative high-performance liquid chromatography photo-diode array (HPLC-PDA) analysis of benzophenones and biflavonoids in eight Garcinia species, J. Food Compos. Anal., 2012, 25, 215–220, DOI:10.1016/j.jfca.2011.10.006.
- K. K. P. Gomes-Copeland, A. S. Lédo, F. T. C. Almeida, B. O. Moreira, D. C. Santos, R. A. F. Santos, J. M. David and J. P. David, Effect of elicitors in Poincianella pyramidalis callus culture in the biflavonoid biosynthesis, Ind. Crops Prod., 2018, 126, 421–425, DOI:10.1016/j.indcrop.2018.10.038.
- S. L. C. Ferreira, R. E. Bruns, H. S. Ferreira, G. D. Matos, J. M. David, G. C. Brandão, E. G. P. Silva, L. A. Portugal, P. S. Reis, A. S. Souza and W. N. L. Santos, Box-Behnken design: An alternative for the optimization of analytical methods, Anal. Chim. Acta, 2007, 597, 179–186, DOI:10.1016/j.aca.2007.07.011.
- P. K. Sahu, R. N. Rao, T. Cecchi, S. Swain, C. S. Patro and J. Panda, An overview of experimental designs in HPLC method development and validation, J. Pharm. Biomed. Anal., 2018, 147, 590–611, DOI:10.1016/j.jpba.2017.05.006.
- G. Civenni, M. G. Iodice, S. F. Nabavi, S. Habtemariam, S. M. Nabavi, C. V. Catapano and M. Daglia, Gallic acid and methyl-3-O-methyl gallate: a comparative study on their effects on prostate cancer stem cells, RSC Adv., 2015, 5, 63800–63806, 10.1039/c5ra07988h.
- S. Kamatham, N. Kumar and P. Gudipalli, Isolation and characterization of gallic acid and methyl gallate from the seed coats of Givotia rottleriformis Griff. and their anti-proliferative effect on human epidermoid carcinoma A431 cells, Toxicol. Rep., 2015, 14, 520–529, DOI:10.1016/j.toxrep.2015.03.001.
- J. M. Landete, Ellagitannins, ellagic acid and their derived metabolites: A review about source, metabolism, functions and health, Food Res. Int., 2011, 44, 1150–1160, DOI:10.1016/j.foodres.2011.04.027.
- C. S. Souza, M. S. Grangeiro, E. P. L. Pereira, C. C. Santos, A. B. Silva, G. P. Sampaio, D. D. R. Figueiredo, J. M. David, J. P. David, V. D. A. Silva, A. M. Butt and S. L. Costa, Agathisflavone, a flavonoid derived from Poincianella pyramidalis (Tul.), enhances neuronal population and protects against glutamate excitotoxicity, Neurotoxicology, 2018, 65, 85–97, DOI:10.1016/j.neuro.2018.02.001.
- S. Chirumbolo and G. Bjørklund, Agathisflavone and GABAA receptors in the biflavone-mediated action on rat primary cortical neurons, Neurotoxicology, 2018, 66, 43–44, DOI:10.1016/j.neuro.2018.03.002.
- H. K. Kim, K. H. Son, H. W. Chang, S. S. Kang and H. P. Kim, Amentoflavone, a Plant Biflavone: A New Potential Anti-inflammatory Agent, Arch. Pharm. Res., 1998, 21, 406–410, DOI:10.1007/BF02974634.
- C. Q. Alves, J. M. David, J. P. David, M. V. Bahia and R. M. Aguiar, Métodos para determinação de atividade antioxidante in vitro em substratos orgânicos, Quim. Nova, 2010, 33, 2202–2210, DOI:10.1590/S0100-40422010001000033.
- J. G. Melo, T. A. S. Araújo, V. T. N. A. Castro, D. L. V. Cabral, M. D. Rodrigues, S. C. Nascimento, E. L. C. Amorim and U. P. Albuquerque, Antiproliferative activity, antioxidant capacity and tannin content in plants of semi-arid Northeastern Brazil, Molecules, 2010, 15, 8534–8542, DOI:10.3390/molecules15128534.
- G. Trevisan, M. F. Rossato, R. Tonello, C. Hoffmeister, J. Z. Klafke, F. Rosa, K. V. Pinheiro, F. V. Pinheiro, A. A. Boligon, M. L. Athayde and J. Ferreira, Gallic acid functions as a TRPA1 antagonist with relevant antinociceptive and antiedematogenic effects in mice, Naunyn-Schmiedeberg's Arch. Pharmacol., 2014, 387, 679–689, DOI:10.1007/s00210-014-0978-0.
- L. A. BenSaad, K. H. Kim, C. C. Quah, W. R. Kim and M. Shahimi, Anti-inflammatory potential of ellagic acid, gallic acid and punicalagin A&B isolated from Punica granatum, BMC Complementary Altern. Med., 2017, 17, 47–56, DOI:10.1186/s12906-017-1555-0.
- P. Chen, F. Chen and B. Zhou, Antioxidative, anti-inflammatory and anti-apoptotic effects of ellagic acid in liver and brain of rats treated by D-galactose, Sci. Rep., 2018, 8, 1465, DOI:10.1038/s41598-018-19732-0.
| This journal is © The Royal Society of Chemistry 2019 |