
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural complexities and sodium-ion diffusion in the intercalates NaxTiS2: move it, change it, re-diffract it†
Dennis Wiedemann *a,
Emmanuelle Suardb and
Martin Lercha
*a,
Emmanuelle Suardb and
Martin Lercha
aTechnische Universität Berlin, Institut für Chemie, 10623 Berlin, Germany. E-mail: dennis.wiedemann@chem.tu-berlin.de
bInstitut Laue-Langevin, 38042 Grenoble, France
First published on 3rd September 2019
Abstract
After momentary attention as potential battery materials during the 1980s, sodium titanium disulphides, like the whole Na–Ti–S system, have only been investigated in a slapdash fashion. While they pop up in current reviews on the very subject time and again, little is known about their actual crystal-structural features and sodium-ion diffusion within them. Herein, we present a short summary of literature on the Na–Ti–S system, a new synthesis route to Na0.5TiS2-3R1, and results of high-temperature X-ray and neutron diffractometry on this polytype, which is stable for medium sodium content. Based thereon, we propose a revision of the crystal structure reported in earlier literature (missed inversion symmetry). Analyses of framework topology, probability-density functions, and maps of the scattering-length density reconstructed using maximum-entropy methods (all derived from neutron diffraction) reveal a honeycomb-like conduction pattern with linear pathways between adjacent sodium positions; one-particle potentials indicate associated activation barriers of ca. 0.1 eV or less. These findings are complemented by elemental analyses and comments on the high-temperature polytype Na0.9TiS2-2H. Our study helps to get a grip on structural complexity in the intercalates NaxTiS2, caused by the interplay of layer stacking and Na–Ti–vacancy ordering, and provides first experimental results on pathways and barriers of sodium-ion migration.
Introduction
Layered lithium-ion conductors are a successful substance class for energy storage, e.g., in modern consumer electronics. Much effort has been put into research on their sodium congeners as possible high-voltage, low-cost alternatives—so far, to no commercial avail. Although alkali transition-metal dichalcogenides are not a recent focus, they pop up in current reviews time and again.1 Probably as analogues of the once front-running lithium intercalates LixTiS2, the sodium compounds NaxTiS2 received some attention in the early to mid-1980s, leading to an overall manageable corpus of literature on the Na–Ti–S system. They have also sparked some commercial interest, manifested in patents, as thermoelectrics and auxiliary materials for cathodes in sodium-ion batteries.2,3As first Na–Ti–S compound, uncharacterised “Na2TiS3” was reported in a process patent to have an X-ray pattern similar to the also uncharacterised “Li2TiS3” and “K2TiS3” in 1960.4 Five years later, Rüdorff described the first layered structure in the system and wrongly assigned it the NaHF2 type [modern prototype according to the Inorganic Crystal Structure Database (ICSD): CrOOH(R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m)]—not yet aware of its hydrogen positions.5 Reliable sodium positions were not established before the first systematic study in 1971.6 Today, five kinds of Na–Ti–S compounds can be distinguished:
m)]—not yet aware of its hydrogen positions.5 Reliable sodium positions were not established before the first systematic study in 1971.6 Today, five kinds of Na–Ti–S compounds can be distinguished:
• The structure of the intercalates NaxTiS2 with 0 < x ≤ 1 consists of 2∞[TiS2]x− sandwich layers with sodium ions in between. The compounds exist in four different structurally elucidated stacking variants: the polytypes 2H, 3R1, 3R2, and 6H (see Table 1 and Fig. 1). With these materials, the remaining article is concerned.
| Polytype | 2H | 3R1 | 3R2 | 6R | |
|---|---|---|---|---|---|
| a Best values;11 reported maximum extents in parentheses.b Within occupied sodium layers.c Corrected for a missed mirror plane,12 originally given as R3.d Corrected in May 2019 (see Table S1),13 formerly ambiguously given as CuCrSe2–AgCrSe2(R3m).e O1 layers are empty. | |||||
| Synonyms | 2H(I) | 3R(I), phase I b | 3R(II), phase I a | 6R(I), phase II | |
| Stability rangea | High-temperature phase | 0.40(2) < x < 0.64(2) (0.35 < x < 0.72) | 0.75(2) < x ≤ 1 (0.70 < x ≤ 1) | 0.15(2) < x < 0.25(2) (0.10 < x < 0.33) | |
| Best characterised representative | x = 1 (ref. 7) | x = 0.55 (ref. 6) | x = 1 (ref. 6) | x = 0.3 (ref. 6 and 8) | |
| Coordination polyhedron9 | Na | [6p] | [6p] | [6ap] | [6p] |
| Ti | [6o] | [6o] | [6o] | [6o] | |
| Stage | 1 | 1 | 1 | 2 | |
| Space-group type | P63/mmc | R3mc | Rm |
Rm |
|
| Structure prototype (ICSD) | NaTiS2 | CuCrSe2d | Delafossite-NaCrS2 | Ag1.5Nb6S12 | |
| Delmas' notation10 | P2 | P3 | O3 | O1–P3e | |
| Anion stacking | AABB | AABBCC | ABCABC | ABABBCBCCACA | |
| Sodium-ion orderb | Disordered over all voids | Ordered in half of the voids | Ordered in half of the voids | Disordered over all voids | |
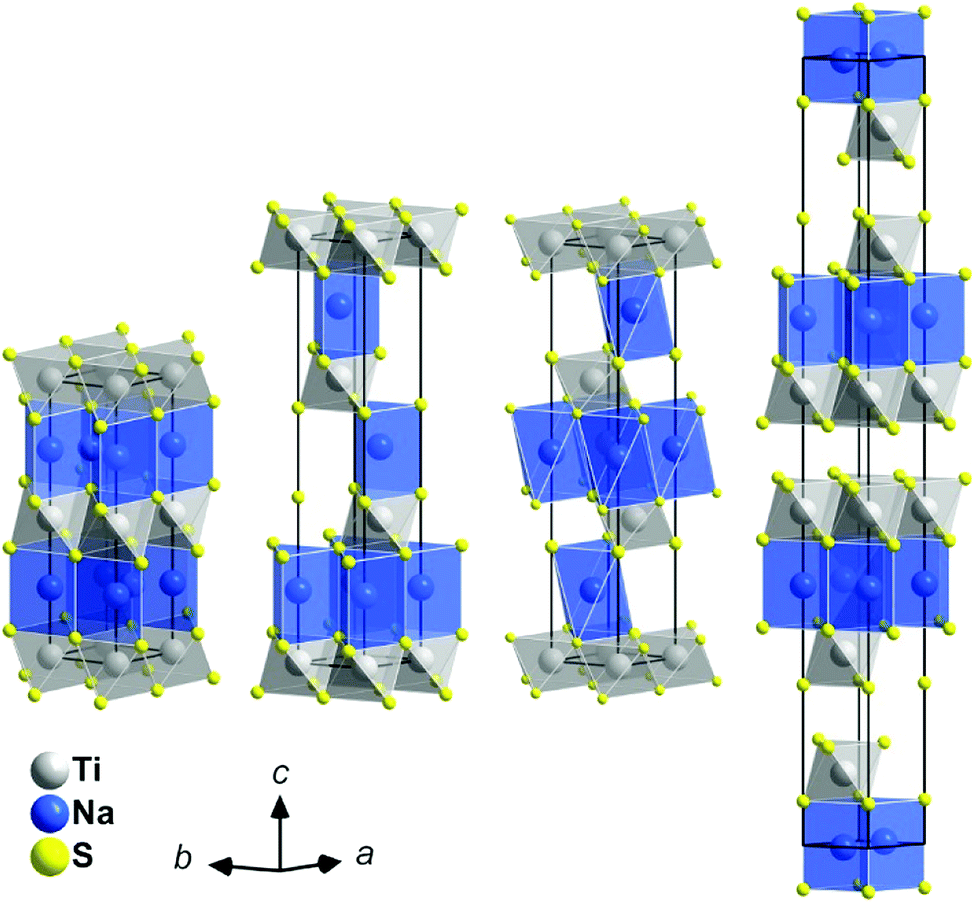 | ||
| Fig. 1 Crystal structures of the NaxTiS2 polytypes; left to right: 2H, 3R1, 3R2, and 6H. Atoms are plotted with arbitrary radii, unit cells are depicted in black. Note: depending on x, sodium positions may not be fully occupied. | ||
• The excess-titanium compounds NaxTi1+δS2 with 0 < δ ≤ ⅓ and 0 < x ≤ 1 − 3δ contain additional titanium ions in intercalation layers, which hinder diffusion of the otherwise mobile sodium ions.14
• Contrary to its first assignment, Na2TiS3 ≡ Na1.Ti0.![[6 with combining macron]](https://www.rsc.org/images/entities/char_0036_0304.gif) S2 exhibits a structure homeotypic to Na2SnS3, not K2TiS3. At closer inspection, it crystallises in a sixfold superstructure of the 3R polytypes with an average structure akin to NaxTiS2-3R2. It represents an OD structure with alternating pure sodium and mixed sodium-titanium layers, constituting a distorted NaCl homeotype.15 Upon heating, Na2TiS3 decomposes to NaTiS2-2H under oxidation of sulphide.8 The compound dubbed “3R′(II)-NaTiS2” most probably is a sodium-deficient variant of Na2TiS3.8,16
S2 exhibits a structure homeotypic to Na2SnS3, not K2TiS3. At closer inspection, it crystallises in a sixfold superstructure of the 3R polytypes with an average structure akin to NaxTiS2-3R2. It represents an OD structure with alternating pure sodium and mixed sodium-titanium layers, constituting a distorted NaCl homeotype.15 Upon heating, Na2TiS3 decomposes to NaTiS2-2H under oxidation of sulphide.8 The compound dubbed “3R′(II)-NaTiS2” most probably is a sodium-deficient variant of Na2TiS3.8,16
• The electrochemically intercalated compounds NaxTiS3 contain sulphide as well as disulphide ions and are formed from TiS3 via disulphide (instead of titanium) reduction and concomitant bond cleavage.17 They have neither been studied ex situ nor structurally characterised.
• Structurally complex Na4TiS4 has only been reported once in a book of poster abstracts. It is said to crystallise in the orthorhombic space group Fdd2 with a = 38.49(2), b = 59.36(3), c = 7.033(2) Å, and Z = 120. The unit cell allegedly contains eight crystallographically independent TiS4 tetrahedra, which are arranged in triple slabs parallel to (010).18
The polytype, which NaxTiS2 assumes, depends on x as well as on temperature. In the past, inconsistencies in reported stability ranges were discussed and bona fide best values were derived acknowledging synthetic (chemical vs. electrochemical intercalation) and analytical differences (diffraction patterns vs. discharge curves).11 The only reported thermally induced phase transformations are the entropy-driven order–disorder transition for small x according to eqn (1) and the irreversible reconstitution of the (at ambient temperature metastable) 2H phase for large x following eqn (2).8 Transformations to the 2H form from either of the 3R polytypes were not observed and an irreversible transition between the latter was attributed to decomposition via sodium loss.
 | (1) |
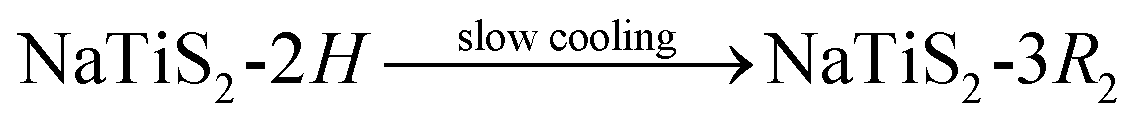 | (2) |
Ordering of sodium ions and vacancies was detected in electrochemically intercalated TiS2 single crystals, where 2 × 2, 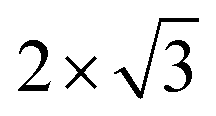 , and
, and  superstructures may occur. For x < 0.11, a stage-3 compound with a
superstructures may occur. For x < 0.11, a stage-3 compound with a 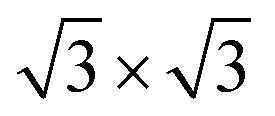 superstructure of a 3R phase containing sodium in trigonal-antiprismatic coordination was discovered but not conclusively characterised.19,20 The existence of such superstructures was later rationalised,21 comprehensively computed and explained.22
superstructure of a 3R phase containing sodium in trigonal-antiprismatic coordination was discovered but not conclusively characterised.19,20 The existence of such superstructures was later rationalised,21 comprehensively computed and explained.22
Data on sodium-ion diffusion in NaxTiS2 are sparse. The chemical diffusivity at ambient temperature is specified as ca. 10−9 cm2 s−1 for 0.25 < x < 0.6 (probably polytype 3R1).23,24 The much higher value of 10−7 to 10−6 cm2 s−1 for x < 0.45, which was only reported once, remains questionable.25 Unfortunately, migration barriers were merely computed for NaTiS2-3R2 (0.19 eV)26 and a hypothetical NaxTiS2 with O1 coordination/layer sequence (1.02 eV).27 Trigonal-antiprismatic coordination, as realised in the polytype 3R2, seems to hinder diffusion, so that diffusivities and migration barriers should be lower and higher in it, respectively, than in the other polytypes.28
Our dealing with these materials is less inspired by prospective application as an ion conductor than by improving the grasp of ion diffusion in them and a comparing them to their lithium congeners. This is why, herein, we report on the revised crystal structure of and sodium-ion diffusion in Na0.5TiS2-3R1 based on high-temperature neutron diffraction (ND). The study comprises topological analyses, the visualisation of diffusion pathways using the probability-density function (PDF) and maps of the scattering-length density (SLD) reconstructed via maximum-entropy methods (MEM), as well as an evaluation of migration barriers using the effective one-particle potential (OPP). Results are complemented by temperature-dependent X-ray diffraction (XRD), elemental analyses, and comments on the related compound Na0.9TiS2-2H.
Experimental
Analytical method
Hydrogen and sulphur contents were determined using a “Thermo Finnigan Flash EA 1112” analyser. Sodium was determined via optical emission spectroscopy with an inductively coupled plasma (ICP-OES) using a “Thermo Fisher Scientific iCAP 6300 Duo” evaluating the emission line at 589.5 nm.The numbers of titanium ions per formula unit (with respect to sodium) were determined via wavelength-dispersive X-ray fluorescence (XRF) measurements on a “PANalytical Axios” spectrometer evaluating the intensities for the Na-Kα and Ti-Kα transitions. Samples were prepared from ca. 100 mg of analyte and 100 mg wax (Hoechst wax) performing uniaxial pressing (PerkinElmer hydraulic hand-press). Intensity ratios and sodium intensities were calibrated against linear regressions for three different mixtures of Na2S and TiS2 with R(Na/Ti) = N(Na) ≈ 0.4, 0.7, and 1.0 to account for instability/correlation of the titanium signal.
Synthetic procedures
Found: H, 1.0(1); Na, 8.8(1); S, 48(1); N(Ti) = 1.0(2). Calc. for Na0.5S2Ti·½H2O: H, 0.8; Na, 8.7; S, 48.4%; N(Ti) = 1.0.
X-ray diffraction
Measurements for phase identification and Rietveld refinement were carried out at ambient temperature on a “PANalytical X'Pert PRO MPD” diffractometer equipped with a “PIXcel” detector using nickel-filtered Cu-Kα radiation in Bragg–Brentano (θ–θ) geometry (see Fig. S1† for details). Temperature-dependent diffractograms were recorded in dinitrogen atmosphere on a “Rigaku SmartLab 3 kW” system using nickel-filtered Cu-Kα radiation.Neutron diffraction
Measurement was carried out at Institut Laue-Langevin (ILL) using the high-resolution two-axis diffractometer D2B with Ge(335)-monochromated constant-wavelength radiation (λ = 1.594 Å) in Debye–Scherrer geometry.29,30 Compacted powder samples were mounted in a vacuum high-temperature furnace inside a vanadium can (d = 13.9 mm, h = 49.8 mm). Measurements were carried out at ambient temperature, 300, 600, and 700 °C. Data were recorded with an array of 128 3He tubes (height: 300 mm), yielding a final range of 0.10° ≤ 2θ ≤ 159.90° with Δ(2θ) = 0.05°. Initial Le-Bail fits and following Rietveld refinements were carried out using JANA2006.31 Neutron data were analytically corrected for absorption (cylindrical sample) and stripped of the irregular onset below 6.5° and, if necessary, cut-off reflections above 155°. Peak profiles were fitted with a pseudo-Voigt function using the Thompson–Cox–Hastings approach (Gaussian parameters U, V, and W; Lorentzian parameter X). A zero-shift correction and an asymmetry correction according to Howard were applied.32 The background was modelled using ten Legendre polynomials interpolating between manually defined points. Relatively weak reflections of a by-phase assuming the space-group type Fmm (probably steel from the sample environment) were treated with an appropriate Le-Bail fit (separate profile parameters).
As a starting point for Rietveld refinement, an atomic model of Na0.55TiS2 was imported from the ICSD and adjusted to reflect the actual cell content and symmetry.33 Anisotropic displacement parameters were refined for all atoms except for Ti1 at 18 °C, the very small displacement of which had to be constrained to isotropy to yield a positive-definite value. For Na1 at 600 and 700 °C, anharmonic displacement was observed. Terms up to the fourth order were tested and only kept in refinement if they were significant (|Cijk| ≥ 3σ[Cijk]) and led to a significant drop in R values. Thus, two additional unique parameters C111(Na1) and C333(Na1) were refined. Towards the end of refinement, z(Na1) was found to be within a 3σ interval of ⅙ (coplanarity of sodium ions) and was fixed at this value, yielding slightly better residuals despite one free parameter less.
Structure graphics were produced using Diamond 4.5 and VESTA.34,35 Refinement results are summarized in Table 2. Diffraction datasets are available in the ILL repository.36 CSD 1942255 to 1942259 contain the supplementary crystallographic data for this paper; these data can be obtained free of charge from FIZ Karlsruhe via https://www.ccdc.cam.ac.uk/structures.
| a I > 3σ(I).b w = 1/[σ2(I) + (0.01I)2]. | ||||
|---|---|---|---|---|
| Formula | Na0.5TiS2 | |||
| Mr/g mol−1 | 123.48 | |||
| θ/°C | 18 | 300 | 600 | 700 |
| Space group | Rm |
|||
| a/Å | 3.43840(8) | 3.45878(5) | 3.47973(6) | 3.48717(7) |
| c/Å | 21.0431(6) | 21.2010(5) | 21.3240(7) | 21.3635(8) |
| V/Å−3 | 215.453(14) | 219.651(9) | 223.610(12) | 224.983(14) |
| Z | 3 | 3 | 3 | 3 |
| Meas./obs.a reflections | 73/69 | 79/74 | 79/71 | 79/71 |
| Rp | 0.0268 | 0.0195 | 0.0178 | 0.0177 |
| wRpb | 0.0359 | 0.0257 | 0.0232 | 0.0230 |
| Rexp | 0.0176 | 0.0176 | 0.0175 | 0.0175 |
| RF | 0.0333 | 0.0320 | 0.0352 | 0.0414 |
| RIa | 0.0520 | 0.0462 | 0.0438 | 0.0464 |
Procrystal void analysis
Procrystal voids in the TiS2x− substructures were analysed using CrystalExplorer 17.5 with default options.37 Models were based on the revised room-temperature structure of Na0.5TiS2-3R1 and structures of NaTiS2-2H, NaTiS2-3R2, and Na0.3TiS2-6R from the ICSD.33 Lower bounds for contiguous void networks were found by successively altering the procrystal density in steps of Δρpro = 0.0001 a.u.MEM reconstruction
Dysnomia 1.0 was used for MEM-reconstruction of SLDs from final structure factors as put out by JANA2006.38 The unit cell was divided into 96 × 96 × 588 voxels. Starting from a uniform intensity prior, the limited-memory Broyden–Fletcher–Goldfarb–Shanno (L-BFGS) algorithm39 was employed with uncertainties augmented by E = 0.5 and relative weights set to λ2 = 1, λn = 0 for n ≥ 4 to avoid overfitting. Final RF/wRF were 0.0342/0.0305 and 0.0376/0.0328 for data acquired at 600 and 700 °C, respectively.OPP calculation
Sodium OPPs were calculated from PDFs as well as from MEM-reconstructed SLDs using CalcOPP 2.0.1.40 In the latter case, the maximal positive SLD found within the sodium layer was set to represent a potential energy of V = 0. For error estimation on PDF-derived data, the Monte-Carlo routines implemented in JANA2006 were employed (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 iterations, final accuracy < 1%).
000 iterations, final accuracy < 1%).
Results and discussion
Synthesis and structure
The intercalates NaxTiS2 can be synthesized from the binary sulphides in a H2S feed. At 700 °C, ca. 3–4% of H2S decompose into H2 and Sn.41 This creates a mildly reductive atmosphere that acts according to eqn (3).| xNa2S + 2TiS2 + xH2 → 2NaxTiS2 + xH2S | (3) |
Compounds of this type are very hygroscopic and may reversibly co-intercalate up to two equivalents of water per formula unit without degradation.42,43 Elemental analyses show that even short handling in air is enough to effect absorption of ca. half an equivalent of water (no significant difference between stored samples with and without neutron diffractometry was observed). At the elevated temperatures examined herein, however, the samples dehydrate completely.
During refinement of the initial model derived from Na0.55TiS2-3R1 against ND data, occupation of a second sodium position at (⅔, ⅓, ∼0.17), strong correlation of the sulphide ions' z coordinates, and refinement instability occurred. A closer look at the structures in the hitherto assigned space group R3m and its supergroup Rm reveals the following differences: the two unique sulphide positions with z = 0.39 and 0.60 in the former correspond to a single one with z = 0.40 in the latter (mismatch Δz = 0.01). The two nearly equally occupied sodium positions with z = 0.17 correspond to a single position that has to be at z = ⅙ to warrant a coplanar arrangement (mismatch Δz = 0.0). Refinement in Rm solved the initial problems (see Fig. 2 for exemplary and Fig. S4–S6† for remaining diffractograms). We found no hint at symmetry lowering at any temperature. While Na0.55TiS2 as synthesised by Rouxel et al.6 might represent an ordered variant, we assume a case of missed symmetry because of the following reasons: due to the perfect overlap of all reflections in R3m that are symmetry equivalent in Rm, only ever-so-subtle intensity differences distinguish one from the other (see Fig. S2†). For 3R1-isotypic K0.6VS2, Bronsema and Wiegers recognised the same problem and could not resolve it via X-ray powder diffraction.44 Furthermore, both sodium positions in R3m are crystal-chemically nearly identical and should not give rise to ordering. We thus conclude that the published structure is to be revised‡.
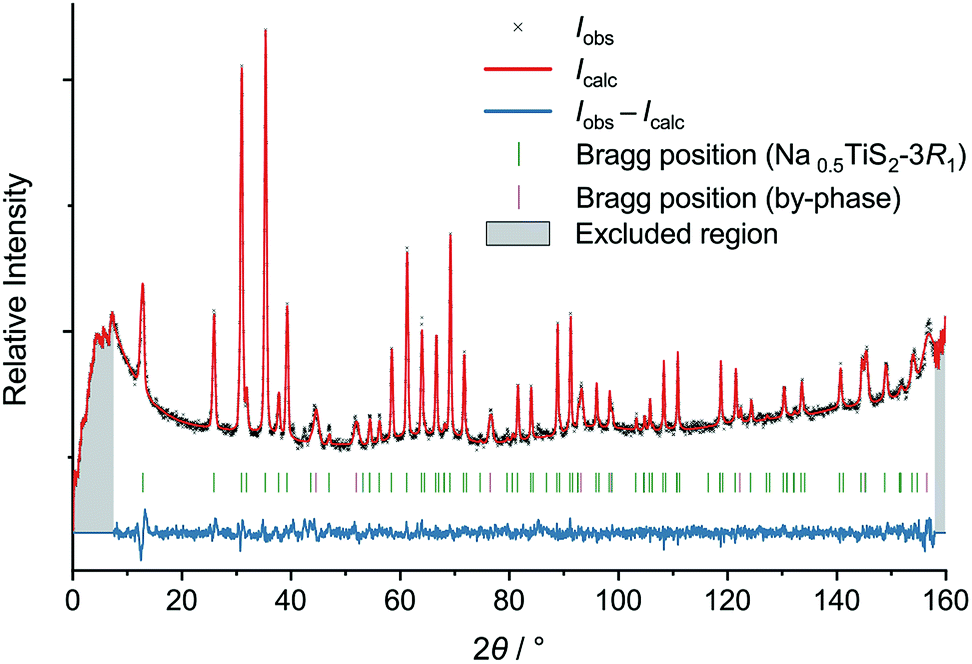 | ||
| Fig. 2 Neutron diffractogram of Na0.5TiS2-3R1 (revised structure) at 700 °C with results of Rietveld refinement. | ||
Temperature-dependent XRD shows that, on heating, a previously stored Na0.5TiS2-3R1 sample loses co-intercalated water up to 100 °C (see Fig. 3, characteristic reflection at ca. 10°). No phase change akin to the 1T–3R transformation§ in the lighter homologue Li0.7TiS2 occurs.45 In dinitrogen atmosphere above 550 °C, decomposition starts (slight reflection-positional shift to higher angles, degradation of intensities, additional small reflections) under concomitant sulphur formation (found in the reaction chamber). While the absence of phase transitions is confirmed by ND, the sample has proven to be stable in a vanadium can surrounded by vacuum up to at least 700 °C. Cell dimensions lie in the range expected for x = 0.5 at ambient temperature and evolve in a roughly linear fashion during heating (cf. Fig. S7†).46 The latter also holds for the equivalent displacement parameters of the titanium and sulphide ions, whereas the one of the sodium ion starts at a value five to six times as large and grows disproportionately (see Fig. S8 and S9†). This is due to supposedly static disorder at room temperature and the onset of anharmonic dynamics (i.e., ion diffusion) between 300 and 600 °C.
 | ||
| Fig. 3 Heat map of uncorrected temperature-dependent X-ray diffractograms of Na0.5TiS2-3R1 in dinitrogen atmosphere. The colouring is based on raw counts; the scale is logarithmic. | ||
Sodium diffusion pathways
We will describe the methods used herein very briefly. A more detailed overview has already been published.47 The pathways refer to thermally activated ion diffusion at temperatures high enough to warrant ample ion mobility.The low and comparable isovalues, at which the voids in the polytypes 2H,¶ 3R1 (our revised structure), and 6R form contiguous paths, indicate that they are similarly well-suited sodium-ion conductors (see Fig. 4). The framework voids are located at the partly occupied sodium positions and connect to a honeycomb-like conduction pattern. The voids in the 3R2 structure also arrange in a honeycomb-like fashion and give no hint at any reason for an ordered occupation of half of them (like the current structural model suggests). The voids connect at a significantly higher isovalue to a carpet-like pattern, which suggests that this polytype is a worse and more isotropic sodium-ion conductor.
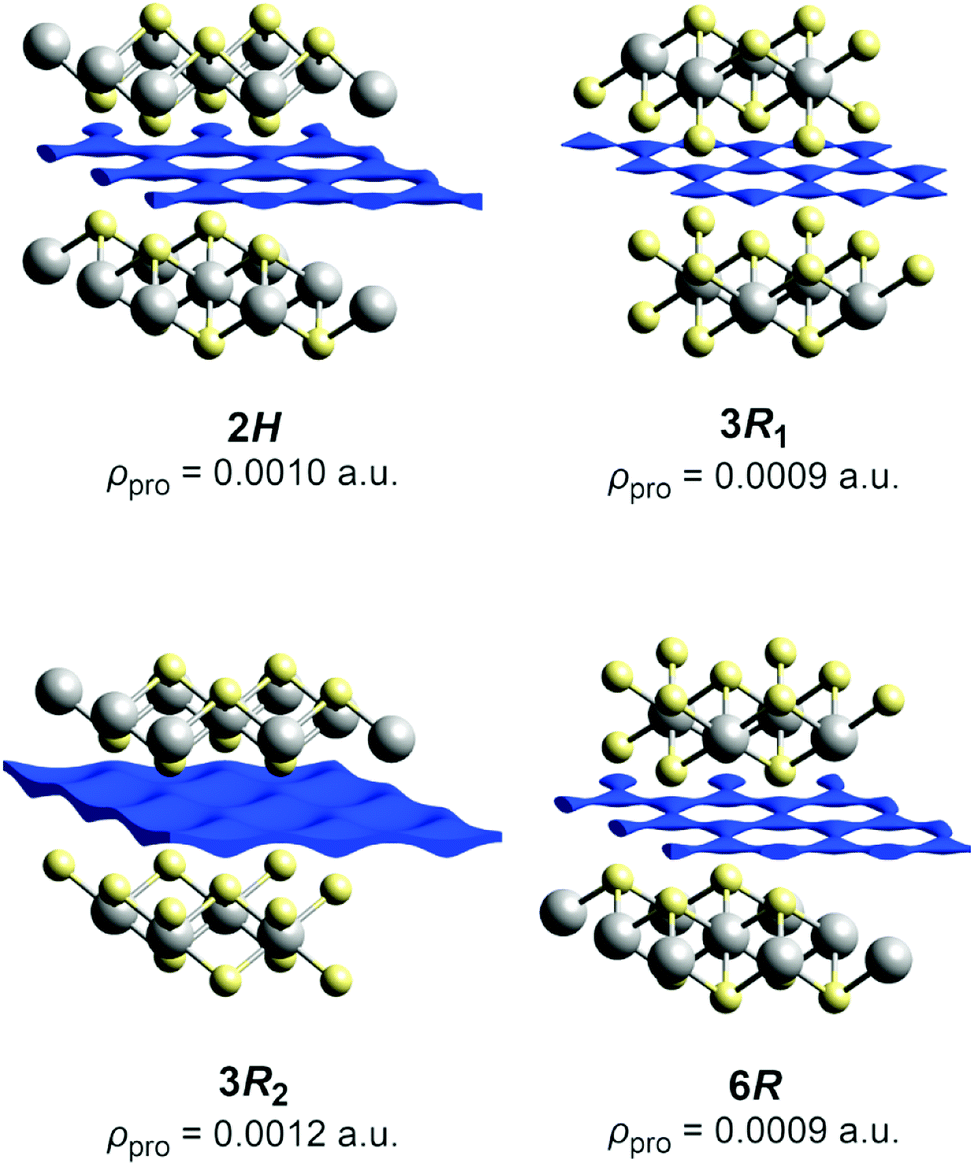 | ||
| Fig. 4 Procrystal void surfaces (blue) in the sodium layers of the TiS2x− frameworks for different polytypes. Atoms are plotted with covalent radii (grey: titanium, yellow: sulphide ions). The depicted isovalue is the minimum for a contiguous surface. | ||
In the case at hand, the sodium ions are displaced along linear pathways between adjacent sodium positions at both relevant temperatures (cf. the similar isosurfaces in Fig. 5). In this way, the honeycomb-like conduction pattern predicted by procrystal void analysis is indeed experimentally reproduced.
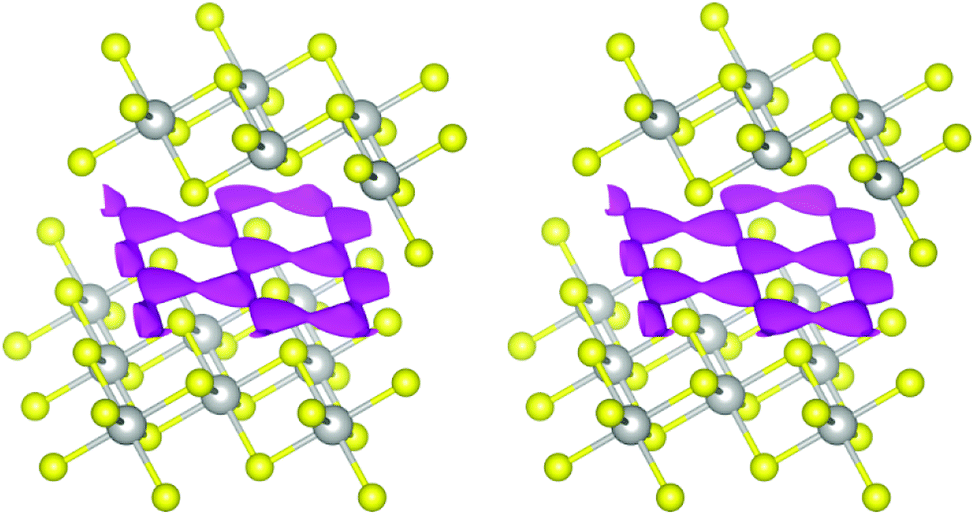 | ||
| Fig. 5 Joint sodium-PDF isosurface of 0.09 Å−3 (pink) in Na0.5TiS2-3R1 at 600 °C (left) and 700 °C (right). Atoms are plotted with covalent radii (grey: titanium, yellow: sulphide ions). | ||
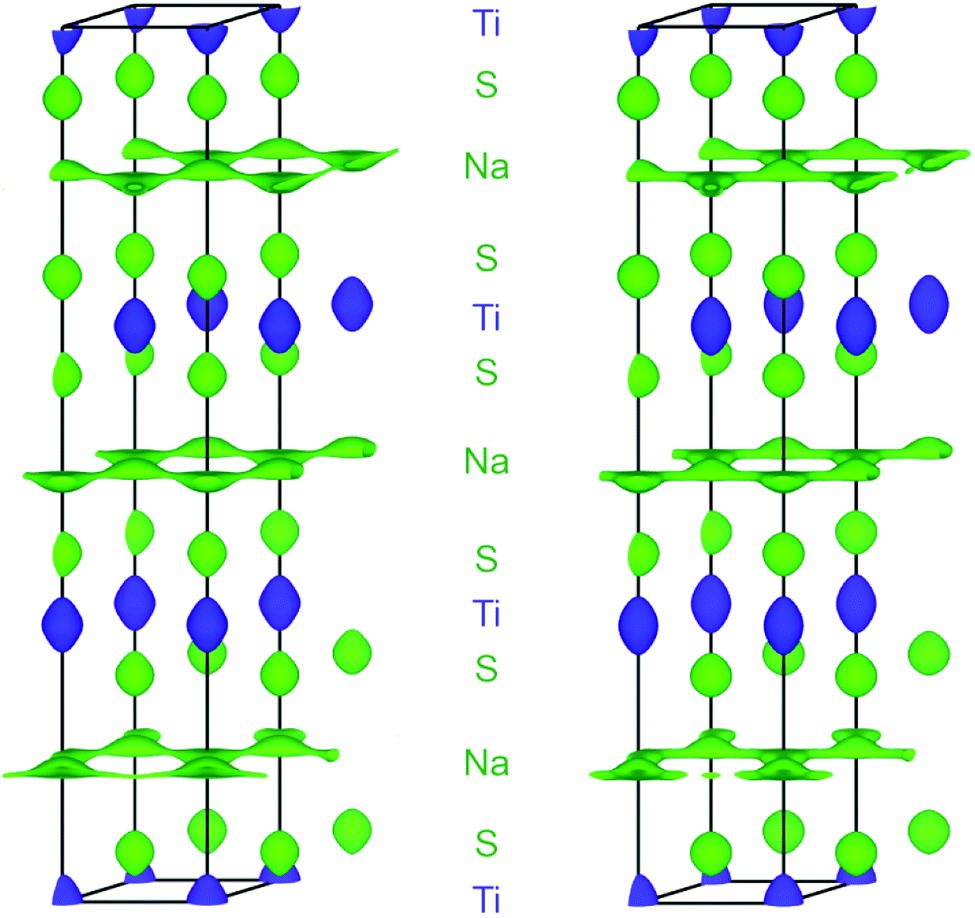 | ||
| Fig. 6 SLD isosurface of ±0.5 fm Å−3 (green: positive, blue: negative) in Na0.5TiS2-3R1 at 600 °C (left) and 700 °C (right) with view roughly along a*. Atomic layers are assigned to elements, unit cells are depicted in black. | ||
In the lighter but more alkali-rich congeners LixTiS2-3R (x = 0.7, 0.9), preferred ion pathways were found between adjacent lithium positions.49 Because of their ordering, however, they correspond to second-neighbours in disordered Na0.5TiS2-3R1.§ As a result, the pathways are not directly comparable. It is noteworthy that, in contrast to LixTiS2-3R, we have not found a significant deviation from coplanarity. We attribute this to the differences in coordination polyhedra (trigonal antiprism vs. prism) and spatial arrangement (interlayer spacing larger by Δd ≈ 0.96 Å, while ionic diameter only larger by 2Δr = 0.52 Å (ref. 50) for the sodium compound).
Migration barriers
For the estimation of the migration energy barrier associated with the found path, we calculated the OPP, which represents the energy landscape experienced by a mobile sodium ion (approximated as an Einstein oscillator that is subject to Boltzmann statistics at the classical limit). The activation energy of the migration (not comprising the defect formation energy) is the OPP at the bottleneck position, i.e., the position of lowest probability along the pathway.The calculation of OPPs from ND-derived PDFs is fairly established,51 but direct derivations from MEM-reconstructed SLDs without any further refinement are scarce.52 The latter is only warranted if an SLD shares basic characteristics of a probability density. Mathematically speaking, a PDF has to be non-negative, Lebesgue-integrable, and normalized to an integral of unity over the whole space. To give a reason for using the direct approach nonetheless, the following points address these issues not in a rigorous but hand-waving way:
• The OPP calculation deals with ratios of input values. Only the non-negative ones are taken into account.
• Numerically sampled SLDs are Lebesgue-integrable.
• For ratios of input values, normalization is unnecessary (the normalizing constant cancels).
Furthermore, the adequacy of the results is ensured by only taking the SLD solely caused by sodium ions (i.e., within the sodium layers) into account.
In agreement with crystal-chemical assumptions, we found the bottleneck of migration at ⅓, ⅙, ⅙—in the middle of a straight line between adjacent sodium positions. The OPPs at this point are summarized in Table 3. Generally, the activation energy does not depend on the temperature as long as only a single migration mechanism is concerned and the structure is stable. For this method, however, this holds only if diffusion is fully activated because the migrating ions themselves are the probe for the potential. This is why slightly lower activation barriers are often found for higher temperatures—as is the case here for the central values—and are thought to be more reliable. Nevertheless, the PDF-derived energies are equal within 1σ. Currently, there is no computational way to estimate the errors on SLD-derived values. Judging by the uncertainties of the PDF-derived barriers, however, the central values are significantly lower, but of the same magnitude. As this is the first study employing both methods, it remains yet unknown if the deviation is systematic. It is, e.g., possible that a restricted set of parameters models parts of the SLD imperfectly (thereby modelling a too low probability density resulting in a too high OPP) or that the SLD-derived OPP carries a considerable error.
| Calculation | At 600 °C | At 700 °C |
|---|---|---|
| From displacement-derived PDF | 0.114(7) | 0.108(8) |
| From MEM-reconstructed SLD | 0.086 | 0.070 |
The topography of the OPP landscape, on the other hand, is similar irrespective of temperature or calculation method (see Fig. 7). As temperature rises, the potential becomes flatter and gives access to a larger area (cf. Fig. 7a/b), but the basic shape is preserved. As the sodium PDF reproduces the features of the SLD (vide supra), the landscapes derived from both resemble each other (cf. Fig. 7c/d). The smaller volume enclosed by the same isosurface, however, shows that the SLD-derived OPP is steeper, therefore stemming from a more localised distribution.
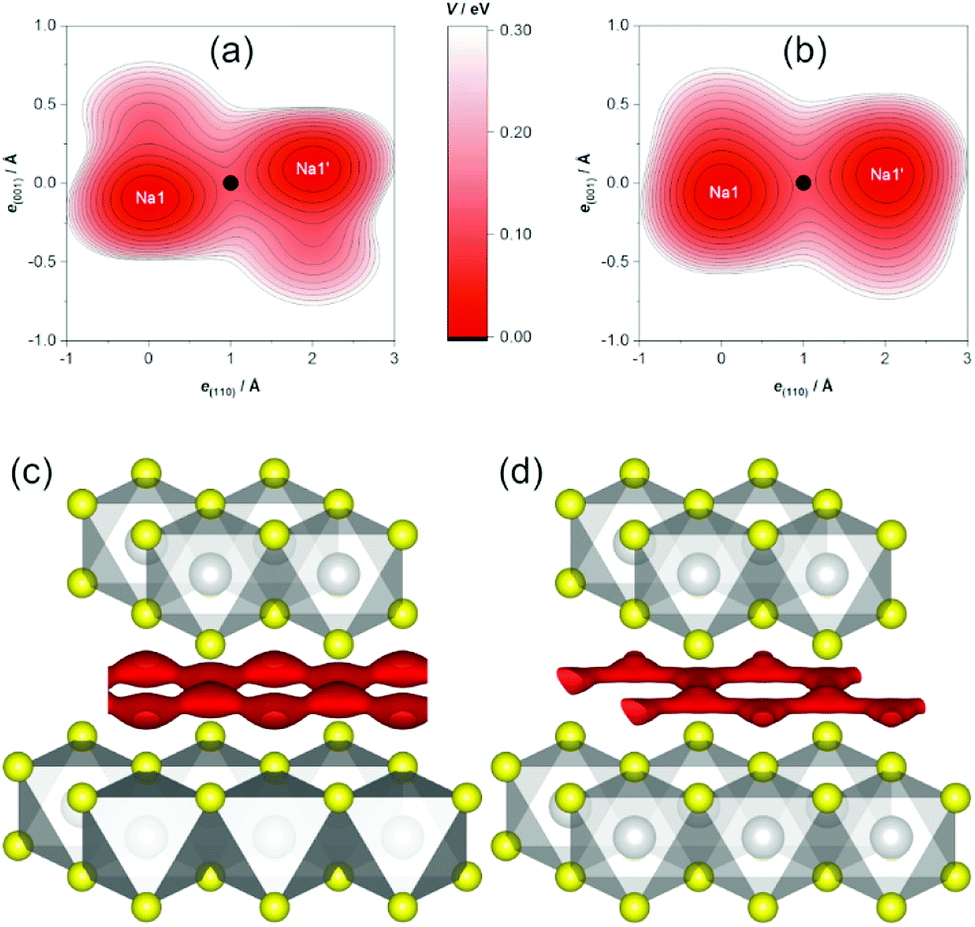 | ||
| Fig. 7 Sodium OPP V in Na0.5TiS2-3R1. Top: Contour plot of PDF-derived OPP within a plane containing two adjacent sodium ions at (a) 600 °C and (b) 700 °C (origin at Na1, bottleneck as black dot, contours: from V0 = 0 on with ΔV = 0.025 eV). Bottom: Isosurface plots of (c) PDF-derived and (d) SLD-derived OPP of V = 0.15 eV at 700 °C (atoms as with covalent radii; grey: titanium, yellow: sulphide ions, red: OPP isosurface). | ||
The experimental barriers are lower than computed for defective NaTiS2-3R2 (0.19 eV)26 and measured for Li0.7TiS2-3R (ca. 0.5 eV).49 Procrystal void analysis already led us to expect that these homeotypic (by alkali occupation) structures be detrimental to alkali-ion conduction (vide supra). In addition, the lower number of vacancies (higher alkali content) in the latter hinders migration.49 For Na0.5TiS2-3R1, low activation barriers corroborate its role as a good sodium-ion conductor. Although a model all-solid-state sodium-ion battery using a NaxTiS2 electrode has been realized,53 problems with reactivity, phase transformations upon de-/intercalation, and mediocre cyclability seem to prohibit its effective use therein.54
Remarks on “Na0.9TiS2-2H”
Our endeavour to explore also the more sodium-rich Na0.9TiS2-2H was hindered by synthesis problems. While XRD for phase identification and ND seemed to indicate a single-phase product of the desired polytype, closer inspection (see Fig. S3†) revealed at least two similar phases, possibly with a more complex (OD) structure. Because of the broader reflections in ND, we found no way of separating the contributions of different phases. A refined (unphysical) single-phase model averaging over all phases was at odds with elemental analysis: The former found only ca. 0.5 sodium ions per formula unit at the crystallographic positions (no significant positive residual SLD) while the latter conformed to the target composition.Furthermore, we found a strong preferred orientation in (001) that agrees well with the plate-like visual appearance of the crystallites in the sample. In a temperature-dependent XRD experiment, we observed the 2H-like structure over the whole stability range. No signs of a transformation into a supposedly stable 3R2-like structure showed above 300 °C, even when heating for a longer time.
Conclusions
We synthesised the intercalate Na0.5TiS2-3R1 from sodium and titanium sulphide in a mildly reducing H2S atmosphere. Our ND and XRD experiments showed that its structure, as reported in the literature, has to be revised: Na0.5TiS2-3R1 assumes the space-group type Rm (not R3m) with disordered (not ordered) sodium ions and one (not two) crystallographically independent sulphide position. Up to ca. 100 °C, the compound reversibly co-intercalates half an equivalent of water. It is stable with respect to phase transformations up to its decomposition temperature.
Sodium-ion diffusion manifests in anharmonic displacement at 600 and 700 °C. In agreement with expectations from topological analysis of the static TiS2x− framework, maps of the MEM-reconstructed SLD and modelled PDFs show a honeycomb-like conduction pattern with linear almost in-plane pathways between adjacent (partly occupied) sodium positions. The bottlenecks of migration are found at the midpoint of said pathways. The associated OPPs at 700 °C indicate low activation barriers of 0.108(8) eV or even 0.070 eV (derived from PDF or SLD, respectively).
Unfortunately, we could not complement these findings with data for Na0.9TiS2-2H. We presume that our synthesis method led to segregation into at least two 2H-like phases—one with lower, one with higher sodium content. Possibly, these are closely related to Na2TiS3 with its OD structure. Bouwmeester et al. had already found such by-phases (re-)forming during high- to mid-temperature syntheses ex elementis with high sodium load.8 The potential of arranging
• pure sodium layers (e.g., in NaTiS2), their defective (e.g., in NaxTiS2 with x < 1), titanium-mixed (e.g., in Na2TiS3), or even as-well-as variants (e.g., in NaxTi1+δS2 with x < 1 − 3δ)
• with their sodium, titanium, and vacancy positions ordered or unordered within one layer
• in an ordered or unordered stacking sequence
accounts for compelling structural complexity in layered sodium titanium sulphides. This is particularly pronounced at the fringes of very high or very low alkali content and makes further exploration, especially of the interplay between sodium mobility and structural disorder, worthwhile.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the Deutsche Forschungsgemeinschaft (SPP 1613, LE 781/13-2) is gratefully acknowledged. The article processing charge was covered by FILL2030, a European Union project within the European Commission's Research and Innovation programme Horizon 2020 under grant agreement no. 731096. We thank Dr Stefan Berendts for XRF analyses and temperature-dependent XRD, Dr Suliman Nakhal for the synthesis of TiS2-1T, Ms Juana Krone for sulphur and hydrogen determination, and Ms Iris Pieper for sodium determination.Notes and references
- J.-Y. Hwang, S.-T. Myung and Y.-K. Sun, Chem. Soc. Rev., 2017, 46, 3529–3614 RSC
.
- H. Imai, Y. Shimakawa, T. Mako and Y. Kubo, JP Pat., 2002-270907, 2002
- R. Zhang, F. Mizuno, C. Ling, M. S. Whittingham, R. Zhang and Z. Chen, US Pat., 20160365577, 2016
- A. Clearfield, US Pat., 3148998, 1960
- W. Rüdorff, Chimia, 1965, 19, 489–499 Search PubMed
- J. Rouxel, M. Danot and J. Bichon, Bull. Soc. Chim. Fr., 1971, 3930–3935 CAS
- R. J. Haange, A. J. A. Bos-Alberink and G. A. Wiegers, Ann. Chim. Sci. Mater., 1978, 3, 201–207 CAS
- H. J. M. Bouwmeester, E. J. P. Dekker, K. D. Bronsema, R. J. Haange and G. A. Wiegers, Rev. Chim. Miner., 1982, 19, 333–342 CAS
- J. Lima-de-Faria, E. Hellner, F. Liebau, E. Makovicky and E. Parthé, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 1–11 CrossRef
- C. Delmas, C. Fouassier and P. Hagenmuller, Physica B+C, 1980, 99, 81–85 CrossRef
- P. Molinie, L. Trichet, J. Rouxel, C. Berthier, Y. Chabre and P. Segransan, J. Phys. Chem. Solids, 1984, 45, 105–112 CAS
- K. Cenzual, L. M. Gelato, M. Penzo and E. Parthé, Acta Crystallogr., Sect. B: Struct. Sci., 1991, 47, 433–439 CrossRef
- R. Hinek, personal communication.
- D. A. Winn, J. M. Shemilt and B. C. H. Steele, Mater. Res. Bull., 1976, 11, 559–566 CrossRef CAS
- P. Colombet, M. Danot, J.-C. Jumas and É. Philippot, C. R. Séances Acad. Sci., Sér. C, 1978, 287, 411–414 CAS
- W. Mark, O. Lindqvist, J.-C. Jumas and É. Philippot, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1974, 30, 2620–2628 Search PubMed
- M. Zanini, J. L. Shaw and G. J. Tennenhouse, J. Electrochem. Soc., 1981, 128, 1647–1650 CrossRef CAS
- K. O. Klepp, Z. Kristallogr. Suppl., 1995, 9, 188 Search PubMed
- T. Hibma, Physica B+C, 1980, 99, 136–140 CAS
- T. Hibma, J. Solid State Chem., 1980, 34, 97–106 CrossRef CAS
- Q.-g. Song, X.-z. Cong, Q.-j. Zhang, W.-l. Mo and Z.-h. Dai, Wuli Xuebao, 2000, 49, 2011–2016 CAS
- J. Vinckevičiūtė, M. D. Radin and A. Van der Ven, Chem. Mater., 2016, 28, 8640–8650 Search PubMed
- K. M. Abraham, Solid State Ionics, 1982, 7, 199–212 CrossRef CAS
- W. B. Johnson and W. L. Worrell, Synth. Met., 1982, 4, 225–248 CAS
- B. A. Marmaduke and L. F. Donaghey, Proc. Intersoc. Energy Convers. Eng. Conf., 1976, 1, 467–470 Search PubMed
- X. Zhang, Z. Zhang, S. Yao, A. Chen, X. Zhao and Z. Zhou, npj Comput. Mater., 2018, 4, 13 CrossRef
- S. N. Li, J. B. Liu and B. X. Liu, J. Power Sources, 2016, 320, 322–331 CrossRef CAS
- B. G. Silbernagel and M. S. Whittingham, Mater. Res. Bull., 1976, 11, 29–36 CrossRef CAS
- A. W. Hewat, Mater. Sci. Forum, 1986, 9, 69–80 CAS
- E. Suard and A. Hewat, Neutron News, 2001, 12, 30–33 CrossRef
- V. Petříček, M. Dušek and L. Palatinus, Z. Kristallogr. - Cryst. Mater., 2014, 229, 345–352 Search PubMed
- C. J. Howard, J. Appl. Crystallogr., 1982, 15, 615–620 CrossRef CAS
- G. Bergerhoff and I. D. Brown, in Crystallographic Databases, ed. F. H. Allen, G. Bergerhoff and R. Sievers, International Union of Crystallography, Chester, U.K., 1987, ch. 2.2, pp. 77–95 Search PubMed
- K. Brandenburg, Diamond 4.5, Crystal Impact – H. Putz & K. Brandenburg GbR, Bonn, Germany, 2018 Search PubMed
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS
- M. Lerch, E. Suard and D. Wiedemann, Pathways of Sodium-Ion Diffusion in NaxTiS2 (x = 0.5, 0.9), 2017, accessed June 2019, DOI:10.5291/ill-data.5-21-1106.
- M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, P. R. Spackman, D. Jayatilaka and M. A. Spackman, CrystalExplorer 17.5, University of Western Australia, Perth, Australia, 2017 Search PubMed
- K. Momma, T. Ikeda, A. A. Belik and F. Izumi, Powder Diffr., 2013, 28, 184–193 CrossRef CAS
- J. Nocedal, Math. Comput., 1980, 35, 773–782 CrossRef
- D. Wiedemann, CalcOPP 2.0.1, Technische Universität Berlin, Berlin, Germany, 2019, DOI:10.5281/zenodo.2530345
- V. E. Kaloidas and N. G. Papayannakos, Int. J. Hydrogen Energy, 1987, 12, 403–409 CrossRef CAS
- R. Schöllhorn and A. Weiss, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1973, 28, 711–715 Search PubMed
- S. N. Patel and A. A. Balchin, J. Mater. Sci., 1985, 20, 917–921 CrossRef CAS
- K. D. Bronsema and G. A. Wiegers, Mater. Res. Bull., 1987, 22, 1073–1080 CrossRef CAS
- D. Wiedemann, S. Nakhal, A. Senyshyn, T. Bredow and M. Lerch, Z. Phys. Chem., 2015, 229, 1275–1288 CAS
- J. Rouxel, in Intercalated Layered Materials, ed. F. Lévy, D. Reidel Publishing, Dordrecht, Netherlands, 1979, pp. 201–250 Search PubMed
- D. Wiedemann, M. M. Islam, T. Bredow and M. Lerch, Z. Phys. Chem., 2017, 231, 1279–1302 CAS
- M. J. Turner, J. J. McKinnon, D. Jayatilaka and M. A. Spackman, CrystEngComm, 2011, 13, 1804–1813 RSC
- D. Wiedemann, M. M. Islam, S. Nakhal, A. Senyshyn, T. Bredow and M. Lerch, J. Phys. Chem. C, 2015, 119, 11370–11381 CrossRef CAS
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef
- H. Boysen, Z. Kristallogr., 2003, 218, 123–131 CAS
- M. Monchak, T. Hupfer, A. Senyshyn, H. Boysen, D. Chernyshov, T. Hansen, K. G. Schell, E. C. Bucharsky, M. J. Hoffmann and H. Ehrenberg, Inorg. Chem., 2016, 55, 2941–2945 CAS
- A. Hayashi, K. Noi, A. Sakuda and M. Tatsumisago, Nat. Commun., 2012, 3, 856 CrossRef PubMed
- H.-S. Ryu, J.-S. Kim, J.-S. Park, J.-W. Park, K.-W. Kim, J.-H. Ahn, T.-H. Nam, G. Wang and H.-J. Ahn, J. Electrochem. Soc., 2013, 160, A338–A343 CAS
Footnotes |
| † Electronic supplementary information (ESI) available: Details on X-ray diffraction, neutron diffraction, and corrections of the ICSD. See DOI: 10.1039/c9ra05690d |
| ‡ The previous assignment of the structure type “CuCrSe2–AgCrSe2(R3m)” to Na0.55TiS2-3R1 complicated a comparison to other structures: several isopointal but alloconfigurational structures were assigned this ill-defined type. It was split correctly in the course of this work (see Table S2†), the result being incorporated in the ICSD since May 2019.13 |
| § The compounds LixTiS2-3R are isotypic to the high-alkali polytype NaxTiS2-3R2, not NaxTiS2-3R1. |
| ¶ Caveat: There had been a transcription error in the former ICSD record that was corrected in 2018. |
| This journal is © The Royal Society of Chemistry 2019 |