
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBioassay-guided purification of sesquiterpenoids from the fruiting bodies of Fomitopsis pinicola and their anti-inflammatory activity†
Shih-Huang Tai‡
a,
Ping-Chung Kuo‡b,
Ching-Che Hungc,
Ying-Hsuan Lin d,
Tsong-Long Hwange,
Sio Hong Lamb,
Daih-Huang Kuof,
Jin-Bin Wug,
Hsin-Yi Hung*b and
Tian-Shung Wu*bf
d,
Tsong-Long Hwange,
Sio Hong Lamb,
Daih-Huang Kuof,
Jin-Bin Wug,
Hsin-Yi Hung*b and
Tian-Shung Wu*bf
aDepartments of Surgery and Anesthesiology, Institute of Biomedical Engineering, National Cheng Kung University, Medical Center and Medical School, Tainan 701, Taiwan
bSchool of Pharmacy, College of Medicine, National Cheng Kung University, Tainan 70101, Taiwan. E-mail: tswu@mail.ncku.edu.tw; z10308005@email.ncku.edu.tw; Fax: +886-6-2740552; Fax: +886-6-2373149; Tel: +886-6-2747538 Tel: +886-6-2353535 ext. 6803
cDepartment of Chemistry, National Cheng Kung University, Tainan 70101, Taiwan
dDepartment of Environmental Sciences, University of California, Riverside, California 92521, USA
eGraduate Institute of Natural Products, College of Medicine, Chang Gung University, Research Center for Industry of Human Ecology, Research Center for Chinese Herbal Medicine, Graduate Institute of Health Industry Technology, College of Human Ecology, Chang Gung University of Science and Technology, Department of Anesthesiology, Chang Gung Memorial Hospital, Taoyuan 333, Taiwan
fDepartment of Pharmacy, College of Pharmacy and Health Care, Tajen University, Pingtung 907, Taiwan
gJen-Li Biotechnology Company, Taichung, Taiwan
First published on 23rd October 2019
Abstract
Twelve undescribed sesquiterpenoids, fomitopins A–L (1–12), were isolated via bioassay-guided purification from the bracket fungus Fomitopsis pinicola (Sw.) P. Karst, and this fungus have been reported to exhibit anti-microbial and anti-inflammatory activities. The structures of 1–12 were elucidated by spectroscopic and spectrometric analyses and their absolute configurations were further confirmed by ECD simulations. Ten isolated compounds were evaluated for their anti-inflammatory potential and compound 11 exhibited the most significant inhibition of superoxide anion generation and elastase release with IC50 values of 0.81 ± 0.15 and 0.74 ± 0.12 μM. These newly purified sesquiterpenoids could be potential candidates for further anti-inflammatory studies.
Introduction
The dried fruiting bodies of different Fomitopsis species have been used in folk medicine and have been reported to be associated with antitumor,1 antibacterial,2 and immunotropic3 effects. Among these species, F. pinicola (Sw.) P. Karst (Fomitopsidaceae), a red banded polypore found on decaying logs including Abies, and Betula, throughout temperate regions, is widely distributed in East Asia and Central Europe.4 A close species, Fomitopsis marginata (previous Polyporous marginata), looks lighter in color and ungulate, while Fomitopsis pinicola is more black and cinnamon.4 Traditionally, F. pinicola is cooked and the decoction used for the treatment of headaches, nausea, liver problems, hemorrhage and inflammation.5 Pharmacological studies revealed the chloroform extract of F. pinicola possesses antitumor activities against hepatoma, lung cancer, colorectal cancer and breast cancer.6 Moreover, alkali extracts rich in polysaccharides exhibited anti-hyperglycemic effects.7,8 In addition, lanostanoid derivatives as well as triterpenes were isolated and the isolated steroids showed antimicrobial activity against Bacillus subtilis in a TLC bioassay.9Inflammation is known to play key roles in many disease conditions, such as Alzheimer's disease, diabetes, infection, psoriasis, wound, colitis, arthritis, atherosclerosis, immune-diseases and cancers.10 Addition of the inflammatory disease costs resulted in more than billion dollars in Global drug markets. In 2005, twelve glycosidic triterpenes isolated from F. pinicola by Yoshikawa et al. exhibited anti-inflammatory activity via cyclooxygenase-2 inhibition. The most potent compound, fomitoside E, showed IC50 value of 0.15 μM, while clinical used NSAID indomethacin had IC50 of 0.6 μM.11 Moreover, ethanol extract of F. pinicola can suppress interferon-γ-induced inflammation marker, IP-10 in a dose-dependent manner.12 Therefore, in continuity of seeking anti-inflammatory agents for new drug development in our program, this study aimed at isolation of anti-inflammatory principals of F. pinicola using bioassay-guided fractionation. Superoxide anion generation and elastase release from human neutrophil were measured as inflammation markers. Normally, under acute inflammation, reactive oxygen species (ROS) and lysosomal enzymes (ex. elastase) will be released to kill microorganism. Once neutrophils persist beyond acute inflammation, chronic inflammation initiate and perpetuate, leading to various diseases.13 Currently, the known chemical constituents from F. pinicola mostly belong to steroids and triterpenoids.5 Minors are terpenoids, sesquiterpenoids and coumarins.5 Other related polypore species, such as Laetiporus sulphureus, Fomes fomentarius, Piptoporus betulinus and Laricifomes officinalis, are also found to rich in triterpenoids and organic acids. Organic solvent extracts from these five species have been evaluated for anti-oxidant, anti-microbial and cytotoxic effects.5 After screening the ethanol extract of F. pinicola as well as six fractions for anti-inflammatory activities, we focused on the purification of fractions 4 to 6 with significant bioactivities. Twelve undescribed sesquiterpenes were found and their structures were elucidated by 1H-, 13C-, 2D-NMR, MS, and ECD spectroscopic and spectrometric analyses. Among these twelve compounds, ten compounds with enough quantity were subjected to further evaluation for anti-inflammatory activities.
Results and discussion
The fruiting bodies of F. pinicola were refluxed with EtOH and the extract was concentrated under reduced pressure to give a dark-brown syrup. The EtOH crude extract was subjected to Diaion HP-20 gel column chromatography to afford six major fractions (FPs 1–6), being submitted for superoxide anion generation and elastase release inhibition assays. The results (Table S103†) showed that FP5 (10 μg) had the best inhibition ability of superoxide anion and FPs 4–6 (10 μg) displayed significant inhibition of elastase release (94, 95, 89%, respectively). Therefore, further isolation was performed mainly on fractions 4–6 (FPs 4–6) and these fractions afforded twelve undescribed sesquiterpenes (1–12). The structures of these compounds were elucidated as described below.Fomitopin A (1) was obtained as colorless needles with a pseudo-molecular formula of C15H18O4Na by HRESIMS (m/z 285.1097 [M + Na]+, calcd for 285.1097), representing seven unsaturation equivalents. The UV (λmax 287, 242 and 216 nm) and IR spectrum (νmax 3420, 3243, 1741 and 1423 cm−1) indicated the absorption of a hydroxyl group, a lactone group and an aromatic unit. The 1H NMR spectrum of 1 (Table 1) showed signals for two aromatic protons at δH 6.74 (1H, s, H-9) and 6.64 (1H, s, H-12), an oxymethylene at δH 4.85 (2H, s, H-15), and two methyl groups at δH 0.93 (3H, s, CH3-13) and 1.10 (3H, s, CH3-14), respectively. The 13C and DEPT-135 combined with HSQC NMR spectrum of 1 revealed 15 signals, of which were one carbonyl at δ 173.6, four aromatic quaternary carbons (δ 122.9, 134.4, 144.7, 152.9), two aromatic tertiary carbons (δ 115.1, 113.0), two quaternary carbons (δ 47.1, 50.7), four methylene carbons (δ 19.7, 29.0, 29.6, 64.4) and two methyl carbons (δ 19.8, 25.1). Analysis of HMBC spectrum (Fig. 3) revealed correlations of Me-13 (δ 0.93) and Me-14 (δ 1.10) to C-3 (δ 29.0), C-5 (δ 29.6), C-2 (δ 50.7) and C-6 (δ 47.1). Me-13 also showed correlation with C-1 (δ 173.6), while Me-14 correlated with C-7 (δ 134.4). Moreover, 2J, 3J-HMBC correlations from H-15 (δ 4.85) to C-9 (δ 115.1), C-10 (δ 122.9) and C-11 (δ 152.9); from H-9 (δ 6.7) to C-7 (δ 134.4), C-8 (δ 144.7), C-11 (δ 152.9) and C-15 (δ 64.4); and from H-12 (δ 6.64) to C-6 (δ 47.1), C-8 (δ 144.7), C-10 (δ 122.9) and C-11 (δ 152.9) constructed the basic skeleton of 1. 1H–1H COSY spectrum of 1 also revealed mutual correlations among H-3 (δ 2.17, 1.76), H-4 (δ 1.97) and H-5 (δ 1.98, 1.86). The relative configuration was determined by X-ray single-crystal diffraction using Cu Kα radiation and the result assigned C-2 and C-6 as S and R configurations, respectively (Fig. 4). These results were further supported by optical rotatory power ([α]25D −109.0). The negative value is the same as that of (−)-herbertenolide,14 while (3aR,9bS)-3a,8,9b-trimethyl-1,3,3a,9b-tetrahydrocyclopenta[c]chromen-4(2H)-one with opposite ring junction has positive value.15 Moreover, the proton spectrum also established the trans relationship between two vicinal methyl group. In order to confirm the absolute configuration of the ring junction, we performed simulation of ECD spectra via Gaussian 09 (Fig. 1). The simulated ECD spectrum showed the same trend as the experimental spectrum that a negative Cotton effect was observed at 249 nm and a positive Cotton effect was observed at 226 nm, which strongly supported 2S, 6R configurations. Therefore, the structure of compound 1 was assigned as shown based on above elucidations (Fig. 2). Although the structure of 1 is simple, there are not many similar sesquiterpenes discovered in nature. The most similar compound is (−)-herbertenolide14 with the same lactone herbertenoid structure, but different substitution on phenyl rings.
| Position | 1a | 2a | 3b | 4a | 5a | 6a | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | |
| a 1H and 13C NMR data (δ) were measured in chloroform-d at 400 and 100 MHz.b 1H and 13C NMR data (δ) were measured in acetone-d6 at 400 and 100 MHz. | ||||||||||||
| 1 | 173.6 | 173.6 | 172.8 | 172.5 | 172.4 | 172.8 | ||||||
| 2 | 50.7 | 50.7 | 51.8 | 51.1 | 51.1 | 50.1 | ||||||
| 3 | 29.0 | 1.76 dd (8.4, 13.9) | 29.0 | 1.76 m | 41.4 | 1.86 d (13.7) | 40.5 | 2.23 dd (7.7, 13.1) | 40.5 | 2.24 dd (5.2, 13.1) | 40.5 | 1.79 d (14.1) |
| 2.17 t (13.9) | 2.17 t (11.2) | 2.43 dd (8.2, 13.7) | 2.34 dd (7.7, 13.4) | 2.35 dd (7.9, 13.1) | 2.68 dd (8.1, 14.1) | |||||||
| 4 | 19.7 | 1.97 m | 19.7 | 1.87 m | 71.0 | 4.70 m | 71.2 | 4.73 qd (1.0, 6.0) | 71.3 | 4.75 qd (1.9, 6.7) | 71.5 | 4.78 qd (2.5, 7.7) |
| 5 | 29.6 | 1.86 t (8.7) | 29.6 | 1.86 m | 41.3 | 2.13 m | 40.7 | 1.90 dd (0.7, 14.4) | 40.7 | 1.91 dd (0.8, 13.5) | 40.9 | 2.04 dd (5.6, 13.3) |
| 1.98 m | 1.93 m | 2.22 dd (9.1, 14.0) | 2.45 dd (8.4, 14.4) | 2.46 dd (9.0, 13.5) | 2.44 dd (7.7, 13.3) | |||||||
| 6 | 47.1 | 47.1 | 47.2 | 46.5 | 46.5 | 47.6 | ||||||
| 7 | 134.4 | 134.3 | 134.3 | 134.1 | 134.1 | 133.7 | ||||||
| 8 | 144.7 | 144.6 | 145.5 | 144.4 | 144.5 | 144.5 | ||||||
| 9 | 115.1 | 6.74 s | 115.3 | 6.71 s | 116.4 | 6.94 s | 115.6 | 6.72 s | 115.3 | 6.74 s | 115.3 | 6.74 s |
| 10 | 122.9 | 120.8 | 124.8 | 120.6 | 123.0 | 123.2 | ||||||
| 11 | 152.9 | 152.9 | 152.4 | 152.9 | 152.9 | 153.0 | ||||||
| 12 | 113.0 | 6.64 s | 112.9 | 6.63 s | 112.3 | 6.62 s | 112.6 | 6.01 s | 112.9 | 6.62 s | 112.8 | 6.63 s |
| 13 | 19.8 | 0.93 s | 19.8 | 0.93 s | 20.2 | 0.87 s | 20.4 | 0.90 s | 20.4 | 0.91 s | 26.0 | 1.15 s |
| 14 | 25.1 | 1.10 s | 25.1 | 1.11 s | 26.6 | 1.28 s | 26.1 | 1.34 s | 26.2 | 1.35 s | 20.6 | 1.09 s |
| 15 | 64.4 | 4.85 s | 71.8 | 4.66 s | 68.5 | 4.53 s | 73.7 | 4.62 s | 64.4 | 4.86 s | 64.4 | 4.86 s |
| 16 | 66.5 | 3.63 q (6.9) | 66.6 | 3.57 q (6.4) | 58.5 | 3.47 s | ||||||
| 17 | 15.0 | 1.29 t (6.9) | 15.5 | 1.2 t (6.4) | ||||||||
| Position | 7a | 8b | 9a | 10a | 11a | 12b | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | |
| 1 | 165.8 | 173.4 | 172.3 | 181.8 | 178.2 | 25.6 | 1.20 s | |||||
| 2 | 54.4 | 51.7 | 50.3 | 53.0 | 55.7 | 44.9 | ||||||
| 3 | 206.5 | 29.8 | 1.66 dd (5.7, 14.3) | 29.0 | 1.80 m | 37.9 | 1.84 t (8.8) | 37.6 | 1.61 m | 41.5 | 1.57 m | |
| 2.08 m | 2.21 m | 2.44 td (2.0, 8.8) | 2.47 m | 1.67 m | ||||||||
| 4 | 33.9 | 2.64 m | 20.2 | 1.94 m | 19.5 | 2.00 m | 20.9 | 1.98 m | 21.7 | 1.82 m | 20.3 | 1.75 m |
| 1.96 m | ||||||||||||
| 5 | 25.1 | 2.16 m | 30.4 | 1.84 t (8.3) | 29.5 | 1.93 m | 36.8 | 1.64 t (9.9) | 38.9 | 1.81 m | 39.7 | 1.77 m |
| 2.28 m | 1.95 m | 2.03 m | 2.55 td (5.9, 9.9) | 2.44 m | 2.53 t (4.6) | |||||||
| 6 | 43.7 | 47.3 | 48.0 | 55.6 | 52.6 | 52.1 | ||||||
| 7 | 131.7 | 125.2 | 144.0 | 157.0 | 147.5 | 146.2 | ||||||
| 8 | 143.9 | 145.4 | 144.7 | 116.7 | 7.05 br s | 115.6 | 6.89 d (2.3) | 147.9 | ||||
| 9 | 115.5 | 6.78 s | 104.6 | 6.55 s | 119.1 | 7.22 s | 160.8 | 155.5 | 118.7 | 6.84 s | ||
| 10 | 123.7 | 144.9 | 118.8 | 118.8 | 125.6 | 118.3 | ||||||
| 11 | 154.5 | 142.5 | 158.9 | 132.6 | 7.44 d (8.6) | 127.5 | 7.11 d (7.4) | 155.1 | ||||
| 12 | 112.5 | 6.74 s | 112.2 | 6.61 s | 114.2 | 6.76 s | 119.7 | 7.07 br d (8.6) | 119.3 | 6.87 dd (2.8, 7.4) | 118.5 | 6.98 s |
| 13 | 16.3 | 1.11 s | 20.1 | 0.89 s | 19.8 | 0.96 s | 23.0 | 0.89 s | 23.3 | 0.86 s | 27.0 | 0.78 s |
| 14 | 25.2 | 1.23 s | 25.7 | 1.03 s | 24.8 | 1.16 s | 26.3 | 1.38 s | 27.0 | 1.34 s | 22.5 | 1.41 s |
| 15 | 64.4 | 4.89 s | 195.4 | 9.83 s | 195.8 | 9.82 s | 61.8 | 4.70 s | 195.0 | 9.75 s | ||
 | ||
| Fig. 1 Simulated ECD spectra of fomitopin A (A) and its isomer (B). Experimental ECD spectrum of fomitopin A (C). | ||
 | ||
| Fig. 2 Structures of fomitopins A–L (1–12). | ||
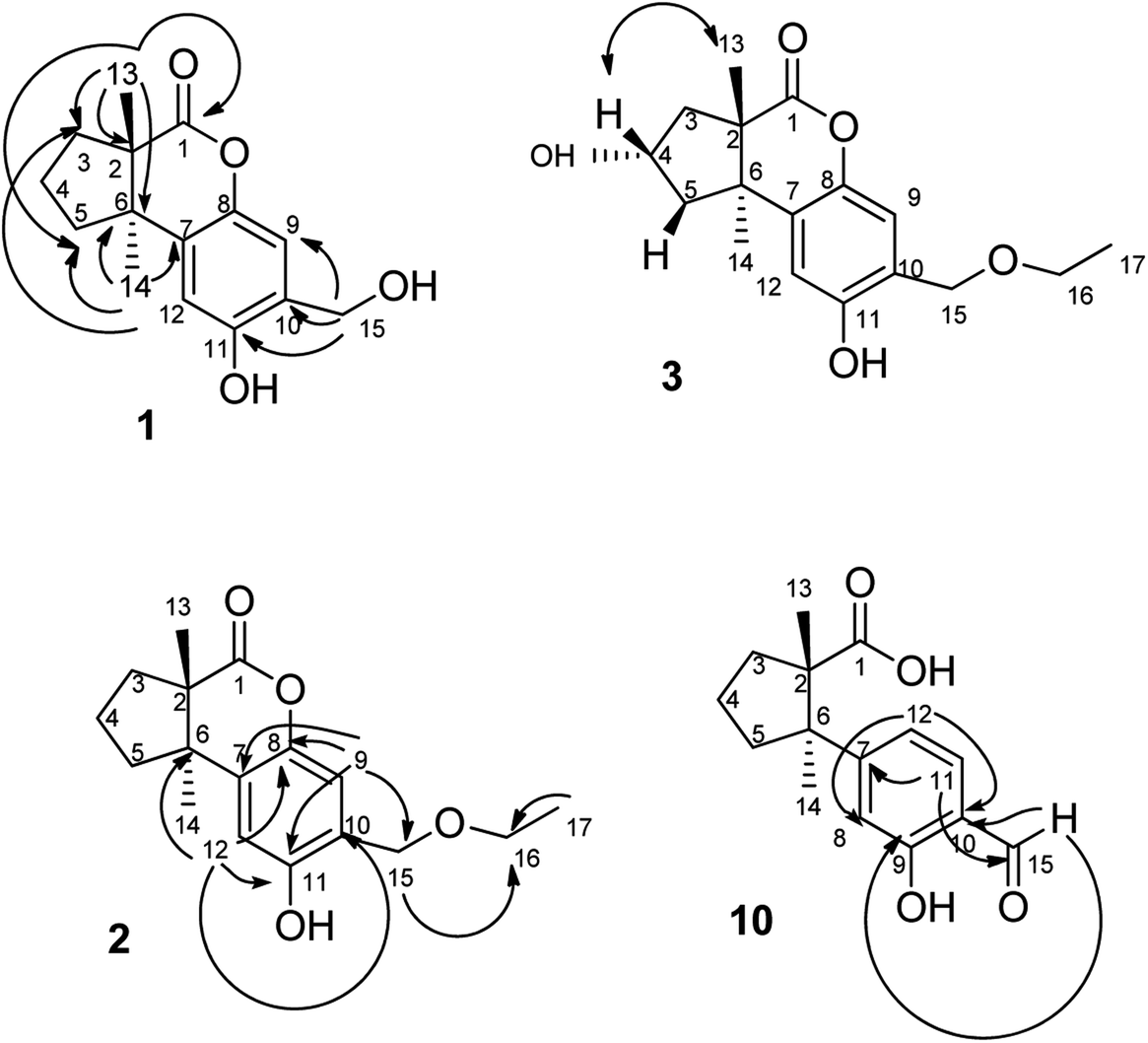 | ||
| Fig. 3 HMBC (→, 1H → 13C) and NOESY (↔, 1H → 1H) correlations of 1, 2, 3 and 10. | ||
 | ||
| Fig. 4 ORTEP drawing of fomitopin A (1). | ||
The UV and IR absorption characteristics of 2–12 were all similar to those of 1. Fomitopin B (2) was obtained as colorless solid and its molecular formula was determined as C17H22O4 by HRESIMS analysis. The 1H and 13C NMR spectral signals of 2 were also similar to those of 1. The only difference was the occurrence of an ethyl group including an oxymethylene at δH 3.63 (2H, q, J = 6.9 Hz, H-16)/δC 66.5 and a methyl group at δH 1.29 (3H, t, J = 6.9 Hz, CH3-17)/δC 15.0. HMBC spectrum of 2 displayed correlations between H-17 (δ 1.29) and C-16 (δ 66.5), and between H-15 (δ 4.66) and C-16 (δ 66.5), suggesting an ethoxy group to be attached at C-15. The ECD spectrum of 2 showed similar pattern with 1, indicating the same trans ring junction and supported the same configurations of 2S and 6R in 2.
The 1H and 13C NMR spectra of fomitopin C (3) showed similar characteristics with those of 2, only with the difference of observation of one oxymethine at δH 4.70 (1H, m, H-4)/δC 71.0. The hydroxyl substitution was evidenced at C-4 according to the 1H–1H COSY correlation between H-4 (δ 4.70) and H-3 (δ 2.43, 1.86), and between H-4 and H-5 (δ 2.22, 2.13). The ECD spectral analysis of 3 revealed similar results to those of 1, assigned as 2S and 6R, respectively. Moreover, the absolute configuration at C-4 can be determined as S via NOE correlations from H-4 (δ 4.70) to Me-13 (δ 0.87) and therefore, the structure of 3 was established as shown. Comparison of 1H and 13C NMR spectra of fomitopin D (4) and 3, the minor differences were found including the disappearance of an oxymethylene signal and downfield shift of the methyl group [δH 3.47 (3H, s, H-16)/δC 58.5]. HMBC spectrum of 4 displayed correlation from H-15 (δ 4.62) to C-16 (δ 58.5), revealing a methoxy substitution at C-15 instead of a methyl group in 3. The ECD and NOESY spectral analysis of 4 determined the same configurations as those of 3 (2S, 4S, 6R).
The HRESIMS analytical data of fomitopins E (5) and F (6) revealed the same molecular formula, and their 1H and 13C NMR spectra were almost identical and very close to those of 4 with the disappearance of a methyl group only, suggesting a free hydroxyl functionality at C-15. The same trans ring junction (2S, 6R) was assigned via the ECD spectra of 5 and 6. Further NOESY experiments of 6 showed correlations from H-4 (δ 4.79) to Me-14 (δ 1.09), determining OH-4 as β orientation, while OH-4 in 5 is α-orientated supported by correlation from H-4 (δ 4.75) to Me-13 (δ 0.91). Therefore, 5 and 6 were epimers and their structures are shown in Fig. 2. Although compound 2 is only one ethoxy different with 1 and compound 3 and 4 are respectively one methoxy or ethoxy different from compound 5, we think they are natural products, not artifacts. The reason is that refluxing condition is around 80–90 °C in weakly acidic condition (ethanol). Thus, SN2 substitution is not favored in acidic condition while SN1 is not favored in primary carbon. Therefore, these compounds originate from plant materials without doubt.
Comparison of 1H and 13C NMR spectra of fomitopin G (7) with those of 5, loss of a 4-hydroxy signal but occurrence of a carbonyl carbon at δ 206.5 were observed. HMBC spectrum of 7 showed correlations from both Me-13 (δ 1.11) and Me-14 (δ 1.23) to C-3 (δ 206.5), and 1H–1H COSY correlations between H-4 (δ 2.64) and H-5 (δ 2.28, 2.16) can be observed, indicating the carbonyl group to be at C-3. The same configurations (2S, 6R) was assigned via ECD spectrum, and fomitopin G (7) was established as shown.
The main skeleton of fomitopin H (8) was similar to 1. The minor differences included loss of a methylene signal and downfield shift of a quaternary carbon in both 1H and 13C NMR spectra, which indicated loss of a methylene group and direct substitution of a hydroxyl on aromatic ring. Analysis of HMBC correlations from H-9 (δ 6.74) to C-7 (δ 125.2), C-8 (δ 145.4), C-11 (δ 142.5), and C-10 (δ 144.9); and from H-12 (δ 6.6) to C-6 (δ 47.3), C-8 (δ 145.4), C-10 (δ 144.9) and C-11 (δ 142.5), confirmed C-10 hydroxyl substitution. ECD spectrum of 8 showed similar pattern with 1 that a negative Cotton effect at 253 nm and a positive Cotton effect at 222 nm were found, indicating the same absolute configurations of 8 with 1. Conclusively, 8 was belonged to an undescribed scaffold norsesquiterpene, and its structure was established as shown.
HRESIMS analysis of fomitopin I (9) revealed a molecular formula of C15H16O4. Its IR spectrum showed the typical band for the presence of aromatic aldehyde at 1659 cm−1. The 1H and 13C NMR spectra of 9 were close to those of 8, with the minor difference of the replacement of phenol group with an aldehyde signal at δH 9.83 (1H, s, H-15)/δC 195.4. 2J, 3J-HMBC correlations from H-9 (δ 7.22) to C-15 (δ 195.4), and from H-15 (δ 9.83) to C-9 (δ 119.1) and C-11 (δ 158.9), evidenced the aldehyde group to be attached at C-10. The ECD spectrum of 9 showed similar pattern with 1. According to the above results, the structure of 9 was determined.
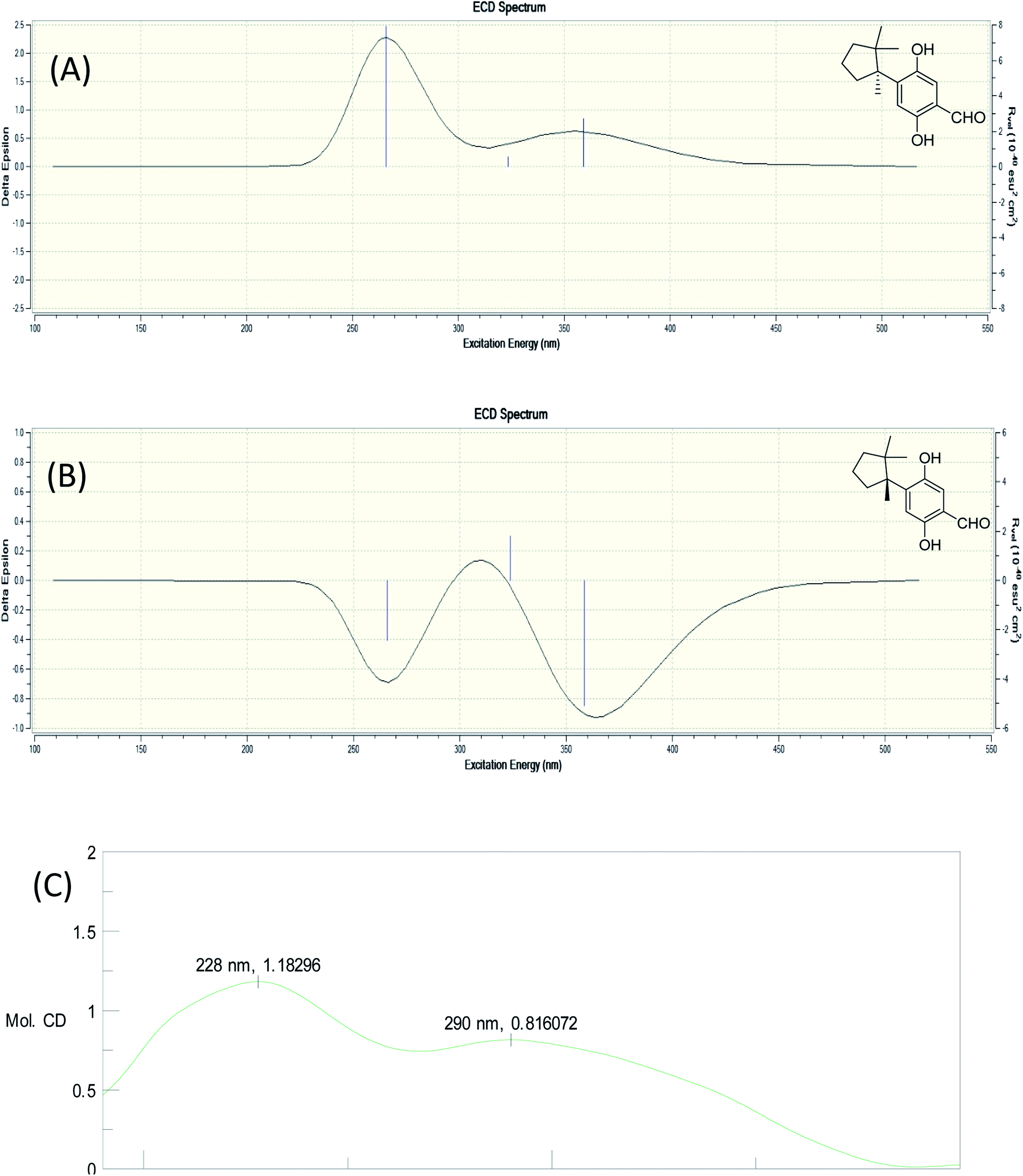
The molecular formula of fomitopin J (10) was established through HRESIMS analysis. Its IR spectrum displayed the characteristic absorption bands of carboxylic acid (1695 cm−1) and aromatic aldehyde (1654 cm−1). The 1H NMR spectrum of 10 only exhibited minor differences from that of 9, including the replacement of two aromatic singlets with one ABX set of signals at δH 7.44 (1H, d, J = 8.6 Hz, H-11), 7.07 (1H, br d, J = 8.6 Hz, H-12), and 7.05 (1H, br s, H-8). In addition, the change of the lactonic carbon with carboxylic acid functionality (δC 178.2) was noticed, suggesting opening of the lactone ring. Further 3J-HMBC correlations from H-8 (δ 7.07) to C-10 (δ 118.8) and C-12 (δ 116.7) supported the substitution pattern of aromatic ring. The trans ring junction was proposed via NOE correlations from Me-13 (δ 0.89) to H-3β (δ 2.44) and H-5β (δ 2.55), and from Me-14 (δ 1.38) to H-3α (δ 1.84) and H-5α (δ 1.64). The 1H and 13C NMR spectra of fomitopin-K (11) were close to those of 1 and the observed differences in 11 were similar to those of 10, suggesting the ring opening of the lactone group to carboxylic acid functionality. The IR spectrum of 11 also revealed the presence of carboxylic acid (1693 cm−1). Further HMBC analysis displayed correlations from H-8 (δ 8.89) to C-6 (δ 52.6), C-9 (δ 155.5), C-10 (δ 125.6) and C-12 (δ 119.3), which also supported the speculation. Simulated ECD of 11 revealed the same pattern as the experimental spectrum that two positive Cotton effects were observed around 230 nm and 280 nm, supporting the configurations of 2S, 6R (Fig. 5). ECD spectrum of 10 revealed the same pattern as those of 11. Therefore, the structures of 10 and 11 were drawn as shown.
 | ||
| Fig. 5 Simulated ECD spectra of fomitopin K (A) and its isomer (B). Experimental ECD spectrum of fomitopin K (C). | ||
Fomitopin L (12) was obtained as colorless solids with a pseudo-molecular formula of C15H20O4Na determined by HRESIMS ([M + Na]+ peak at m/z 271.1306, calcd for 271.1304), representing six unsaturation equivalents. The UV and IR characteristics of 12 revealed the lack of a carboxylic acid functionality as compared to 10. The 1H, 13C, and HSQC NMR spectra revealed two aromatic protons at δH 6.84 (1H, s, H-9)/δC 118.7 and 6.98 (1H, s, H-12)/δC 118.5; one aldehyde at δH 9.75 (1H, s, H-15)/δC 195.0; three methyls at δH 1.20 (3H, s, Me-1)/δC 25.6, δH 0.78 (3H, s, Me-13)/δC 27.0, and δH 1.41 (3H, s, Me-14)/δC 22.5; four aromatic quaternary carbons at δ 118.3, 146.2, 147.9, 155.1 and two quaternary carbons at δ 44.9, 52.1, respectively. In the HMBC spectrum, 2J, 3J-HMBC correlations were observed from Me-1 (δ 1.20) to C-2 (δ 44.9), C-3 (δ 41.5), C-5 (δ 39.7), C-6 (δ 52.1) and C-13 (δ 27.0); from Me-13 (δ 0.78) to C-2 (δ 44.9), C-3 (δ 41.5), C-5 (δ 39.7), and C-6 (δ 52.1); from Me-14 (δ 1.41) to C-2 (δ 44.9), C-3 (δ 41.5), C-5 (δ 39.7), C-6 (δ 52.1), and C-7(δ 144.0). In addition, 1H–1H COSY correlations between H-3 (δ 1.67, 1.57) and H-4 (δ 1.75), and between H-4 (δ 1.75) and H-5 (δ 2.53, 1.77) indicated that a 1-methyl-2,2-dimethylcyclopentyl moiety was connected to the aromatic ring. The aromatic substituted pattern was also confirmed by HMBC correlations of H-9 (δ 7.22) to C-7 (δ 146.2), C-8 (δ 147.9), C-11 (δ 155.1), and C-15 (δ 195.0); of H-12 (δ 6.76) to C-6 (δ 52.1), C-8 (δ 147.9), C-10 (δ 118.3), and C-11 (δ 155.1); and of H-15 (δ 9.83) to C-9 (δ 118.7), C-10 (δ 118.3) and C-11 (δ 155.1), respectively. Comparison between the simulated ECD spectra of 12 and its isomer, it indicated the R configuration of 12 (Fig. 6). Therefore, the structure of 12 was established as shown.
 | ||
| Fig. 6 Simulated ECD spectra of fomitopin L (A) and its isomer (B). Experimental ECD spectrum of fomitopin L (C). | ||
The anti-inflammatory activities of the isolates were evaluated in a human neutrophil cell model. Ten isolated compounds with enough quantities were submitted for their inhibition on the production of superoxide anion and elastase in human neutrophils with formyl-L-methionyl-L-leucyl-L-phenylalanine/cytochalasin B (fMLP/CB) activation (Table 2).16 Among the tested compounds, 11 exhibited the most potent anti-inflammatory activity with IC50 of 0.81 ± 0.15 μM for inhibition of superoxide anion generation and IC50 of 0.74 ± 0.12 μM for inhibition of elastase release. Compound 12 and 10 also exhibited moderate inhibition of superoxide anion generation with IC50 of 1.72 ± 0.71 and 1.66 ± 0.81 μM, respectively, compared with the reference compound LY294002 (IC50 0.4 ± 0.02 μM). Among all the compounds, we found the structures with either carboxylic acid (10, 11) or aldehyde (9, 12) would possess good anti-inflammatory activities. Superoxide anion generation and elastase release are markers for inflammations. The different inhibition ability of compounds between these two markers implied the compounds may affect different signal pathways upon the production or release of these two markers. For example, the neutrophil activating agent in this study is fMLP, which can activate fMLP receptor on neutrophil and subsequent p38 mitogen-activated protein kinase (MAPK) signaling pathway to trigger both inflammation markers' production and release.17 Other mechanisms, such as elevation of intracellular cAMP level, is believed to suppress the activation of neutrophil.18 Further mechanism studies are needed to address the difference of inhibitory activity. Although the structure of compounds isolated are simple, these kind of sesquiterpenes are rare in the literature and several types of skeletons are discovered in this study. Further, their absolute configuration were solved via X-ray crystal structure and simulated ECD and their anti-inflammatory activities were screened, all of which contributes to the progress of natural products development. Moreover, the newly characterized sesquiterpenes can be potential leads for the further anti-inflammatory studies.
| Compound | IC50a (μM) | |
|---|---|---|
| Superoxide anion generation | Elastase release | |
| a Concentration necessary for 50% inhibition. The results are presented as the means ± SEM (n = 3–5).b A phosphatidylinositol-3-kinase inhibitor was used as a positive control. | ||
| 1 | >10 | >10 |
| 2 | >10 | >10 |
| 4 | >10 | >10 |
| 6 | >10 | >10 |
| 7 | >10 | >10 |
| 8 | >10 | >10 |
| 9 | 4.02 ± 1.07 | 6.65 ± 0.68 |
| 10 | 1.66 ± 0.81 | 4.60 ± 0.26 |
| 11 | 0.81 ± 0.15 | 0.74 ± 0.12 |
| 12 | 1.72 ± 0.71 | 3.09 ± 0.45 |
| LY294002b | 0.4 ± 0.02 | 1.5 ± 0.3 |
Experimental section
General experimental procedures
Melting points were recorded on a Yanaco MP-S3 micromelting point measuring apparatus without correction. Optical rotations were measured using a JASCO P-2000 digital polarimeter. UV spectra were examined at room temperature on a U-0080-D UV-Vis spectrophotometer. IR spectra were obtained with a PerkinElmer FT-IR Spectrum RX I spectrophotometer. 1H and 13C NMR spectra were recorded on a Bruker AV III 400 NMR spectrometer. Chemical shifts are shown in δ values (ppm) with tetramethylsilane as an internal standard. The ESIMS and HRESIMS were taken on a Bruker APEX II FT-MS spectrometer (positive-ion mode). ECD spectra were obtained on a JASCO J-720 spectrometer. X-ray single crystal diffraction was measured in National Chung Hsing University with a Bruker D8 VENTURE diffractometer with a Photon 100 CMOS detector system equipped with a Cu Incoatec IμS microfocus source (λ = 1.54178 Å). Column chromatography (CC) was performed on silica (70–230 mesh and 230–400 mesh, Merck) and Diaion HP-20 (Mitsubishi) gels. Preparative thin-layer chromatography (TLC) was conducted on Merck precoated silica gel 60 F254 plates, using UV light to visualize the spots. High-performance liquid chromatography (HPLC) was performed on a Shimadzu LC-20AT series pumping system equipped with a Shimadzu SPD-20A UV-Vis detector, and a SIL-10AF autosampling system at ambient temperature and a RP-18 column (Ascentis C18, 5 μm, 10 mm × 25 cm).Fungus material
The fruiting bodies of Fomitopsis pinicola (Sw.) P. Karst (Fomitopsidaceae) were collected in July 2012 at Qilai Mountain in Hualien, Taiwan. The fungus material was identified by Prof. Jin-Bin Wu, Institute of Pharmaceutical Chemistry and Department of Pharmacy, China Medical University, Taichung, Taiwan. A voucher specimen (TSWu-20130711) was deposited in the School of Pharmacy, National Cheng Kung University, Taiwan.Extraction and isolation
The fresh fruiting bodies (13.7 kg) were cut into small pieces and consecutively refluxed with ethanol for 8 hours for three times by Jen Li biotech company. The ethanol was removed and evaporated in vacuo to yield 3320 g dried extract. The water layer was evaporated in vacuo (260 g dried extract). One kilogram of the ethanol extract was subjected to column chromatography on Diaion HP-20 with a step-gradient of water-methanol to give 6 fractions (FP-1–FP-6). And 10 g of ethanol extract was separated at the same condition and was sent to test the bioassay. The bioactive fractions were FP4–FP6. Repeated silica gel column chromatography of the FP-4 fraction (4.64 g) eluted with chloroform–methanol (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) or isopropyl ether–methanol (7:1 or 19:1) system yielded four sesquiterpenoids, fomitopin-D (4, 26.7 mg), fomitopin-G (7, 7.3 mg), fomitopin-E (5, 17.11 mg) and fomitopin-F (6, 4.39 mg). The FP-5 fraction (15.01 g) eluted with chloroform–acetone (15:1) yielded fomitopin-A (1, 28.55 mg). Subfraction of FP-5 was chromatographed over silica gel eluted with isopropyl alcohol to give fomitopin-H (8, 22.3 mg). Another subfraction of FP-5 eluted with chloroform–methanol (15:1) to give fomitopin-K (11, 9.8 mg). Fomitopin-C (3, 1.2 mg) was obtained from subfraction of FP-5 via semipreparative HPLC using RP-18 column and methanol–water (40:60) system (retention time: 20.0 min). The last fraction (FP-6, 500 g) eluted with isopropyl ether–acetone yielded 11 subfractions (FP-6-1–P-6-11). FP-6-2 was chromatographed over silica gel with isopropyl ether–acetone (15:1) and then isopropyl ether–chloroform–isopropyl alcohol (10:19:1) system to obtain fomitopin-I (9, 2.0 mg) and fomitopin B (2, 2.6 mg). Fomitopin-J (10, 9.1 mg) was obtained from subfraction of FP-6-10 via semipreparative HPLC using RP-18 column and methanol–water (80:20 with 0.1% formic acid) system (retention time: 6.0 min). Fomitopin-L (12, 1.1 mg) was also obtained from this FP-6-2-5 via thin-layer chromatography using chloroform–methanol (15:1).
ε) 287 (1.7), 242 (2.8), 216 (3.2) nm; ECD (MeOH) (mol. CD) 249 (−21.1), 226 (+13.8) nm; IR (KBr) νmax 3420, 3243, 2972, 1741, 1423, 1377, 1171, 1073, 879, 754 cm−1; 1H NMR and 13C NMR, see Table 1; HRESIMS m/z 285.1097 ([M + Na]+ calcd for C15H18O4Na, 285.1097).ε): 287 (1.2), 241 (2.3), 216 (3.0) nm; ECD (MeOH) (Mol. CD) 249 (−4.9), 224 (2.6) nm; IR (KBr) νmax 3373, 2967, 2879, 1768, 1741, 1587, 1425, 1175, 1098, 755 cm−1; 1H NMR and 13C NMR: see Table 1; HRESIMS m/z 313.1407 ([M + Na]+ calcd 313.1410).ε): 287 (1.2), 241 (1.9), 217 (3.0) nm; ECD (MeOH) (mol. CD) 253 (−6.1), 222 (3.7) nm; IR (KBr) νmax 3362, 2928, 1760, 1587, 1427, 1381, 1267, 1178, 1015, 755 cm−1; 1H NMR and 13C NMR: see Table 1; HRESIMS m/z 329.1358 ([M + Na]+ calcd 329.1359).ε): 287 (1.1), 242 (1.7), 214 (2.6) nm; ECD (MeOH) (mol. CD) 251 (−9.9), 222 (5.6) nm; IR (KBr) νmax 3309, 2964, 2829, 1766, 1759, 1748, 1627, 1496, 1427, 1264, 1174, 1080, 755 cm−1. 1H NMR and 13C NMR: see Table 1; HRESIMS m/z 315.1200 ([M + Na]+ calcd 315.1202).ε) 287 (1.6), 243 (2.7), 215 (3.3) nm; ECD (MeOH) (Mol. CD) 251 (−10.0), 222 (4.6) nm; IR (KBr) νmax 3708, 2969, 2873, 1755, 1427, 1278, 1173, 1075, 1014, 754 cm−1; 1H NMR and 13C NMR: see Table 1; HRESIMS m/z 301.1045 ([M + Na]+ calcd 301.1046).ε) 287 (1.0), 242 (1.8), 212 (3.3) nm; ECD (MeOH) (mol. CD) 251 (−8.0), 222 (3.9) nm; IR (KBr) νmax 3706, 2957, 2845, 1755, 1427, 1280, 1174, 1084, 1014, 755 cm−1; 1H NMR and 13C NMR: see Table 1; HRESIMS m/z 301.1045 ([M + Na]+ calcd 301.1046).ε) 286 (1.2), 237 (2.7), 215 (3.3) nm; ECD (MeOH) (mol. CD) 252 (−3.3), 222 (1.4) nm; IR (KBr) νmax 3381, 2930, 2877, 1778, 1747, 1427, 1282, 1174, 1080, 756 cm−1.1H NMR and 13C NMR: see Table 1; HRESIMS m/z 299.0887 ([M + Na]+ calcd 299.0889).ε) 291 (1.6), 248 (1.2), 214 (3.6) nm; ECD (MeOH) (mol. CD) 253 (−6.8), 222 (2.9) nm; IR (KBr) νmax 3386, 2967, 2931, 1746, 1628, 1509, 1444, 1138, 1099, 756 cm−1; 1H NMR and 13C NMR: see Table 1; HRESIMS m/z 271.0938 ([M + Na]+ calcd 271.0940).ε) 339, 258, 231 nm; ECD (MeOH) (mMol. CD) 240 (−6.8), 216 (2.5) nm; IR (KBr) νmax 2962, 2930, 1775, 1659, 1576, 1432, 1378, 1223, 1142, 1068, 883 cm−1; 1H NMR and 13C NMR: see Table 1; HRESIMS m/z 283.0940 ([M + Na]+ calcd 283.0940).ε) 329 (1.4), 263 (3.5), 217 (3.3) nm; ECD (MeOH) (mol. CD) 270 (1.3) nm; IR (KBr) νmax 3684, 2963, 2879, 1695, 1654, 1561, 1506, 1458, 1064, 757 cm−1; 1H NMR and 13C NMR: see Table 1; HRESIMS m/z 285.1098 ([M + Na]+ calcd 285.1097).ε) 276 (2.2), 230 (3.4), 218 (2.4) nm; ECD (MeOH) (mol. CD) 283 (0.5), 231 (1.3) nm; IR (KBr) νmax 3328, 2970, 1693, 1623, 1579, 1509, 1417, 1379, 1209, 757 cm−1; 1H NMR and 13C NMR: see Table 1; HRESIMS m/z 287.1255 ([M + Na]+ calcd 287.1253).ε) 270 (0.6), 219 (3.6) nm; ECD (MeOH) (mol. CD) 290 (0.8), 228 (1.2) nm; IR (KBr) νmax 3472, 2955, 2929, 2879, 1666, 1617, 1440, 1410, 1246, 1132 cm−1; 1H NMR and 13C NMR: see Table 1; HRESIMS m/z 271.1306 ([M + Na]+ calcd 271.1304).
:1) or isopropyl ether–methanol (7:1 or 19:1) system yielded four sesquiterpenoids, fomitopin-D (4, 26.7 mg), fomitopin-G (7, 7.3 mg), fomitopin-E (5, 17.11 mg) and fomitopin-F (6, 4.39 mg). The FP-5 fraction (15.01 g) eluted with chloroform–acetone (15:1) yielded fomitopin-A (1, 28.55 mg). Subfraction of FP-5 was chromatographed over silica gel eluted with isopropyl alcohol to give fomitopin-H (8, 22.3 mg). Another subfraction of FP-5 eluted with chloroform–methanol (15:1) to give fomitopin-K (11, 9.8 mg). Fomitopin-C (3, 1.2 mg) was obtained from subfraction of FP-5 via semipreparative HPLC using RP-18 column and methanol–water (40:60) system (retention time: 20.0 min). The last fraction (FP-6, 500 g) eluted with isopropyl ether–acetone yielded 11 subfractions (FP-6-1–P-6-11). FP-6-2 was chromatographed over silica gel with isopropyl ether–acetone (15:1) and then isopropyl ether–chloroform–isopropyl alcohol (10:19:1) system to obtain fomitopin-I (9, 2.0 mg) and fomitopin B (2, 2.6 mg). Fomitopin-J (10, 9.1 mg) was obtained from subfraction of FP-6-10 via semipreparative HPLC using RP-18 column and methanol–water (80:20 with 0.1% formic acid) system (retention time: 6.0 min). Fomitopin-L (12, 1.1 mg) was also obtained from this FP-6-2-5 via thin-layer chromatography using chloroform–methanol (15:1).
ε) 287 (1.7), 242 (2.8), 216 (3.2) nm; ECD (MeOH) (mol. CD) 249 (−21.1), 226 (+13.8) nm; IR (KBr) νmax 3420, 3243, 2972, 1741, 1423, 1377, 1171, 1073, 879, 754 cm−1; 1H NMR and 13C NMR, see Table 1; HRESIMS m/z 285.1097 ([M + Na]+ calcd for C15H18O4Na, 285.1097).ε): 287 (1.2), 241 (2.3), 216 (3.0) nm; ECD (MeOH) (Mol. CD) 249 (−4.9), 224 (2.6) nm; IR (KBr) νmax 3373, 2967, 2879, 1768, 1741, 1587, 1425, 1175, 1098, 755 cm−1; 1H NMR and 13C NMR: see Table 1; HRESIMS m/z 313.1407 ([M + Na]+ calcd 313.1410).ε): 287 (1.2), 241 (1.9), 217 (3.0) nm; ECD (MeOH) (mol. CD) 253 (−6.1), 222 (3.7) nm; IR (KBr) νmax 3362, 2928, 1760, 1587, 1427, 1381, 1267, 1178, 1015, 755 cm−1; 1H NMR and 13C NMR: see Table 1; HRESIMS m/z 329.1358 ([M + Na]+ calcd 329.1359).ε): 287 (1.1), 242 (1.7), 214 (2.6) nm; ECD (MeOH) (mol. CD) 251 (−9.9), 222 (5.6) nm; IR (KBr) νmax 3309, 2964, 2829, 1766, 1759, 1748, 1627, 1496, 1427, 1264, 1174, 1080, 755 cm−1. 1H NMR and 13C NMR: see Table 1; HRESIMS m/z 315.1200 ([M + Na]+ calcd 315.1202).ε) 287 (1.6), 243 (2.7), 215 (3.3) nm; ECD (MeOH) (Mol. CD) 251 (−10.0), 222 (4.6) nm; IR (KBr) νmax 3708, 2969, 2873, 1755, 1427, 1278, 1173, 1075, 1014, 754 cm−1; 1H NMR and 13C NMR: see Table 1; HRESIMS m/z 301.1045 ([M + Na]+ calcd 301.1046).ε) 287 (1.0), 242 (1.8), 212 (3.3) nm; ECD (MeOH) (mol. CD) 251 (−8.0), 222 (3.9) nm; IR (KBr) νmax 3706, 2957, 2845, 1755, 1427, 1280, 1174, 1084, 1014, 755 cm−1; 1H NMR and 13C NMR: see Table 1; HRESIMS m/z 301.1045 ([M + Na]+ calcd 301.1046).ε) 286 (1.2), 237 (2.7), 215 (3.3) nm; ECD (MeOH) (mol. CD) 252 (−3.3), 222 (1.4) nm; IR (KBr) νmax 3381, 2930, 2877, 1778, 1747, 1427, 1282, 1174, 1080, 756 cm−1.1H NMR and 13C NMR: see Table 1; HRESIMS m/z 299.0887 ([M + Na]+ calcd 299.0889).ε) 291 (1.6), 248 (1.2), 214 (3.6) nm; ECD (MeOH) (mol. CD) 253 (−6.8), 222 (2.9) nm; IR (KBr) νmax 3386, 2967, 2931, 1746, 1628, 1509, 1444, 1138, 1099, 756 cm−1; 1H NMR and 13C NMR: see Table 1; HRESIMS m/z 271.0938 ([M + Na]+ calcd 271.0940).ε) 339, 258, 231 nm; ECD (MeOH) (mMol. CD) 240 (−6.8), 216 (2.5) nm; IR (KBr) νmax 2962, 2930, 1775, 1659, 1576, 1432, 1378, 1223, 1142, 1068, 883 cm−1; 1H NMR and 13C NMR: see Table 1; HRESIMS m/z 283.0940 ([M + Na]+ calcd 283.0940).ε) 329 (1.4), 263 (3.5), 217 (3.3) nm; ECD (MeOH) (mol. CD) 270 (1.3) nm; IR (KBr) νmax 3684, 2963, 2879, 1695, 1654, 1561, 1506, 1458, 1064, 757 cm−1; 1H NMR and 13C NMR: see Table 1; HRESIMS m/z 285.1098 ([M + Na]+ calcd 285.1097).ε) 276 (2.2), 230 (3.4), 218 (2.4) nm; ECD (MeOH) (mol. CD) 283 (0.5), 231 (1.3) nm; IR (KBr) νmax 3328, 2970, 1693, 1623, 1579, 1509, 1417, 1379, 1209, 757 cm−1; 1H NMR and 13C NMR: see Table 1; HRESIMS m/z 287.1255 ([M + Na]+ calcd 287.1253).ε) 270 (0.6), 219 (3.6) nm; ECD (MeOH) (mol. CD) 290 (0.8), 228 (1.2) nm; IR (KBr) νmax 3472, 2955, 2929, 2879, 1666, 1617, 1440, 1410, 1246, 1132 cm−1; 1H NMR and 13C NMR: see Table 1; HRESIMS m/z 271.1306 ([M + Na]+ calcd 271.1304).Single-crystal X-ray diffraction analysis and crystallographic data for compound 1
The diffraction intensity data for compound 1 were acquired on a Bruker D8 Venture with a Photon 100 CMOS detector system equipped with a Cu INCOATEC IμS microfocus source (λ = 1.54178 Å). The data were collected by Bruker APEX2 software. The data reductions were conducted with Bruker SAINT. Structure solutions and refinements were performed with the SHELXTL program package. The crystal structures of compound 1 was drawn by ORTEP (Fig. 4).Crystallographic data of 1
C15H18O4, formula weight 262.29, space group P212121, a = 6.6564(2) Å, b = 10.3704(3) Å, c = 17.9622(6) Å, α = β = γ = 90°, V = 1239.92(7) Å3, Z = 4, Dcalcd = 1.405 mg m−3, crystal dimensions 0.45 × 0.36 × 0.32 mm3 were used for measurements on a Bruker D8 VENTURE diffractometer with a graphite monochromator (Φ/ω scans, 2θmax = 72.19°), Cu Kα radiation. The total number of independent reflections measured was 2382, of which 4664 were observed. Final indices: R1 = 0.0375, wR2 = 0.0975, S = 1.022.The crystallographic data for the structures of fomitopin A has been deposited with the Cambridge Crystallographic Data Centre (deposit numbers CCDC 1573301).
All experiments were performed in accordance with the Guidelines of the Institutional Review Board at Chang Gung Memorial Hospital, and experiments were approved by the ethics committee at Chang Gung Memorial Hospital. Informed consents were obtained from human participants of this study.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This study is sponsored by the Ministry of Science and Technology, Taiwan. Authors are also thankful to Chang Gung Memorial Hospital (CMRPD1B0281–3, CMRPF1D0442–3, CMRPF 1F0011–3, CMRPF1F0061–3 and BMRP450 granted to T.-L. H.) for the partial financial support for the present research.References
- G. Bhattarai, Y. H. Lee, N. H. Lee, I. K. Lee, B. S. Yun, P. H. Hwang and H. K. Yi, Biol. Pharm. Bull., 2012, 35, 1711–1719 CrossRef CAS.
- M. Popova, B. Trusheva, M. Gyosheva, I. Tsvetkova and V. Bankova, Fitoterapia, 2009, 80, 263–266 CrossRef CAS.
- C. H. Hwang, B. U. Jaki, L. L. Klein, D. C. Lankin, J. B. McAlpine, J. G. Napolitano, N. A. Fryling, S. G. Franzblau, S. H. Cho, P. E. Stamets, Y. Wang and G. F. Pauli, J. Nat. Prod., 2013, 76, 1916–1922 CrossRef CAS PubMed.
- J.-E. Haight, G. A. Laursen, J. A. Glaeser and D. L. Taylor, Mycologia, 2016, 108, 925–938 CrossRef CAS.
- U. Grienke, M. Zöll, U. Peintner and J. M. Rollinger, J. Ethnopharmacol., 2014, 154, 564–583 CrossRef.
- D. Choi, S.-S. Park, J.-L. Ding and W.-S. Cha, Biotechnol. Bioprocess Eng., 2007, 12, 516 CrossRef CAS.
- S. I. Lee, J. S. Kim, S. H. Oh, K. Y. Park, H. G. Lee and S. D. Kim, J. Med. Food, 2008, 11, 518–524 CrossRef CAS.
- W.-S. Cha, J.-L. Ding, H.-J. Shin, J.-S. Kim, Y.-S. Kim, D. Choi, H.-D. Lee, H.-B. Kang and C.-W. Lee, Korean J. Chem. Eng., 2009, 26, 1696–1699 CrossRef CAS.
- A. C. Keller, M. P. Maillard and K. Hostettmann, Phytochemistry, 1996, 41, 1041–1046 CrossRef CAS.
- M. T. Heneka, M. J. Carson, J. E. Khoury, G. E. Landreth, F. Brosseron, D. L. Feinstein, A. H. Jacobs, T. Wyss-Coray, J. Vitorica, R. M. Ransohoff, K. Herrup, S. A. Frautschy, B. Finsen, G. C. Brown, A. Verkhratsky, K. Yamanaka, J. Koistinaho, E. Latz, A. Halle, G. C. Petzold, T. Town, D. Morgan, M. L. Shinohara, V. H. Perry, C. Holmes, N. G. Bazan, D. J. Brooks, S. Hunot, B. Joseph, N. Deigendesch, O. Garaschuk, E. Boddeke, C. A. Dinarello, J. C. Breitner, G. M. Cole, D. T. Golenbock and M. P. Kummer, Lancet Neurol., 2015, 14, 388–405 CrossRef CAS.
- K. Yoshikawa, M. Inoue, Y. Matsumoto, C. Sakakibara, H. Miyataka, H. Matsumoto and S. Arihara, J. Nat. Prod., 2005, 68, 69–73 CrossRef CAS.
- J.-J. Cheng, C.-Y. Lin, H.-S. Lur, H.-P. Chen and M.-K. Lu, Process Biochem., 2008, 43, 829–834 CrossRef CAS.
- K. M. Pietrosimone and P. Liu, World J. Transl. Med., 2015, 4, 60–68 CrossRef.
- A. Matsuo, S. Yuki and M. Nakayama, J. Chem. Soc., Perkin Trans. 1, 1986, 701–710 RSC.
- T. Eicher, F. Servet and A. Speicher, Synthesis, 1996, 863–870 CrossRef CAS.
- P. C. Kuo, H. Y. Hung, C. W. Nian, T. L. Hwang, J. C. Cheng, D. H. Kuo, E. J. Lee, S. H. Tai and T. S. Wu, J. Nat. Prod., 2017, 80, 1055–1064 CrossRef CAS.
- D. A. Partrick, E. E. Moore, P. J. Offner, D. R. Meldrum, D. Y. Tamura, J. L. Johnson and C. C. Silliman, Arch. Surg., 2000, 135, 219–225 CrossRef CAS.
- T. L. Hwang, S. H. Yeh, Y. L. Leu, C. Y. Chern and H. C. Hsu, Br. J. Pharmacol., 2006, 148, 78–87 CrossRef CAS.
- A. E. Nugroho and H. Morita, J. Nat. Med., 2014, 68, 1–10 CrossRef CAS.
- S.-C. Yang, P.-J. Chung, C.-Y. Kuo, M.-F. Hung, Y.-T. Huang, W.-Y. Chang, Y.-W. Chang, K.-H. Chan and T.-L. Hwang, J. Immunol., 2013, 190, 6511–6519 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1573301. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra05899k |
| ‡ The authors contribute equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |