DOI:
10.1039/C9RA05831A
(Paper)
RSC Adv., 2019,
9, 27789-27799
Combining a ligand photogenerator and a Ru precatalyst: a photoinduced approach to cross-linked ROMP polymer films†
Received
27th July 2019
, Accepted 21st August 2019
First published on 4th September 2019
Abstract
Although metathesis photoinduced catalysis is now well established, there is little development in thin film preparation using photochemically activated ring-opening metathesis polymerization (ROMP). Herein, a N-heterocyclic carbene (NHC) photogenerator (1,3-bis(mesityl)imidazolium tetraphenylborate) is combined with an inactive metathesis catalyst ([RuCl2(p-cymene)]2) to generate under UV irradiation an active catalyst (p-cymene)RuCl2 (NHC), that is capable of producing in a single step cross-linked copolymer films by ROMP of norbornene with dicyclopentadiene. The study shows that the photoinitiated catalytic system can be optimized by increasing the yield of photogenerated NHC through a sensitizer (2-isopropylthioxanthone), and by choosing [RuI2(p-cymene)]2 as precatalyst to provide a long-term photolatency. The cross-linked polymer structure is investigated by a range of techniques including gel content measurement, FT-IR and solid-state 13C NMR spectroscopy, TGA and DSC, which reveal a cross-linking mechanism proceeding through both metathesis and olefin coupling.
Introduction
As one of the most valuable tools for the generation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds, olefin metathesis has been exploited in many useful ways in the field of polymers, including the synthesis of complex architectures,1 pharmaceutical products,2 or biodegradable copolymers.3 In all these cases, progress has been mainly driven by the development of advanced and well-defined metal alkylidene complexes, like the notorious Grubbs and Schrock catalysts.4,5 Ligand fine-tuning has resulted in a portfolio of catalysts featuring a broad range of reactivity, functional group tolerance and bench stability. In this respect, a major breakthrough took place in 1999 with the introduction of N-heterocyclic carbene (NHC) ligands in the place of the conventional phosphine compounds.6,7 In particular, sterically bulky and strong electron donating NHCs have proven to be highly effective, resulting in NHC-coordinated ruthenium complexes with longer life-time and superior catalytic activity.6,8 However, the main developments in olefin metathesis catalysis have responded more to the needs of synthetic chemistry (as the above mentioned examples show), but have been less useful for the preparation of polymer materials. Ring-opening metathesis polymerization (ROMP) of cyclic olefins is currently the main type of olefin metathesis polymerizations.6,9 Polymers with a broad spectrum of properties have been achieved through ROMP of norbornene (NB), cyclopentene, cyclooctene or dicyclopentadiene (DCPD). However, the use of ROMP polymers has been limited so far to the manufacturing of specific structural and engineering materials. For example, polycyclooctene (Vestenamer®) and polyNB (Norsorex®) elastomers are employed after vulcanization as shock absorbers and anti-grip materials. In addition, hydrogenated polyNB (Zeonex®) is a high performance optical thermoplastic, while polyDCPD (Metton® or Telene®) is known as a prime thermoset with mechanical properties similar to engineering thermoplastics.10 One reason for the limited applications of these polymers can be found in their restrictive processing methods. For example, polyDCPD is prepared essentially by reaction injection molding (RIM), which requires separate monomer and catalyst compartments, efficient mixing and titration heads, nitrogen inerting of the mold, etc.11–13
C double bonds, olefin metathesis has been exploited in many useful ways in the field of polymers, including the synthesis of complex architectures,1 pharmaceutical products,2 or biodegradable copolymers.3 In all these cases, progress has been mainly driven by the development of advanced and well-defined metal alkylidene complexes, like the notorious Grubbs and Schrock catalysts.4,5 Ligand fine-tuning has resulted in a portfolio of catalysts featuring a broad range of reactivity, functional group tolerance and bench stability. In this respect, a major breakthrough took place in 1999 with the introduction of N-heterocyclic carbene (NHC) ligands in the place of the conventional phosphine compounds.6,7 In particular, sterically bulky and strong electron donating NHCs have proven to be highly effective, resulting in NHC-coordinated ruthenium complexes with longer life-time and superior catalytic activity.6,8 However, the main developments in olefin metathesis catalysis have responded more to the needs of synthetic chemistry (as the above mentioned examples show), but have been less useful for the preparation of polymer materials. Ring-opening metathesis polymerization (ROMP) of cyclic olefins is currently the main type of olefin metathesis polymerizations.6,9 Polymers with a broad spectrum of properties have been achieved through ROMP of norbornene (NB), cyclopentene, cyclooctene or dicyclopentadiene (DCPD). However, the use of ROMP polymers has been limited so far to the manufacturing of specific structural and engineering materials. For example, polycyclooctene (Vestenamer®) and polyNB (Norsorex®) elastomers are employed after vulcanization as shock absorbers and anti-grip materials. In addition, hydrogenated polyNB (Zeonex®) is a high performance optical thermoplastic, while polyDCPD (Metton® or Telene®) is known as a prime thermoset with mechanical properties similar to engineering thermoplastics.10 One reason for the limited applications of these polymers can be found in their restrictive processing methods. For example, polyDCPD is prepared essentially by reaction injection molding (RIM), which requires separate monomer and catalyst compartments, efficient mixing and titration heads, nitrogen inerting of the mold, etc.11–13
To unlock the potential of ROMP polymer materials, more efficient and “smarter” catalysts are crucial, especially to extend the shaping of ROMP polymers as film, particle or fiber, but also, to foster more versatile, energy-efficient and facile process conditions.14–20 To address those challenges, latent olefin metathesis catalysts have emerged that are able to release active species in situ and “on demand” by external stimulation. Heat,14–16 mechanical force,17 and radiation18–20 are the most common stimuli. A separate category concerns catalysts activated after the addition of an external chemical agent.21–23 Among these different options, the activation brought about by visible or ultraviolet irradiation is particularly attractive.24 Firstly, it allows activation of the catalyst at ambient temperature, and the use of storable and ready-to-use formulations combining together the catalyst and the monomer(s). Secondly, as a standard product of a photoactivated polymerization, thin films based on cross-linked ROMP polymer make possible the development of novel UV-curable coatings or adhesives. Thirdly, unlike the other triggers, light allows the reaction to be spatially-controlled, opening the way to applications in photolithography.23,25
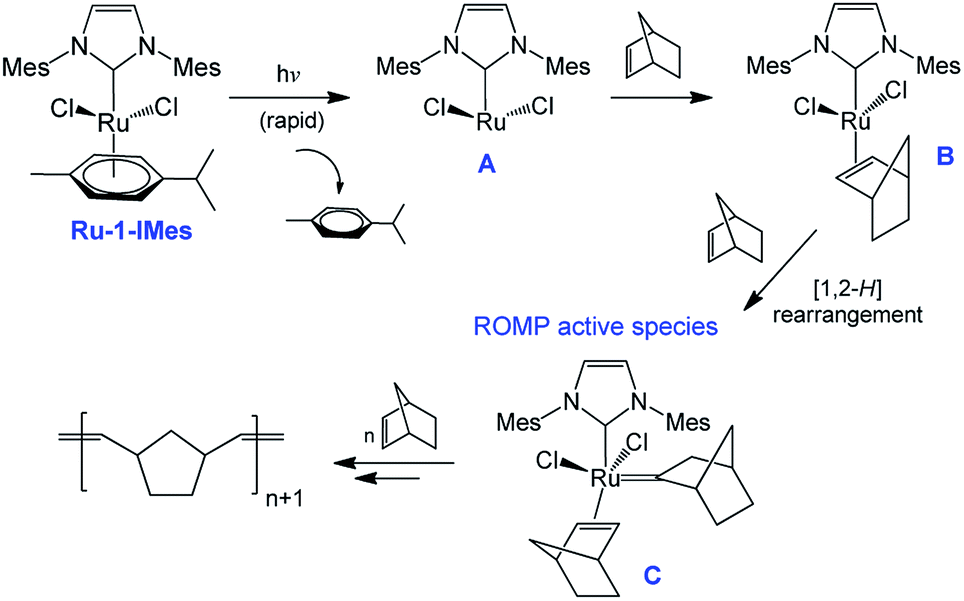
In the field of photochemically activated olefin metathesis, three main approaches have been proposed to promote activation of the catalyst that all involve the photochemical reaction of one ligand of a transition metal complex.13 The two most reported routes rely on ligand photodissociation26,27 and ligand cis-to-trans photoisomerization.18,20 The third route, which is the least investigated, is the focus of the present study. In the latter, the active olefin metathesis catalyst is produced from an inactive and non-absorbing precatalyst after reaction with a ligand generated in situ by a second photosensitive compound (ligand photogenerator). This methodology was first introduced by Grubbs et al. in 2009 by using a Ru–alkylidene complex ligated by acetylacetonate (acac) as precatalyst and a photoacid generator (PAG) based on triphenylsulfonium chloride.21 Upon irradiation at 254 nm, HCl was released, and the subsequent displacement of acac by Cl− resulted in an active metathesis catalyst. A similar method was also employed by Piers et al. using a ruthenium carbide and Ph3S triflate (OTf).22 Recently, we replaced the PAG with a NHC photogenerator and used a stable, commercially available dimeric Ru(II) complex as metathesis inactive catalyst (Scheme 1).28,29 Our tandem system made up of 1,3-bis(mesityl)imidazolium tetraphenylborate IMesH+BPh4− (1) as NHC progenitor and [RuCl2(p-cymene)]2 precatalyst (Ru-1) proved to be very efficient at ROMP of NB. In comparison with the examples discussed above, a safer UVA radiation (320–400 nm) was employed, and no ligand displacement occurred. The cleavage of the Ru-chlorido bridges of Ru-1 was rather enthalpically driven through the addition of 2 equivalents of free NHC IMes, that were photogenerated in situ from 1. As a result, the presumed active catalyst (Ru-1-IMes) features one p-cymene cycle, two chlorido ligands and one IMes ligand. First introduced by Hermann et al.,30 this class of RuCl2(p-cymene)NHC catalyst has been extensively studied by Noels et al. for the ROMP of NB and cyclooctene.31–33
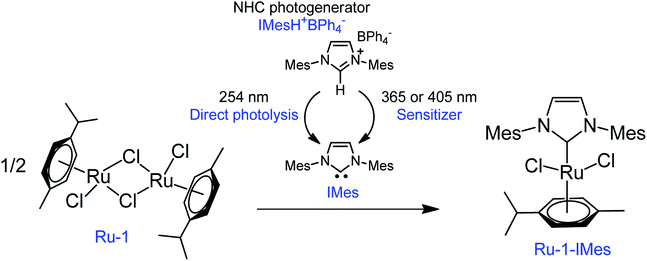 |
| | Scheme 1 A mixture of NHC photogenerator (IMesH+BPh4−, 1) and an inactive dimeric Ru(II) complex (Ru-1) results after irradiation in an active olefin metathesis photocatalyst (Ru-1-IMes). The NHC (IMes) can be photogenerated through two distinct routes: direct photolysis or sensitized reaction using an electronically excited electron acceptor (sensitizer). | |
In this contribution, the main objective is to demonstrate the utility of this tandem approach for the photoinduced preparation of cross-linked ROMP polymer films. To this purpose, we report first our efforts to improve the photocatalytic system by addressing a number of issues: optimizing the yield of photogenerated IMes, ensuring a long-term photolatency, identifying the active metathesis catalyst and the initiating species, and finally, collecting some preliminary results on photoROMP in solution of common cyclic olefin monomers such as NB. In a second part, a range of copolymer films was prepared by photoinduced ROMP copolymerization between NB and DCPD using the photocatalytic mixture based on 1/ITX/Ru-1. The coatings were prepared in a single step from a one-pot formulation, which is close to the conditions of a conventional UV-curing process. The cross-linked copolymer structure was thoroughly investigated by gel content measurement, FT-IR and solid-state 13C NMR spectroscopy. The thermal properties of these polymer films were also studied through TGA and DSC.
Experimental
Materials
Dicyclopentadiene (DCPD, >97%, TCI), 2-norbornene (NB, >99%, TCI), 5-ethylidene-2-norbornene (ENB, >98%, TCI), vinyl ethyl ether (>98%, TCI), 2-isopropylthioxanthone (ITX, analytical standard, Aldrich), 1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene (IMes, 97%, Aldrich) and 1,3-di-p-tolylcarbodiimide (CDI, 96%, Aldrich) were used as received without purification. 1,3-bis(2,4,6-trimethylphenyl)imidazolium tetraphenylborate (IMesH+BPh4−, 1) was prepared following a procedure published elsewhere.28 Dichloro(p-cymene)ruthenium(II) dimer (Ru-1, 98%), diiodo(p-cymene)ruthenium(II) dimer (Ru-2, 98%) and dichlorodi-μ-chlorobis[1,2,3,6,7,8-η-2,7-dimethyl-2,6-octadiene-1,8-diyl] diruthenium(IV) (Ru-3, 98%) were bought from Strem Chemicals. Dichloromethane-d2 (CD2Cl2, 99.5% D, Eurisotop), tetrahydrofuran-d8 (THF-d8, 99.5% D, Eurisotop) and other HPLC grade solvents such as ethanol, acetonitrile and tetrahydrofuran (THF) received from VWR were distilled and stored over molecular sieve (4 Å) under nitrogen atmosphere before use.
Syntheses and procedures
Quantification of IMes yield. Quantitative conversion of IMes into IMes-carbodiimide (CDI) adduct was described elsewhere.29 The irradiation sources include a medium pressure Xe–Hg lamp (75 mW cm−2, Hamamatsu, LC-9588/01A), a LED@365 nm (65 mW cm−2, Hammamatsu LC-L1V3) and a LED@405 nm (100 mW cm−2, Thorlabs M405L2). The irradiances were determined by using an optical power meter PM100D from Thorlabs.
Assessment of catalyst photolatency. In a typical experiment, NB (47 mg, 510 equiv.), ITX (0.63 mg, 2.5 equiv.) and ruthenium pre-catalyst Ru-1 (0.62 mg, 1 equiv.) were placed in a borosilicate NMR tube containing 0.5 mL of CD2Cl2. All solutions were stored in dark. After a fixed time, a 1H-NMR spectrum was recorded to determine the extent of monomer conversion and cis/trans ratio of the polymer possibly formed.
Photoactivated ROMP in solution. To a borosilicate NMR tube was added 47 mg of NB (510 equiv.), 0.63 mg of ITX (2.5 equiv.), 3 mg of 1 (5 equiv.), 0.62 mg of Ru-1 (1 equiv.), then 0.5 mL of CD2Cl2. The solutions were irradiated in a circular LED photoreactor. For this, a LED strip was wrapped around a quartz cylinder (ϕint = 60 mm) emitting a monochromatic light centered at 365 nm (SMD3528-60LED/Meter, Lightingwill, 6.5 mW cm−2) or 405 nm (SMD2835-60LED/Meter, Banggood, 4.7 mW cm−2). After 1 min irradiation, 1H-NMR spectra were recorded to determine monomer conversion and cis/trans ratio. Excess amount of vinyl ethyl ether (0.1 mL) was added to terminate the polymerization. Polymers were collected through precipitation in acetone and washed 3 times with CH2Cl2/acetone. SEC measurements were performed after drying under air for 48 h. For measurements of real-time kinetics by RT-FTIR (see characterization section), the same NB/ITX/1/Ru-1 mixture was introduced into an IR liquid cell (Specac® Omni cell) containing two sandwich CaF2 windows. A 1 mm Teflon spacer in the middle of pellets was used to fix the specimen thickness. Samples were then exposed synchronously to an IR beam and an UV irradiation resource equipped with a light guide. Two types of LED were used: a LED@365 nm and a LED@405 nm (see above for details).
Preparation of cross-linked ROMP polymer films. A mixture of NB and DPCD was prepared at different NB concentrations ranging from 25 to 75 mol%. Ru-1 (9 mg, 1 equiv.), ITX (9.3 mg, 2.5 equiv.) and 1 (45 mg, 5 equiv.) were first dissolved in CH2Cl2 (2 mL), then the monomer mixture (510 equiv.) was added and left 10 min under stirring to obtain a homogeneous solution. The mixture was then poured into a Teflon mold (20 × 85 × 0.35 mm) covered with a borosilicate glass plate. Clamps were used to maintain the glass plate in order to prevent air entry as well as evaporation of solvent and monomers. The weight of mold containing the reactive mixture was determined before irradiation to be used subsequently for gravimetry measurements. The mold was placed under a LED panel (365 nm, 5.5 mW cm−2, Top UV Curing Light) for 1 h. The wet gel was removed from the mold and left to dry overnight at ambient atmosphere. Residual monomers and solvent were removed by drying under vacuum at 50 °C until the film reached a constant weight. The films exhibited an average film thickness of 0.25 mm.
Characterization methods
All 1H-NMR spectra were recorded in appropriate deuterated solvents with tetramethylsilane (TMS) as the internal reference on a Varian 300. All chemical resonances were documented in parts per million (ppm) relative to the residual THF-d8 (δ 1.72 ppm and 3.58 ppm), and CD2Cl2 (δ 5.32 ppm). Peaks multiplicities in 1H-NMR spectra were abbreviated as s (single), d (double), t (triplet), m (multiplet), br (broad).
Molecular weights of polyNB and polyENB were determined using a size exclusion chromatography (SEC) instrument from a Varian apparatus equipped with TOSOHAAS TSK gel columns and a refractive index detector. The mobile phase was THF at a flow rate of 1 mL min−1; the injected volume was 100 μL. The MALS instrument was normalized using a THF solution of polystyrene standard.
Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) experiments were performed using, respectively, a TGA/DSC3+ (Metter Toledo) and a DSC1 (Metter Toledo) under nitrogen atmosphere. For TGA analysis, the samples (8–15 mg) were heated from 30 °C to 600 °C at a heating rate of 10 °C min−1. For DSC analysis, the samples (3 mg) were scanned from – 80 °C to 250 °C at a heating rate of 10 °C min−1. The glass transition temperatures were determined in the second run.
To determine gel content, a polymer specimen (∼50 mg) was placed into a 5 mL vial containing 4 mL of CH2Cl2. After magnetic stirring for 24 h at room temperature, the solvent was removed and insoluble component was dried under vacuum at 40 °C until weight constant. The gel content was finally measured.
13C CP MAS NMR spectra were recorded by using an Advance II 400 MHz Bruker Spectrometer (Ascend™ magnet) (B0 = 9.4 T) operating at 100.64 MHz and controlled by TOPSPIN 3.2 software. The solid films were grounded into fine powders prior to measurement. Experiments were performed at ambient temperature with a 4 mm double resonance MAS Bruker probe at a speed of 12 kHz, a 13C π/2 pulse duration of 5.6 μs, a contact time of 1 ms and a recycle delay of 2 s. Chemical shifts reported thereafter are relative to tetramethylsilane and the deconvolution of the experimental spectra were carried out with the DMfit software.34 The fraction of pendant cyclopentene rings involved in olefin coupling reaction can be determined semi-quantitatively from the 13C CP MAS NMR spectrum. DCPD contains initially 4sp2 and 6sp3 carbons. The ratio of sp2-C to sp3-C does not change when cyclopentene reacts by ROMP. Conversely, olefin coupling leads to the replacement of 2sp2-C by 2sp3-C. This change can be used to estimate the fraction of cyclopentene reacting via olefin coupling (x) in polyDCPD film (eqn (1)):35
| |
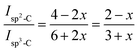 | (1) |
where
Isp2-C and
Isp3-C are the integrated areas of the signals associated to olefinic carbons (125–130 ppm) and aliphatic carbons (30–60 ppm), respectively. This equation was changed to adapt to our specific copolymer structure resulting from copolymerization of NB and DCPD (
eqn (2)):
| |
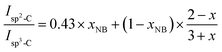 | (2) |
where 0.43 is the value of the experimental

in polyNB (calculated from sample NB-100), and
xNB is the molar fraction of NB in the NB/DCPD monomer mixture.
FTIR spectra were recorded with a Nicolet 8700 spectrophotometer (Thermo Scientific) using a mercury cadmium telluride detector. Two modes were used, transmission for monitoring the real-time kinetics of photoROMP in solution and attenuated total reflectance (ATR) for characterization of polymer films. All the spectra were recorded in the range 4000–600 cm−1. The real-time measurements used a scanning velocity of 11.45 cm s−1 corresponding to a resolution of 0.964 cm−1. Monomer conversion (Conv.) was determined grounded on the changing of absorption area At at 1566 cm−1 (NB, CC stretch), 3047 cm−1 (DCPD, sp2 = CH) (eqn (3)):
| |
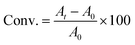 | (3) |
where
A0 and
At are the integrated absorbance of the characteristic vibrational modes before and after UV radiation for
t min. To check reproducibility, each experiment was performed at least three times.
Results and discussion
Study and optimization of the tandem photocatalytic system
NHC photogeneration and structure of the active ROMP catalyst. There are two possible photochemical pathways to deprotonate the IMesH+ imidazolium cation in 1, and thereby to generate the targeted NHC IMes (Scheme 1). Both approaches rely on the photochemistry of tetraphenylborate counter-anion: a direct photolysis of BPh4−,36 or a sensitized reaction involving an additional electron acceptor species such as isopropylthioxanthone (ITX).28,29 Direct photolysis was carried out by exposure of 1 in THF-d8 (0.03 M) to 254 nm, while the sensitized reaction was conducted in similar conditions with a mixture of 1 and ITX (1/1 equiv.) upon irradiation at 365 nm or 405 nm (thanks to the higher wavelength absorption of the sensitizer). The precise reaction mechanisms in both cases have been commented on elsewhere.28,29,37,38 Table 1 compares the efficiency of the two methods in terms of NHC generated after 5 min irradiation.39 Clearly, 1 irradiated alone at 254 nm (entry a) shows less activity in comparison with the combination of 1 and ITX at 365 nm (entry b) and 405 nm (entry c). Only 11% IMes was liberated in direct photolysis versus 29% (405 nm) and 67% (365 nm) in the sensitized reaction. This suggests that the photoinduced route using the 2-component system 1/ITX is more efficient. To account for the poorer NHC yield after the direct photolysis, we rely on the fact that IMes has a non-negligible absorption band at 254 nm (ε254 = 9.1 × 103 M−1 cm−1), while it does not absorb in the UVA range. This could reduce the amount of photons absorbed by 1, and undermine the yield of the photolysis reaction. Therefore, the mixture 1/ITX was employed as NHC photogenerator in the rest of the study.
Table 1 Effect of excitation wavelength on the release of free NHC IMes in THF-d8 ([1] = 0.03 M, [ITX] = 0.03 M) after 5 min of irradiation
| Entry |
NHC photogenerator |
Irradiation wavelengtha (nm) |
IMes yieldb (%) |
| Irradiances values: 65 mW cm−2 (365 nm), 100 mW cm−2 (405 nm) and 75 mW cm−2 (254 nm). NHC yield was obtained indirectly after addition of 1,3-di-p-tolylcarbodiimide (CDI) ([CDI] = 0.03 M, 1 equiv.) to the as-irradiated medium. The stable IMes-CDI adduct is formed, which can be precisely quantified by 1H-NMR.29 |
| a |
1 |
254 |
11 |
| b |
1/ITX |
365 |
67 |
| c |
1/ITX |
405 |
29 |
Having proved the aptitude of 1 to act as NHC precursor under irradiation (in combination with a sensitizer), we studied the subsequent reaction of IMes with [RuCl2(p-cymene)]2 dimer Ru-1 to form an active ROMP catalyst.28,29 For this, we performed a control experiment in which Ru-1 was reacted with free IMes in THF-d8 at room temperature. An orange solution formed spontaneously, and subsequent 1H-NMR analysis (performed without prior isolation) established that the expected catalyst structure (p-cymene)RuCl2(IMes) was formed in high yield (see Fig. S1 in ESI†). The spectrum found was indeed in full agreement with previous literature data.40 This suggests that Ru-1-IMes could be actually the active catalytic species when the tandem system NHC photogenerator/Ru precatalyst is irradiated. However, all attempts to characterize by 1H-NMR the photogenerated active catalyst have failed, preventing to confirm our hypothesis. The spectra of the catalytic reaction mixture 1/Ru-1/ITX show after irradiation too many signals, preventing the unequivocal identification of the chelated NHC–Ru complex (see Fig. S2 in ESI†).
Catalyst photolatency. Photochemical latency is a very important characteristic for practical implementation of a photocatalytic system. Consequently, the catalytic activities of Ru-1 (in the presence of ITX, but without 1) has been tested with exclusion of light by using standard ROMP substrates such as NB, and using CD2Cl2 as a solvent. Results are presented in Table 2. The photochemical latency of Ru-1 was found to be poor since 18% conversion (entry a) was obtained after 4 h at room temperature. ROMP could proceed by a decoordination of the η6-arene ligand to afford a highly unsaturated ruthenium center that evolves into a propagating alkylidene after addition of a NB molecule.41,42 In agreement with this hypothesis, no premature reaction took place with ENB (entry b) due to its higher steric hindrance which limits the reaction with the ruthenium center. In the search of other ruthenium pre-catalysts able to ensure a complete photolatency, Ru-1 was substituted with [RuI2(p-cymene)]2 (Ru-2) and [RuCl2(COD)]2 (Ru-3, with COD = 1,5-cyclooctadiene) to probe separately the effects of the halogen and the ligand in the Ru dimer (see structures in Scheme 2). Remarkably, Ru-2 in the presence of ITX afforded almost no conversion after 4 h (entry c), and even after 168 h (entry d). We carefully checked that the addition of 1 to Ru-2/ITX did not increase the conversion, illustrating the minor role played by the NHC precursor in the instability issue. Conversely, the Ru-3 complex exhibited nearly no latency since 90% conversion was reported after 4 h (entry e). These results show that the size of the halogen is a crucial parameter affecting the equilibrium for olefin binding.43,44 As expected, a larger halogen than chloride such as iodide disfavors olefin binding due to steric hindrance, resulting in a better latency. In all instances, a low content of cis double bond (σcis = 23–30%) was determined in the polymer structure, which is consistent with the literature data for ROMP relying on ruthenium dimers as catalyst.45,46 In conclusion, Ru-2 is the best precatalyst candidate.
Table 2 Photolatency results of Ru dimer/ITX/monomer mixture in CD2Cl2 ([Monomer] = 1 M). All the samples were stored in the dark (Ru dimer/ITX/monomer = 1/2.5/510 equiv.)
| Entry |
Precatalyst |
Monomer |
Reaction time (h) |
Conv.a (%) |
σcisa |
| Determined by 1H-NMR. |
| a |
Ru-1 |
NB |
4 |
17.6 |
30 |
| b |
Ru-1 |
ENB |
4 |
0 |
n.d |
| c |
Ru-2 |
NB |
4 |
<1.0 |
n.d |
| d |
Ru-2 |
NB |
168 |
1.4 |
23 |
| e |
Ru-3 |
NB |
4 |
90 |
24 |
 |
| | Scheme 2 Chemical structure of alternative ruthenium dimers used as precatalyst. | |
Photoactivated ROMP in solution. The catalytic activity of the three-component system Ru precatalyst/1/ITX (1/5/2.5 equiv.) under UV irradiation (365 nm, 1 min) was thus evaluated for the ROMP of NB (Table 3) in CD2Cl2, using a NMR tube as reaction flask. As expected, the irradiation of a solution devoid of Ru dimer resulted in no reaction (entry a). By contrast, a small conversion was observed when Ru-1 precatalyst was irradiated alone (1%, entry b) or in presence of ITX (7%, entry c). This result can be reconciled with the poor latency of Ru-1 noted earlier, and suggests that the η6-arene ligand displacement could be enhanced by a photochemical activation. The vacant site created on the Ru center can form subsequently an active Ru complex after the addition of a monomer molecule.31,41
Table 3 Photopolymerization results of NB using Ru dimer/1/ITX photoinitiating system in CD2Cl2 ([NB] = 1 M, 510 equiv.). Irradiation conditions: 365 nm, 6.5 mW cm−2, 1 min
| Entry |
Precatalyst |
Pre-catalyst/1/ITX (equiv.) |
Conv.a (%) |
σcisb |
Mn-thc (kg mol−1) |
Mn-SECd (kg mol−1) |
Đd |
Determined by 1H-NMR. Calculated from 1H-NMR spectrum. Theoretical number average molar mass 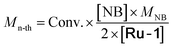 . Experimental number average molar mass Mn-th obtained by SEC. Irradiation at 405 nm, 4.7 mW cm−2, 1 min. n.d. means not determined due to the absence of reaction or the formation of an insoluble polymer. . Experimental number average molar mass Mn-th obtained by SEC. Irradiation at 405 nm, 4.7 mW cm−2, 1 min. n.d. means not determined due to the absence of reaction or the formation of an insoluble polymer. |
| a |
Ru-1 |
0/5/2.5 |
0 |
n.d |
n.d |
n.d |
n.d |
| b |
Ru-1 |
1/0/0 |
1 |
9 |
0.36 |
n.d. |
n.d. |
| c |
Ru-1 |
1/0/2.5 |
7 |
23 |
1.60 |
n.d. |
n.d. |
| d |
Ru-1 |
1/5/2.5 |
78 |
52 |
18.69 |
146.8 |
3.02 |
| e |
Ru-1 |
1/5/5 |
94 |
52 |
22.57 |
108.4 |
1.96 |
| fe |
Ru-1 |
1/5/2.5 |
22 |
65 |
5.27 |
63.4 |
2.05 |
| g |
Ru-2 |
1/5/2.5 |
69 |
52 |
16.43 |
447.2 |
1.59 |
| h |
Ru-3 |
1/5/2.5 |
67 |
48 |
16.10 |
n.d. |
n.d. |
A much higher catalytic activity was achieved in presence of 1 (entry d) with 78% conversion being reached after 1 min. This suggests the successful formation of a highly active Ru-arene complex, which is likely to be Ru-1-IMes. The content of cis double bond (σcis = 52%) was similar to that obtained with previous systems and supports this assumption.41,47 Upon increasing the proportion of ITX from 2.5 equiv. to 5 equiv., a significant increase of conversion was noted (94%, entry e) which correlates with a higher amount of NHC released and thus of generated Ru-1-IMes. This result is also confirmed by the lower molecular weight (Mn-SEC) of the resulting polymer using 5 equiv. of ITX as compared to the one obtained with 2.5 equiv. In contrast, irradiation at 405 nm instead of 365 nm led to a much lower conversion (22%, entry f). This can be rationalized on the basis of a lower IMes yield at higher excitation wavelength (vide supra). For the polymer produced with the tandem approach precatalyst/NHC (entries d–f), the molecular weight distributions were monomodal (SEC data), suggesting that only one mechanism is involved in the catalysis. However, high molecular weights (Mn-SEC) and rather broad polydispersities (Đ) were obtained, which reflect a poor control of the initiation step. The source of the problem is the ill-defined initiation mechanism and above all, the poor initiation efficiency resulting in only a small number of propagating centers produced in solution. This issue has been widely reported with such class of NHC-coordinated Ru complex.31,32,48,49 The effect of the precatalyst structure was also examined. Upon replacing Ru-1 by Ru-2, the rate of polymerization slightly decreased (69%, entry g). By the same reasoning used above to explain latency, it was expected that catalyst activity would decrease from Cl to I. In this case, there are consequently less backbiting reactions, resulting in polymer with a higher molecular weight and a fairly narrow dispersity index. Ru-3 precatalyst (entry h) led to similar conversions, but the polymer was not soluble in THF, indicating a very high molecular weight polymer and an even lower initiation efficiency. It can be concluded that Ru-1 achieves the best results in terms of NB conversion, but lacks of latency in presence of this monomer. Conversely, Ru-2 exhibits a much better photolatency but at the expense of decreasing reactivity. Our photocatalytic system was also active in the ROMP of ENB. The results are summarized in Table S1.†
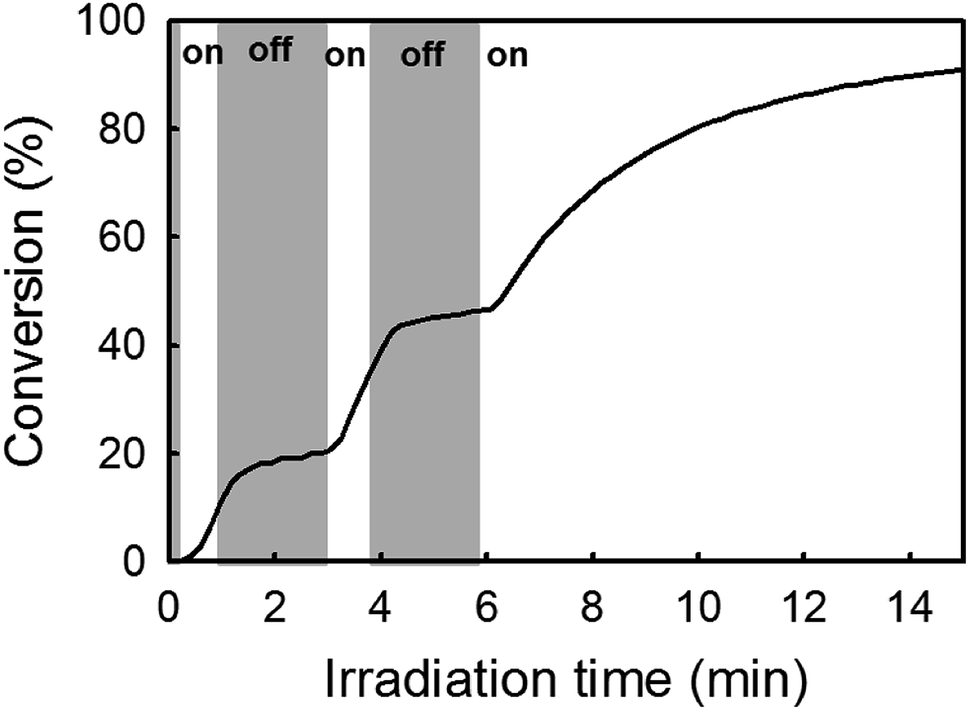
Mechanism of catalyst activation. As mentioned before, our approach relies on the in situ formation of an active Ru-arene complex from reaction between [RuCl2(p-cymene)]2 Ru-1 and the photogenerated IMes ligand. However, the presumed NHC-coordinated catalyst Ru-1-IMes identified in this study as the active metathesis catalyst lacks a suitable metal–carbene fragment to initiate ROMP. The literature reports an ill-defined mechanism involving p-cymene decoordination and a subsequent reaction between the resultant highly unsaturated ruthenium and a cyclic olefin as being accountable for the formation of active species.31,49 This last section questions the role(s) played by irradiation (365 nm) in this mechanism. As established earlier, light has a clear role in the generation of NHC. Bench stability experiments also demonstrate a second role of light in the disengagement of the η6-arene ligand. To explore other possible effect on polymerization mechanism, NB conversion kinetics was measured in situ by real-time FTIR spectroscopy. For this, the reactive monomer mixture was introduced in a liquid IR cell. IR spectra were acquired continuously during UV exposure (LED spotlight 365 nm) in order to construct a conversion–time curve. Fig. 1 shows a typical conversion–time curve while alternating between periods of radiation exposure (white area) and dark (grey area). Surprisingly from the mechanistic hypothesis, a temporal control of polymer chain growth was demonstrated through intermittent exposure of the reaction to UV light since an almost stop of polymerization was observed when light was switched off. This result is in apparent contradiction with the hypothesis of a formation of an active Ru catalyst whose activity could be preserved without irradiation. While almost no conversion occurred in the dark (off periods), polymerization could be re-initiated by re-exposure to light. This has been attributed to the fact that radiation might promote the multiple insertion of new NB units into the propagating centre.33,41,50
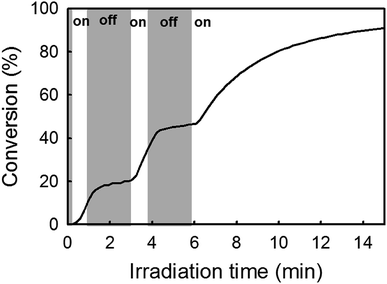 |
| | Fig. 1 Conversion–time curve for the ROMP of NB in solution in CH2Cl2: light off (grey area), light on (white area). NB/Ru-1/1/ITX = 510/1/5/0.5 equiv. Irradiation conditions: LED@365 nm, 65 mW cm−2. | |
Based on this observation, we propose a putative reaction mechanism (Scheme 3) for the ROMP photoinitiation. First, the active complex RuCl2(arene)(NHC) Ru-1-IMes is subjected to the rapid loss of p-cymene ring upon photochemical activation to form an unsaturated Ru species A.33,41,50 As a subsequent step, the insertion of NB into the metal center occurs to form the Ru complex RuCl2(IMes)(NB) B. Then a sigmatropic [1,2-H] rearrangement leads to the formation of a ruthenium–alkylidene complex C (initiating species).51 The next steps follow the typical chain-growth polymerization mechanism involving the metallacyclization and cycloreversion steps, with the particularity that the insertion of new cyclic olefin molecules could be facilitated by light.
 |
| | Scheme 3 Proposed photoactivation mechanism of Noels' catalyst. | |
Preparation of cross-linked ROMP polymer films
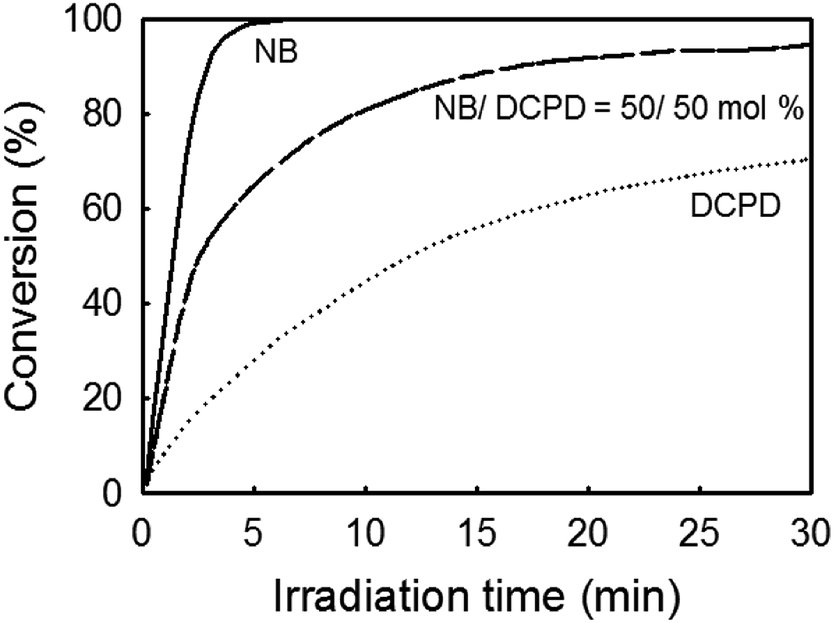
Scouting experiments. For the preparation of UV-cured films by photoactivated ROMP, emphasis has been made on DCPD because this monomer is able to form a cross-linked structure with high stiffness and impact resistance.52 To assess to which extent DCPD could react with our photocatalytic system (Ru-1/1/ITX), preliminary tests were carried in CH2Cl2 by monitoring the photopolymerization kinetics using real-time FTIR. Despite its lack of complete latency, Ru-1 was preferred because of its higher reactivity. Fig. 2 shows a typical conversion kinetics of DCPD, together with that of NB. As expected, DCPD exhibited a much slower polymerization rate compared to NB.53–55 To increase reactivity and achieve a cross-linked structure unattainable with NB only, we proposed a copolymerization of DCPD with NB.13,54 As shown on the same plot, a mixture of DCPD/NB (50/50 mol%) resulted in intermediate polymerization rates and almost full conversion after 30 min irradiation.
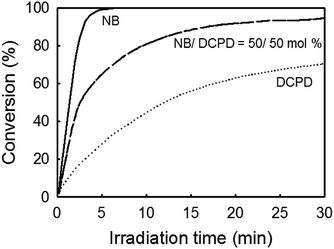 |
| | Fig. 2 Photopolymerization kinetic of NB, DCPD and NB/DCPD (50/50 mol%) using photocatalyst system Ru-1/1/ITX in CH2Cl2 (Monomer/Ru-1/1/ITX = 510/1/5/2.5 equiv.). | |
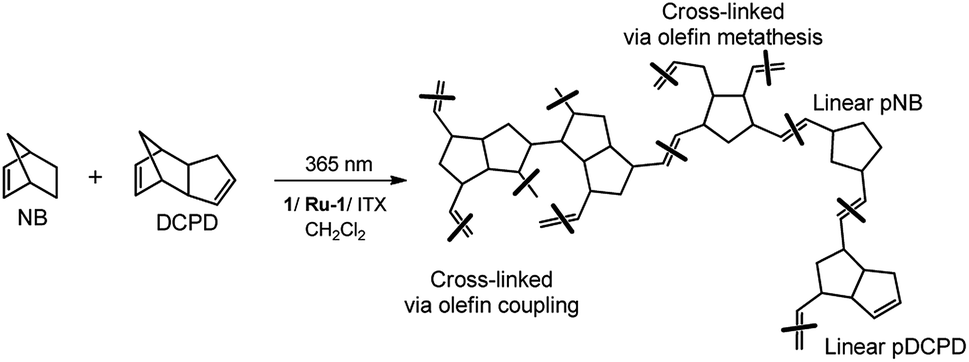
Given the potential of this approach, a range of different (co)polymer films were prepared from a mixture of NB and DCPD (NB-25: 25/75 mol%; NB-50: 50/50 mol% and NB-75: 75/25 mol%). For comparison, two control experiments using NB (NB-100) and DCPD (DCPD-100) alone were also performed under similar conditions. As can be seen in Fig. 3, a mixture of monomer and photocatalyst dissolved in CH2Cl2 was placed in a Teflon mold, covered with a glass slide to avoid solvent or monomer evaporation, and irradiated for 1 h under a LED panel (365 nm, 5.5 mW cm−2) in order to reach a high monomer conversion. The swollen polymer films were then dried at ambient conditions, placed under vacuum, then characterized using a range of spectroscopic techniques (FTIR-ATR, 13C CP MAS), gravimetry, gel content, DSC and TGA. All these techniques are intended to give insight into the structure of the polymer, and in particular the possibility of cross-linking. With regard to this last point, Scheme 4 shows the various possible structures arising from the copolymerization of DCPD with NB. Because the cyclopentene-derived double bond is much less reactive than the norbornenyl double-bond, we assume that the two monomers DCPD and NB could copolymerize first, resulting in the formation of a linear backbone. The cyclopentene-derived double bonds can react subsequently to form a cross-linked network. It has been established that the pendant cyclopentene ring from DCPD monomer may react via two possible pathways: ROMP and olefin addition through radical coupling.35,56,57
 |
| | Fig. 3 Photographs of: (a) general set up for the preparation of cross-linked films and (b) polymeric films (i) NB-100, (ii) NB-75, (iii) NB-50, (iv) NB25 and (v) DCPD-100. | |
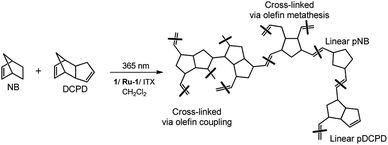 |
| | Scheme 4 Set of chemical structures arising from a ROM copolymerization of NB and DCPD. | |
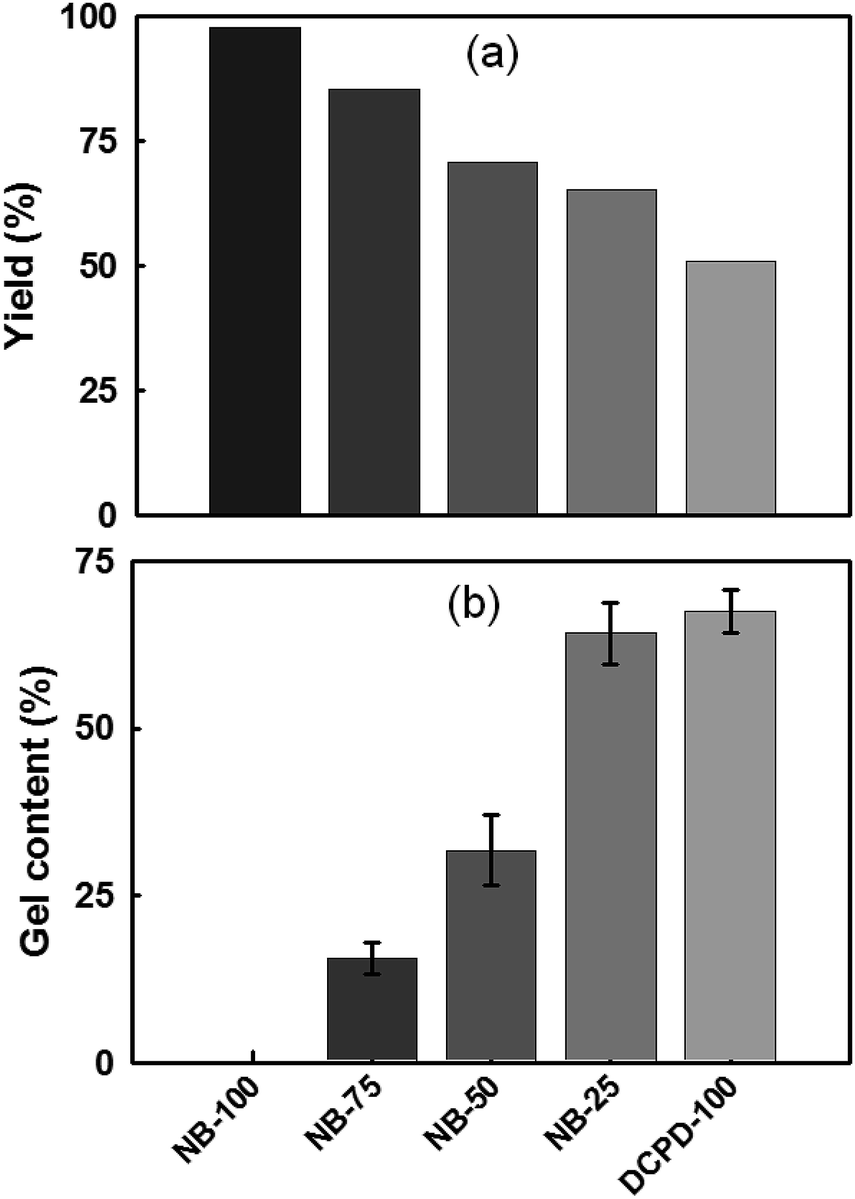
Polymer yield and gel content. Polymer yield and gel contents were determined for the NB/DCPD-based films containing different NB loadings (Fig. 4). As expected, owing to the lower relative reactivity of DCPD compared to that of NB monomer, there is a gradual decrease of the polymer yield at higher concentration in DCPD (Fig. 4a). Indeed, the reaction is almost quantitative for NB-100 (yield > 98%) while the yield is minimal for DCPD-100 (50%). Between these two homopolymers, the hybrid samples display a continuous range of intermediate values. To assess the possible formation of cross-linked network, the gel fraction of the cured specimens was also evaluated. NB-100 film, devoid of DCPD, shows consistently no gel content, in agreement with the formation of linear chains (Fig. 4b). The gel fraction of the films steadily increases with addition of more DCPD, but never reaches 100 wt%. This result proves that our photocatalytic system allows the cross-linking of a fraction of cyclopentene rings. This is in contrast with the 1st generation Grubbs catalysts which are effective only at polymerizing the norbornene ring, thus undermining the production of cross-linked materials.52,58,59
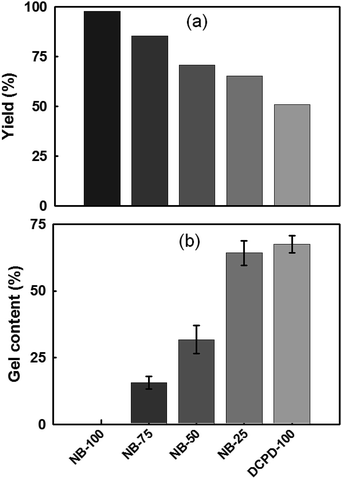 |
| | Fig. 4 Characterization of UV-irradiated NB/DCPD specimens: (a) polymer yield, and (b) gel content. | |
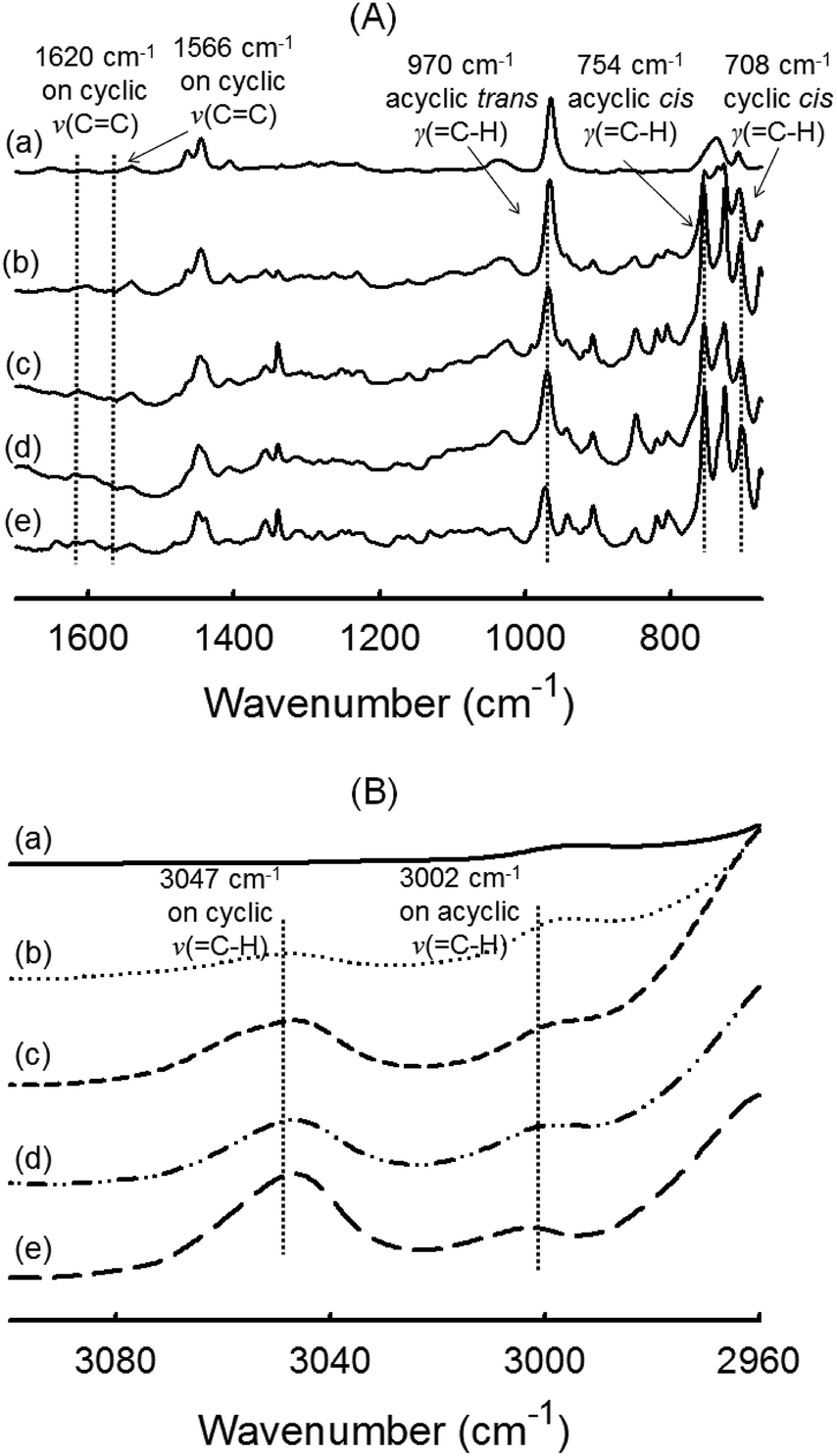
FTIR spectroscopy. Turning to chemical structure of the polymer network, the same series of five films was analyzed by ATR-FTIR spectroscopy. In Fig. 5A, all spectra show the absence of signal at 1566 cm−1 ascribed to the alkene CC stretching vibration of norbornenyl groups (present indistinctly in NB or DCPD).28,60 This suggests that norbornenyl rings were either completely reacted during the ROMP or removed upon drying (unreacted monomer). Because cyclopentene has a lower ring strain than NB, the frequency of the CC stretching vibration is decreased and generally arises at 1610–1650 cm−1. The ν(CC) of the internal double bond of cyclopentene (which is anyway a weakly intense band) is not visible in our system regardless of DCPD content. To estimate the amount of residual cyclopentene rings, the specific out-of-plane deformation vibration at ∼710 cm−1 was considered. Clearly visible at 708 cm−1, its presence supports an incomplete cross-linking process.57 In addition, the signals at 970 cm−1 and 754 cm−1 were attributed to the deformation vibrations of C–H bonds on trans and cis acyclic double bonds, respectively.57,61 They indicate that the chain backbone is made up of a mixture of both trans- and cis-disubstituted alkene –(CHCH)– in accordance with a ROMP mechanism. The C–H stretching region at 3000–3100 cm−1 is also informative because it is not overlapped by alkane C–H stretching absorption occurring below 3000 cm−1. As shown in Fig. 5B, two broad signals at 3047 cm−1 and 3002 cm−1 were found in the spectra of polymer films containing DCPD, that are readily ascribed to the C–H stretching of –CHCH– vinylene groups. The band at 3047 cm−1 can be associated to cyclic olefins (residual cyclopentene) while the second signal at 3002 cm−1 reflects acyclic olefin (cyclopentene reacted by ROMP).35,60,62 In conclusion, the FTIR data of DCPD-based films show a partial cross-linking by reaction of the cyclopentene ring, which occurs through a metathesis mechanism. The possibility of cross-linking via olefin coupling of the cyclopentene ring is examined in the next section that deals with the use of 13C solid state NMR to characterize the polymer films.
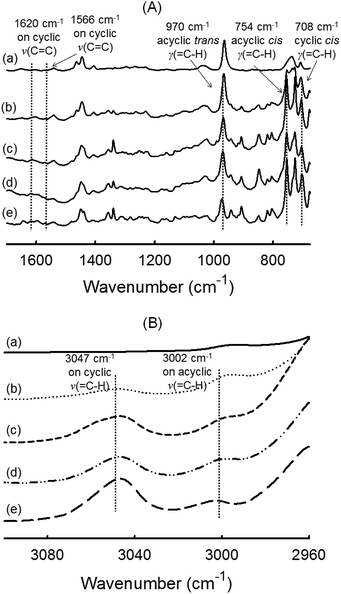 |
| | Fig. 5 FTIR-ATR spectra of polymer films obtained by photoROMP at 365 nm: (a) NB-100, (b) NB-75, (c) NB-50, (d) NB-25 and (e) DCPD-100: (A) expanded spectra in a range of 675–1700 cm−1 and (B) expanded spectra in a range of 2960–3100 cm−1. | |
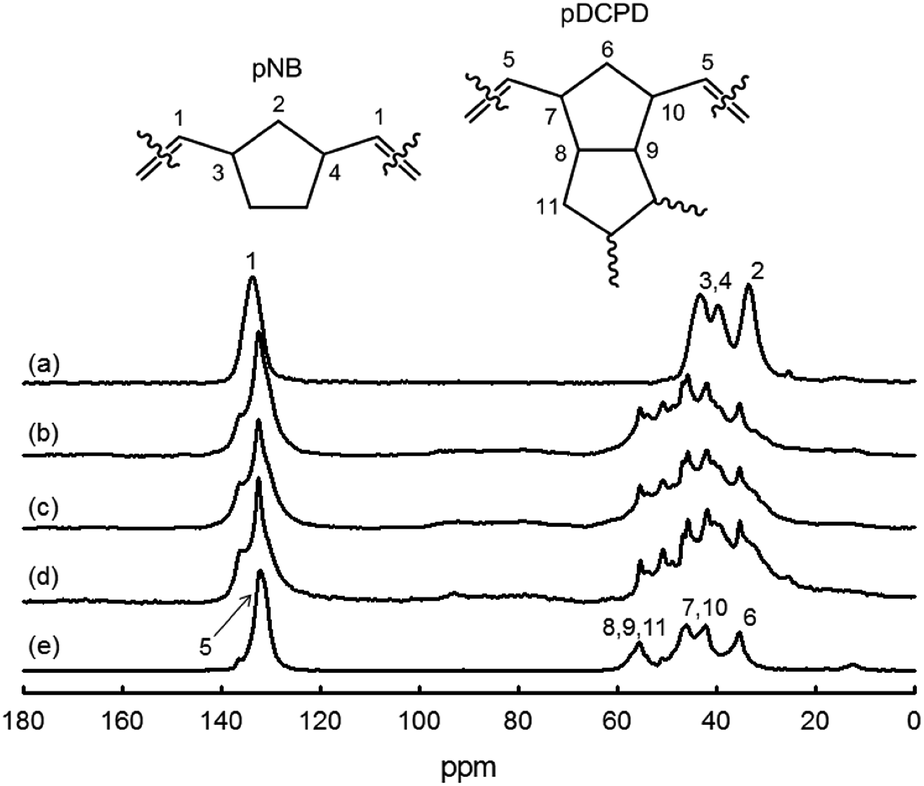
13C CP MAS NMR. In Fig. 6, the 13C CP MAS spectra of the different NB/DCPD samples display strong similarities. The region at ∼130 ppm represents olefinic carbons (sp2-C) while the signals at 30–60 ppm are reflective of aliphatic carbons (sp3-C). As mentioned above (Scheme 4), the CC double bonds of cyclopentene ring can undergo either metathesis or olefin coupling reactions. Of interest is that 13C CP MAS NMR can be used to estimate the contribution of olefin coupling. Indeed, occurrence of olefin coupling is manifested by the conversion of two sp2 carbons (from cyclopentene) into two sp3 carbons. Consequently, the ratio of integrated area of sp2-C and sp3-C can give access to the fraction of cyclopentene involved in olefin coupling (see Experimental section for calculation details).35,63 Our calculations reveal a significant contribution of olefin coupling which ranges from 11 to 34%. Olefin coupling of cyclopentene ring tends to increase with a higher concentration of NB: 11% (DCPD-100) <22% (NB-25) <26% (NB-50) <34% (NB-75). This trend can be rationalized on the basis that ROMP of NB is known to be highly exothermic and that olefin coupling is activated by heat.56
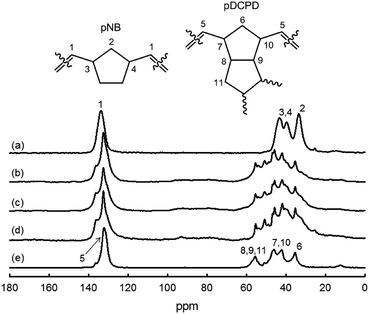 |
| | Fig. 6 13C CP MAS NMR spectra of NB/DCPB films: (a) NB-100, (b) NB-75, (c) NB-50, (d) NB-25 and (e) DCPD-100. | |
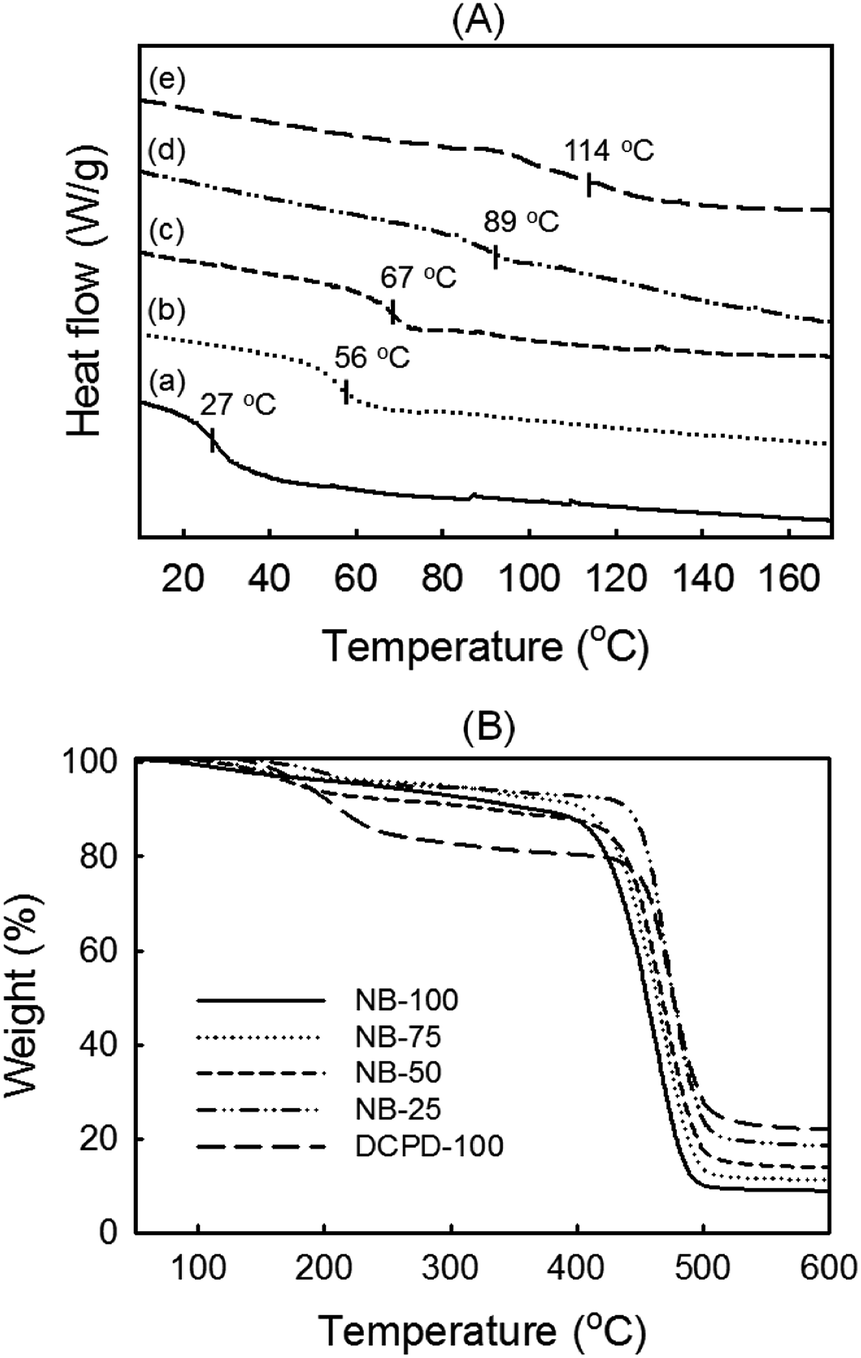
Thermal analysis. The same series of NB/DCPD-based specimens was analyzed by DSC and TGA under N2. As shown in Fig. 7A, the DSC thermograms indicate a strong impact of DCPD content on the glass transition temperature (Tg). As expected, the lowest Tg value was found for pure polyNB (NB-100); the value of 27 °C is fully consistent with literature data.64,65 Upon increasing the content of DCPD, higher Tg values were gradually obtained, with a maximum at 114 °C found for DCPD-100. This tendency is likely to be caused by the increase of cross-linking density as more cyclopentene rings react. In the literature, the Tg of polyDCPD generally ranges from 53–165 °C.52,53 Hence, our intermediate Tg value agrees with a significant fraction of non-reacted cyclopentene rings as evidenced by FTIR and solid-state NMR. The TGA traces in Fig. 7B are much less dependent on the DCPD concentration and show systematically two distinct decomposition regions. The gradual weight loss during the first decomposition stage arising at 200 °C is suggestive of low-molecular weight chains decomposition. Because of its sluggish polymerization rates, pure polyDCPD is likely to contain a higher fraction of short linear chains. This drives a weight loss at low temperature that is significantly more marked than for NB-samples. During the second decomposition stage taking place at 400–500 °C, all curves exhibited a serious decline in the weight loss, illustrating the degradation of the main polymer chains.52,66 DCPD-100 showed the greatest thermal stability whereas a slight and progressive decrease was noted upon increasing NB loading. This is consistent with the drop of cross-linking density.66 Also noteworthy is that a single main decomposition peak is observed regardless of NB content, suggesting a good homogeneity of the copolymer network. All samples left a charcoal residue, the highest amount (23%) was achieved with DCPD-100.
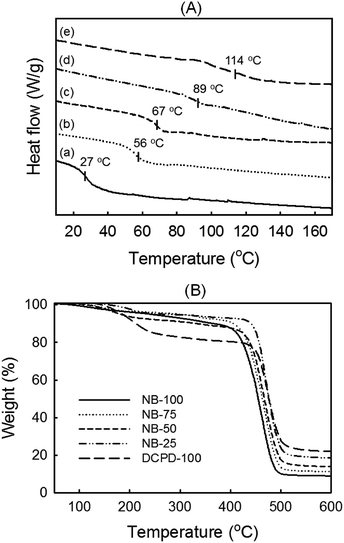 |
| | Fig. 7 Thermal characterization of NB/DCPD samples performed under nitrogen environment NB-100 (a), NB-75 (b), NB-50 (c), NB-25 (d) and DCPD-100 (e). (A) DSC traces and (B) TGA thermograms. | |
Conclusions
We have demonstrated that cross-linked ROMP polymer films can be produced under UV irradiation through the copolymerization of NB with DCPD. The approach for photoactivated olefin metathesis used a NHC photogenerator (1/ITX) combined with an inactive Ru precatalyst (Ru-1, Ru-2 and Ru-3). An active NHC-coordinated Ru-arene complex was presumed to form by reaction between the ruthenium precatalyst and the photogenerated NHC IMes. Irradiation at 365 nm was also proved to have a critical role in the formation of the active metathesis species by promoting the decoordination of the p-cymene ligand and the multiple insertions of new NB units into the propagating centre. The films were homogeneous, and the possibility of cross-linking was attested by the measurement of gel contents (10–70%) and the rising of Tg value when increasing DCPD loading. FTIR data showed a complete conversion of norbornenyl group in both monomers, but an incomplete ring-opening of the cyclopentene pendant group from DCPD. This lower ring-strained cyclic olefin is subjected to metathesis but also to olefin coupling reactions as evidenced by 13C solid-state NMR. We anticipate that photoROMP of NB and DCPD derivatives will enable the development of new cross-linked coatings with chemical, thermal and mechanical properties currently not attainable with conventional radical photopolymerization.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial support by French National Research Agency (ANR program: DS0304 2016, contract number: ANR-16-CE07-0016) is gratefully acknowledged. We are grateful to Dr Olivier Soppera, Dr Julien Poly, Dr Aissam Airoudj and Mr Stephan Knopf for their support in analysis of results.
Notes and references
- J. B. Matson and R. H. Grubbs, J. Am. Chem. Soc., 2008, 130, 6731–6733 CrossRef CAS PubMed.
- X. Bantreil and S. P. Nolan, Nat. Protoc., 2011, 6, 69–77 CrossRef CAS PubMed.
- K. J. Arrington, J. B. Waugh, S. C. Radzinski and J. B. Matson, Macromolecules, 2017, 50, 4180–4187 CrossRef CAS.
- P. Schwab, M. B. France, J. W. Ziller and R. H. Grubbs, Angew. Chem., Int. Ed., 1995, 34, 2039–2041 CrossRef CAS.
- R. R. Schrock, J. S. Murdzek, G. C. Bazan, J. Robbins, M. DiMare and M. O'Regan, J. Am. Chem. Soc., 1990, 112, 3875–3886 CrossRef CAS.
- M. Scholl, S. Ding, C. W. Lee and R. H. Grubbs, Org. Lett., 1999, 1, 953–956 CrossRef CAS PubMed.
- J. Huang, E. D. Stevens, S. P. Nolan and J. L. Petersen, J. Am. Chem. Soc., 1999, 121, 2674–2678 CrossRef CAS.
- S. B. Garber, J. S. Kingsbury, B. L. Gray and A. H. Hoveyda, J. Am. Chem. Soc., 2000, 122, 8168–8179 CrossRef CAS.
- C. W. Bielawski and R. H. Grubbs, Angew. Chem., Int. Ed., 2000, 39, 2903–2906 CrossRef CAS.
- W. A. Green, J. Mol. Catal. A: Chem., 2004, 213, 39–45 CrossRef.
- Z. Yao, L. Zhou, B. Dai and K. Cao, J. Appl. Polym. Sci., 2012, 125, 2489–2493 CrossRef CAS.
- J. Asrar, J. Appl. Polym. Sci., 1993, 47, 289–293 CrossRef CAS.
- A. E. Goetz and A. J. Boydston, J. Am. Chem. Soc., 2015, 137, 7572–7575 CrossRef CAS PubMed.
- A. Ben-Asuly, E. Tzur, C. E. Diesendruck, M. Sigalov, I. Goldberg and N. G. Lemcoff, Organometallics, 2008, 27, 811–813 CrossRef CAS.
- C. Slugovc, D. Burtscher, F. Stelzer and K. Mereiter, Organometallics, 2005, 24, 2255–2258 CrossRef CAS.
- M. Barbasiewicz, A. Szadkowska, R. Bujok and K. Grela, Organometallics, 2006, 25, 3599–3604 CrossRef CAS.
- A. Piermattei, S. Karthikeyan and R. P. Sijbesma, Nat. Chem., 2009, 1, 133–137 CrossRef CAS PubMed.
- A. Ben-Asuly, A. Aharoni, C. E. Diesendruck, Y. Vidavsky, I. Goldberg, B. F. Straub and N. G. Lemcoff, Organometallics, 2009, 28, 4652–4655 CrossRef CAS.
- A. Aharoni, Y. Vidavsky, C. E. Diesendruck, A. Ben-Asuly, I. Goldberg and N. G. Lemcoff, Organometallics, 2011, 30, 1607–1615 CrossRef CAS.
- Y. Ginzburg, A. Anaby, Y. Vidavsky, C. E. Diesendruck, A. Ben-Asuly, I. Goldberg and N. G. Lemcoff, Organometallics, 2011, 30, 3430–3437 CrossRef CAS.
- B. K. Keitz and R. H. Grubbs, J. Am. Chem. Soc., 2009, 131, 2038–2039 CrossRef CAS PubMed.
- A. Y. Khalimon, E. M. Leitao and W. E. Piers, Organometallics, 2012, 31, 5634–5637 CrossRef CAS.
- C. Theunissen, M. A. Ashley and T. Rovis, J. Am. Chem. Soc., 2019, 141, 6791–6796 CrossRef CAS PubMed.
- O. Eivgi and N. Lemcoff, Synthesis, 2018, 50, 49–63 CrossRef CAS.
- R. A. Weitekamp, H.
A. Atwater and R. H. Grubbs, J. Am. Chem. Soc., 2013, 135, 16817–16820 CrossRef CAS PubMed.
- D. Wang, K. Wurst, W. Knolle, U. Decker, L. Prager, S. Naumov and M. R. Buchmeiser, Angew. Chem., Int. Ed., 2008, 47, 3267–3270 CrossRef CAS PubMed.
- D. Wang, K. Wurst and M. R. Buchmeiser, Chem.–Eur. J., 2010, 16, 12928–12934 CrossRef CAS PubMed.
- J. Pinaud, T. K. H. Trinh, D. Sauvanier, E. Placet, S. Songsee, P. Lacroix-Desmazes, J.-M. Becht, B. Tarablsi, J. Lalevée, L. Pichavant, V. Héroguez and A. Chemtob, Chem.–Eur. J., 2018, 24, 337–341 CrossRef CAS PubMed.
- T. K. H. Trinh, J.-P. Malval, F. Morlet-Savary, J. Pinaud, P. Lacroix-Desmazes, C. Reibel, V. Héroguez and A. Chemtob, Chem.–Eur. J., 2019, 25, 9242–9252 CAS.
- W. A. Herrmann, M. Elison, J. Fischer, C. Köcher and G. R. J. Artus, Chem.–Eur. J., 1996, 2, 772–780 CrossRef CAS.
- A. Tudose, A. Demonceau and L. Delaude, J. Organomet. Chem., 2006, 691, 5356–5365 CrossRef CAS.
- L. Delaude, M. Szypa, A. Demonceau and A. F. Noels, Adv. Synth. Catal., 2002, 344, 749 CrossRef CAS.
- L. Delaude, A. Demonceau and A. F. Noels, Chem. Commun., 2001, 986–987 RSC.
- D. Massiot, F. Fayon, M. Capron, I. King, S. L. Calvé, B. Alonso, J.-O. Durand, B. Bujoli, Z. Gan and G. Hoatson, Magn. Reson. Chem., 2002, 40, 70–76 CrossRef CAS.
- D. P. Mohite, S. Mahadik-Khanolkar, H. Luo, H. Lu, C. Sotiriou-Leventis and N. Leventis, Soft Matter, 2013, 9, 1516–1530 RSC.
- J. D. Wilkey and G. B. Schuster, J. Org. Chem., 1987, 52, 2117–2122 CrossRef CAS.
- X. Sun, J. P. Gao and Z. Y. Wang, J. Am. Chem. Soc., 2008, 130, 8130–8131 CrossRef CAS PubMed.
- X. Sun, Development of tetraphenylborate-based photobase generators and sacrificial polycarbonates for radiation curing and photoresist applications, PhD thesis, Carleton University, 2009.
- A. V. Zhukhovitskiy, J. Geng and J. A. Johnson, Chem.–Eur. J., 2015, 21, 5685–5688 CrossRef CAS PubMed.
- L. Jafarpour, J. Huang, E. D. Stevens and S. P. Nolan, Organometallics, 1999, 18, 3760–3763 CrossRef CAS.
- Y. Zhang, D. Wang, P. Lönnecke, T. Scherzer and M. R. Buchmeiser, Macromol. Symp., 2006, 236, 30–37 CrossRef CAS.
- A. Hafner, A. Mühlebach and P. A. van der Schaaf, Angew. Chem., Int. Ed., 1997, 36, 2121–2124 CrossRef CAS.
- E. L. Dias, S. T. Nguyen and R. H. Grubbs, J. Am. Chem. Soc., 1997, 119, 3887–3897 CrossRef CAS.
- M. S. Sanford, J. A. Love and R. H. Grubbs, J. Am. Chem. Soc., 2001, 123, 6543–6554 CrossRef CAS PubMed.
- L. Delaude, A. Demonceau and A. F. Noels, Macromolecules, 2003, 36, 1446–1456 CrossRef CAS.
- A. Demonceau, A. F. Noels, E. Saive and A. J. Hubert, J. Mol. Catal., 1992, 76, 123–132 CrossRef CAS.
- L. Delaude, A. Demonceau and A. F. Noels, Curr. Org. Chem., 2006, 10, 203–215 CAS.
- M. Ahr, C. Thieuleux, C. Copéret, B. Fenet and J.-M. Basset, Adv. Synth. Catal., 2007, 349, 1587–1591 CrossRef CAS.
- P. Alvarez, J. Gimeno and E. Lastra, Organometallics, 2002, 21, 5678–5680 CrossRef CAS.
- C. S. Day and D. E. Fogg, Organometallics, 2018, 37, 4551–4555 CrossRef CAS.
- D. Wang, J. Unold, M. Bubrin, W. Frey, W. Kaim and M. R. Buchmeiser, ChemCatChem, 2012, 4, 1808–1812 CrossRef CAS.
- S. Saha, Y. Ginzburg, I. Rozenberg, O. Iliashevsky, A. Ben-Asuly and N. Gabriel Lemcoff, Polym. Chem., 2016, 7, 3071–3075 RSC.
- J. Chen, F. P. Burns, M. G. Moffitt and J. E. Wulff, ACS Omega, 2016, 1, 532–540 CrossRef CAS PubMed.
- G. Yang, T. C. Mauldin and J. K. Lee, RSC Adv., 2015, 5, 59120–59130 RSC.
- K. Nomura and X. Hou, Dalton Trans., 2016, 46, 12–24 RSC.
- T. A. Davidson, K. B. Wagener and D. B. Priddy, Macromolecules, 1996, 29, 786–788 CrossRef CAS.
- G. Raptopoulos, G. C. Anyfantis, D. Chriti and P. Paraskevopoulou, Inorg. Chim. Acta, 2017, 460, 69–76 CrossRef CAS.
- S. J. Cantrill, R. H. Grubbs, D. Lanari, K. C.-F. Leung, A. Nelson, K. G. Poulin-Kerstien, S. P. Smidt, J. F. Stoddart and D. A. Tirrell, Org. Lett., 2005, 7, 4213–4216 CrossRef CAS PubMed.
- L. Gong, K. Liu, E. Ou, F. Xu, Y. Lu, Z. Wang, T. Gao, Z. Yang and W. Xu, RSC Adv., 2015, 5, 26185–26188 RSC.
- Q. Sun, S. Ma, Z. Ge and Y. Luo, High Perform. Polym., 2017, 29, 931–936 CrossRef CAS.
- M. J. Abadie, M. Dimonie, C. Couve and V. Dragutan, Eur. Polym. J., 2000, 36, 1213–1219 CrossRef CAS.
- S. Saha, Y. Ginzburg, I. Rozenberg, O. Iliashevsky, A. Ben-Asuly and N. Gabriel Lemcoff, Polym. Chem., 2016, 7, 3071–3075 RSC.
- A. Bang, D. Mohite, A. M. Saeed, N. Leventis and C. Sotiriou-Leventis, J. Sol-Gel Sci. Technol., 2015, 75, 460–474 CrossRef CAS.
- R. F. Ohm and T. M. Vial, J. Elastomers Plast., 1978, 10, 150–162 CrossRef CAS.
- A. M. Spring, D. Maeda, M. Ozawa, K. Odoi, F. Qiu, K. Yamamoto and S. Yokoyama, Polym. Bull., 2015, 72, 503–521 CrossRef CAS.
- M. R. Kessler and S. R. White, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2373–2383 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra05831a |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence c,
Patrick Lacroix-Desmazes
c,
Patrick Lacroix-Desmazes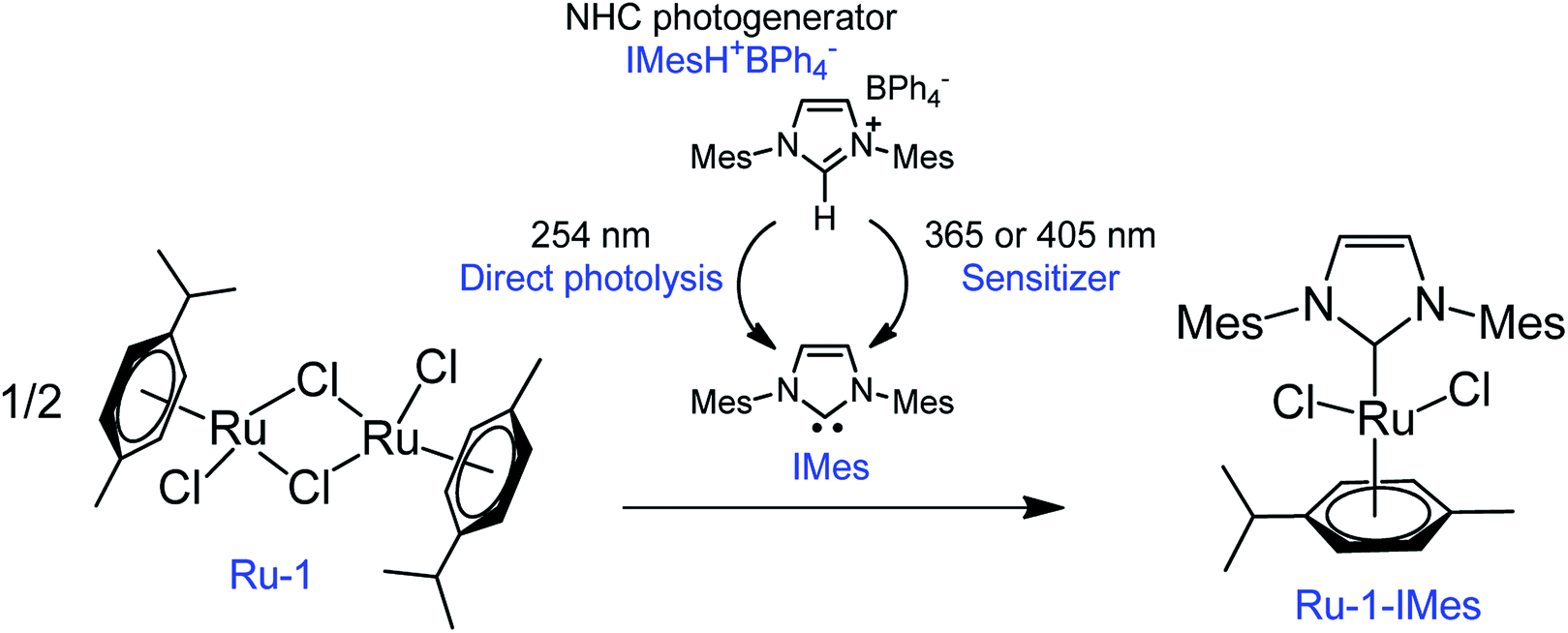
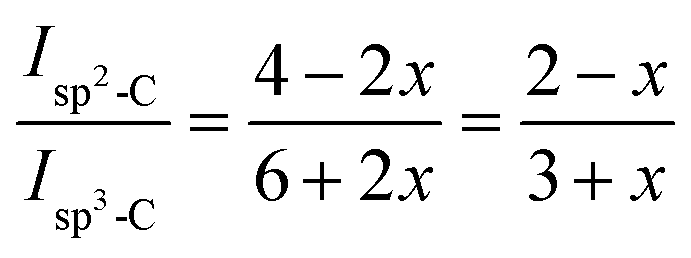
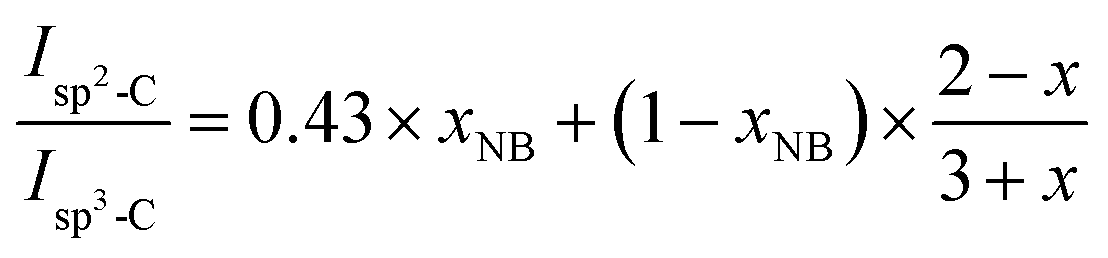
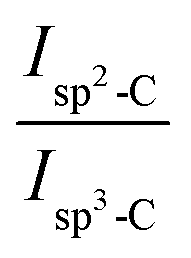 in polyNB (calculated from sample NB-100), and xNB is the molar fraction of NB in the NB/DCPD monomer mixture.
in polyNB (calculated from sample NB-100), and xNB is the molar fraction of NB in the NB/DCPD monomer mixture.
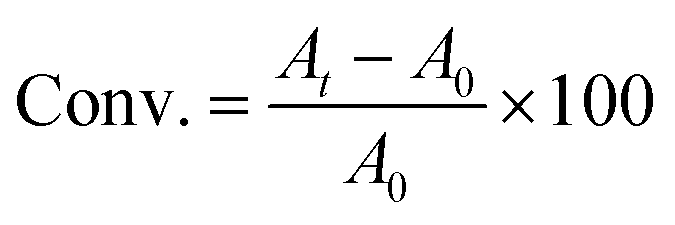

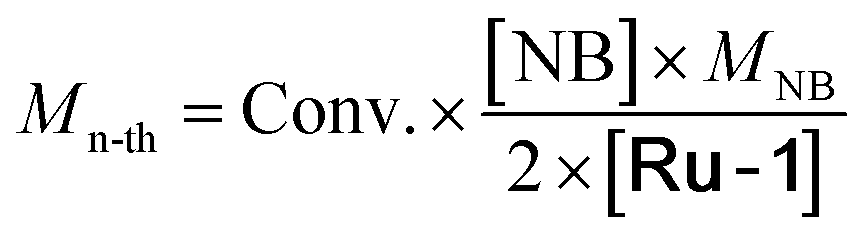 .d Experimental number average molar mass Mn-th obtained by SEC.e Irradiation at 405 nm, 4.7 mW cm−2, 1 min. n.d. means not determined due to the absence of reaction or the formation of an insoluble polymer.
.d Experimental number average molar mass Mn-th obtained by SEC.e Irradiation at 405 nm, 4.7 mW cm−2, 1 min. n.d. means not determined due to the absence of reaction or the formation of an insoluble polymer.