
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of solution pH on degradation of atrazine during UV and UV/H2O2 oxidation: kinetics, mechanism, and degradation pathways†
Yucan Liu*a,
Kai Zhu *b,
Miaomiao Sua,
Huayu Zhuc,
Jianbo Lua,
Yuxia Wangd,
Jinkun Donga,
Hao Qina,
Ying Wanga and
Yan Zhanga
*b,
Miaomiao Sua,
Huayu Zhuc,
Jianbo Lua,
Yuxia Wangd,
Jinkun Donga,
Hao Qina,
Ying Wanga and
Yan Zhanga
aSchool of Civil Engineering, Yantai University, Yantai 264005, China. E-mail: liuyucan@ytu.edu.cn; Tel: +86 0535 6902606
bCollege of Resources and Environment, Linyi University, Linyi 276000, China. E-mail: zhukai@lyu.edu.cn
cSchool of Chemistry & Chemical Engineering, Linyi University, Linyi 276000, China
dSchool of Environmental and Municipal Engineering, North China University of Water Resources and Electric Power, Zhengzhou, 450046, China
First published on 4th November 2019
Abstract
The kinetics, degradation mechanism and degradation pathways of atrazine (ATZ) during sole-UV and UV/H2O2 processes under various pH conditions were investigated; the effects of UV irradiation time and H2O2 dose were also evaluated. A higher reaction rate was observed under neutral pH conditions in the UV only process. For the UV/H2O2 process, a higher reaction rate was observed in acidic solution and the degradation rate of ATZ firstly increased with the increase of concentration of H2O2 and then decreased when H2O2 concentration exceeded 5 mg L−1. In addition, qualitative and quantitative analyses of oxidation intermediates of ATZ in aqueous solution during the sole-UV and UV/H2O2 processes were conducted using UPLC-ESI-MS/MS. Ten kinds of dechlorinated intermediates were detected during sole-UV treatment under all five pH conditions. In contrast, the speciation of intermediates in the UV/H2O2 process varied dramatically with solution pH. Based on the analysis of ATZ oxidation intermediates, ATZ degradation pathways under different pH conditions were proposed for the sole-UV and UV/H2O2 processes. The results showed that the main degradation reactions of ATZ included dechlorination-hydroxylation, dechlorination-dealkylation, de-alkylation, deamination-hydroxylation, alkylic-oxidation of lateral chains, dehydrogenation-olefination, dechlorination-hydrogenation, dechlorination-methoxylation and dehydroxylation.
1. Introduction
Atrazine(2-chloro-4-(ethylamino)-6-(isopropylamino)-s-triazine, ATZ), the main representative of the chlorinated triazines, has been widely used in agricultural and forestry fields.1,2 Due to its high solubility, low adsorbance on soil particles and low biodegradability in water, ATZ could migrate easily to underground water via infiltration or to surface water by runoff.3,4 Therefore, ATZ is frequently detected in surface and ground waters. As a possible carcinogen and endocrine disrupting chemical (EDC), numerous studies have reported the negative impact of ATZ on animals and humans.5–7 Its chlorinated metabolites were also reportedly EDCs. The occurrence of ATZ in underground and surface waters has attracted growing attention from the public and scientific agencies, moreover, ATZ is classified as a hazardous substance of the Water Framework Directive in Europe's priority list.8 In order to ensure drinking water quality, European countries have adopted a maximum contaminant level (MCL) of 1.0 μg L−1 for ATZ, and the United States Environmental Protection Agency (US EPA) set a drinking water limit of 3 μg L−1.9Due to the inefficiency of conventional water/wastewater treatment processes in removing ATZ,10,11 comprehensive efforts have been made to develop effective physical, chemical and biological methods, such as adsorption,12,13 membrane separation14,15 and oxidation processes.16–19
The most popular and environmentally friendly oxidation process is photo-oxidation, and numerous studies have reported the photo-degradation of ATZ in water and wastewaters.20 Dechlorination reaction during photo-oxidation process could reduce the negative impact of ATZ and its degradation products since the toxicity of ATZ is related to the chlorine group.21,22 It is well known that the chlorine in ATZ can be replaced by hydroxyl group (–OH) under sole-ultraviolet (sole-UV) light irradiation or combined processes with oxidants or catalysts during photocatalytic oxidation process.23–25 The typical degradation pathway of ATZ during photo-oxidation process is the cleavage of C–Cl bond, giving rise to dechloro-hydrogenated products.23 In addition, extensive attention has been devoted to the photocatalytic oxidation processes with oxidants, such as O3, H2O2 and persulfate (S2O82−), in recent years.26 These composite processes exhibits excellent oxidation performance of ATZ in water due to the generation of highly reactive species.27
The removal efficiency of ATZ during the photo-oxidation process depends on experimental parameters, such as the type of UV light source, the composition of the solution, and the speciation and concentrations of oxidants or catalysts.28 However, even under optimal conditions, the complete mineralization of ATZ in water could rarely occurred during photo-oxidation process because of the special chemical properties of the s-triazine ring.29 Hence, the photo-oxidation reaction of ATZ is a complicated process including chloro-dealkylation, dechlorination, dechloro-dealkylation, alkylic-oxidation, delamination-hydroxylation and olefination, etc.30 Torrents et al. explored the ATZ degradation by direct and nitrate-mediated hydroxy radical photo-oxidation processes, irradiation of ATZ solution in the presence of nitrate leading to 23% ATZ conversion, generated 14% of 2-hydroxy-4-acetamido-6-ethylamino-s-triazine (OIET) and cyanuric acid, 9% of chloro-dealkylated and chloro-alkyloxidized products.31 In addition, Ta et al. proposed four plausible oxidation pathways of ATZ in microwave-assisted photolytic process, including dealkylation, dechlorination-hydroxylation, alkylic-oxidation and delamination-hydroxylation.32
Previous studies showed that organic pollutants in different dissociation forms have unique physicochemical properties and toxicity that depend on the composition of the aquatic medium.33,34 The variation of photolytic rate with solution pH is an elusive phenomenon, since the specific photochemical reactions of different organics that may exist in the aqueous solution at a given pH value.35 Although numerous studies focus on the degradation kinetics and mechanism of ATZ have been carried out, few papers systematically reported the effect of solution pH on degradation products of ATZ and its degradation pathways during photo-oxidation process. In the present paper, degradation kinetics of ATZ under different solution pH and H2O2 dose are elucidate; photo-degradation intermediates of ATZ are analysed qualitatively and quantitatively in order to propose the possible photo-degradation pathways of ATZ under different solution pH conditions.
2. Materials and methods
2.1 Materials
ATZ (purity > 98%) was supplied by Sigma-Aldrich Corporation (Bellefonte, PA, USA). Methanol (HPLC grade) and acetonitrile (HPLC grade) was obtained from Merck Chemicals (Darmstadt, Germany). Hydrogen peroxide (30%, w/w) and sulfuric acid (98%) were obtained from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). Sodium dihydrogen phosphate, disodium hydrogen phosphate, sodium hydroxide and sodium tetraborate pentahydrate were purchased from Xi'an Chemicals Ltd (Xi'an, China). ATZ solutions were prepared with ultrapure water from an Elga Purelab Ultra Analytic system (Bucks, UK) with a resistance of 18.2 MΩ cm.2.2 Photocatalytic oxidation experiments
ATZ oxidation experiments were conducted in an annular photochemical reactor (V = 300 mL) with a low pressure mercury UV (LPUV) lamp (monochromatic emission at 253.7 nm (emission spectrum of the lamp shown in Fig. S1†), Tepro Co., Ltd., Guangdong, China) positioned inside a quartz glass well on the axis (schematic diagram shown in Fig. S2†). The average UV fluence rate was 0.58 mW cm−2 calculated according to previous study.36 The photon flux into the solution from the LPUV lamp was set at 1.18 × 10−7 einstein per s detected with iodide–iodate chemical actinometer.37 During oxidation process, working solution, containing 5 mg L−1 ATZ, was under vigorous magnetic stirring at thermostatically controlled temperature 20 (±0.5) °C. Prior to UV irradiation, the LPUV lamp was ignited for 30 min to obtain a stable output.The concentration of H2O2 solution was 5, 15, 30 and 50 mg L−1 in UV/H2O2 process, respectively. Solution pH value was buffered to 4.0, 5.5, 7.0, 8.5 and 10.0 using 2 mmol L−1 phosphate and/or borate buffers, adjusted by small amounts of 1 mol L−1 sulfuric acid or sodium hydroxide solution. Ascorbic acid was added into samples to quench the H2O2 before analysis. Each experiment was performed in triplicate, and the error bars in figures are represented as the standard error of the mean.
2.3 Analytical methods and intermediates identification
The concentration of H2O2 was determined by the I3− method described by Klassen et al.38 UV absorbance and absorption spectra of ATZ and its products in aqueous solutions were measured using a UV-vis spectrophotometer (DR5000, HACH, Loveland, CO, USA).The qualitative and quantitative analyses of ATZ and its intermediates or by-products were carried out using ultra-performance liquid chromatography-electrospray ionization mode-triple quadrupole mass spectrometry (UPLC-ESI-MS/MS) coupled with an ACQUITY™ UPLC BEH C8 column (2.1 mm × 100 mm × 1.7 μm) (Waters, USA). A detailed description of the instrument and its operation parameters is presented in ESI (see Text S1).†
The degradation rate of ATZ was calculated from eqn (1):
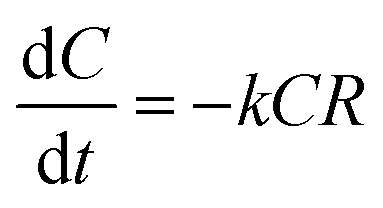 | (1) |
The semi-log graphs of the ATZ concentration versus UV irradiation time were calculated from eqn (2):
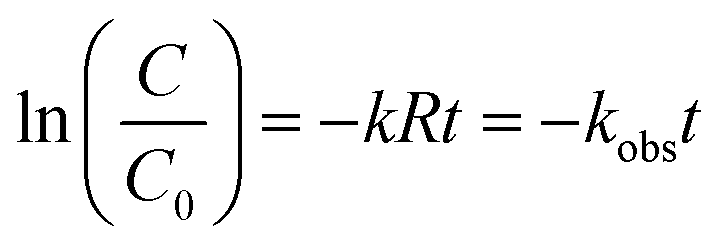 | (2) |
3. Results and discussion
3.1 Oxidation kinetic, intermediates and possible pathway in sole-UV process
Direct photolysis of water molecule, generated ·OH and hydrated electron, can only be achieved at wavelengths lower than 190 nm.39 Therefore, the degradation of ATZ by sole-UV was probably due to the absorption of UV photons with subsequent direct photolysis.40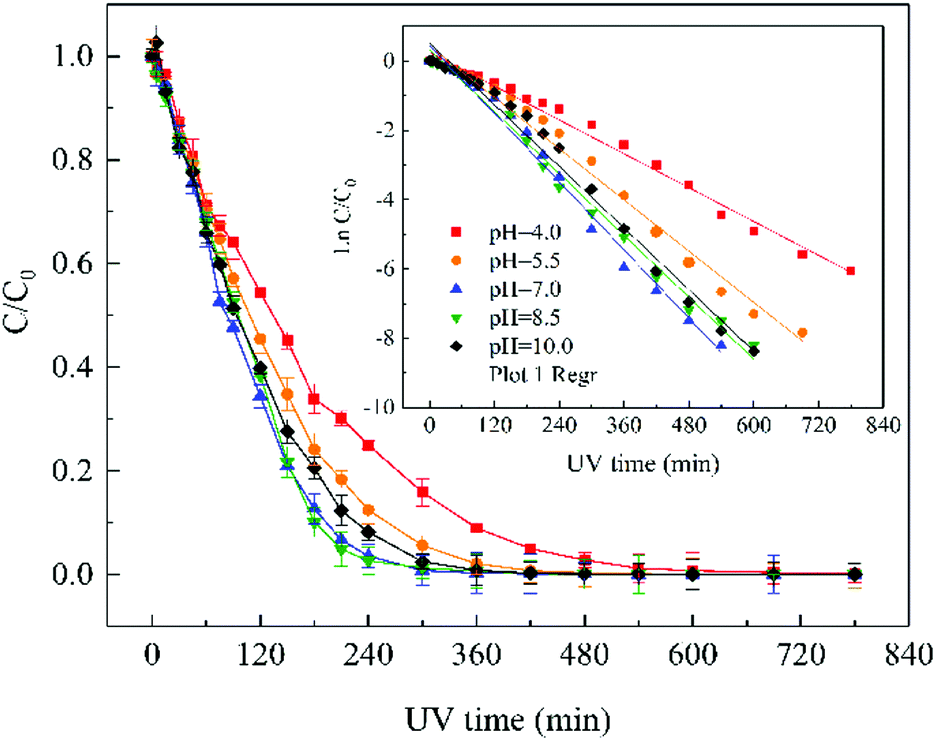 | ||
| Fig. 1 Effect of solution pH on ATZ removal efficiency. Raw ATZ solution: 5 mg L−1. | ||
The semi-log graph of ATZ concentration versus irradiation time under different solution pH conditions presented straight lines (inset of Fig. 1), indicating that the degradation of ATZ during sole-UV process followed pseudo-first-order reaction kinetic.30 Table S1† summarizes the observed reaction rate constant kobs and the linear correlation coefficient (r2) under different pH conditions. The kobs value was 0.0076, 0.0113, 0.0150, 0.0140 and 0.0134 min−1 with initial pH values as 4.0, 5.5, 7.0, 8.5 and 10.0, respectively. The kobs value at initial pH of 7.0 is 0.0.0150 min−1, which was approximately 2 times of that at initial pH of 4.0. Apparently, higher reaction rate was observed under neutral pH condition in sole-UV process.
The abovementioned tendency can be attributed to the protonated species in acidic solutions. Many studies have proven that the distribution of diazinon species may influence the UV light absorbance and photolytic properties. The monochromatic absorption coefficients at 254 nm of diazinon solution at acidic conditions was found to be significantly higher than those at alkaline conditions.41 Our previous research also found that the ratio of protonated diazinon in aqueous solution is affected by the pH value of solution.42
The distribution of protonated and deprotonated ATZ species as a function of solution pH was shown in Fig. S3.† The difference of molar absorption coefficient (ε) and quantum yield (Φ) of ATZ solution at different pH may be the main reason for the difference of ATZ degradation rate. When pH < pKa, the protonated species of ATZ is the dominant species, while the deprotonated species is the dominant when pH > pKa. Therefore, the different dissociation forms of ATZ under different pH conditions lead to the different degradation rates of ATZ.
MS and MS/MS spectra analyses at ESI+ and ESI− mode were conducted in order to analyse the molecular structure of these intermediates. Retention time (RT) and MS spectral in formation in full scan modes of the above 11 chromatographic peaks are shown in Table S2,† and the m/z of these intermediates were used to deduce molecular weight (MW). Moreover, to obtain further information on the structure of the intermediates of ATZ, collision induced dissociation (CID) experiments were also conducted. The collision energy for each intermediate was optimized in the range from 10 to 30 eV. Argon was used as collision gas and its flow rate was 0.12 mL min−1. The precursor ions of these intermediates in daughter scan mode are shown in Table 1, and the detailed information on the fragments in MS/MS spectra for each of these precursor ions are shown in Fig. S5–S14.†
| Name | ESI model | MS fragment ions (m/z) | Precursor ions (m/z) | MW (Da) | Formula | Proposed structure | Chemical name |
|---|---|---|---|---|---|---|---|
| P1 | ESI+ | 112, 85, 70, 68 | 154 | 153 | C6H11N5 | 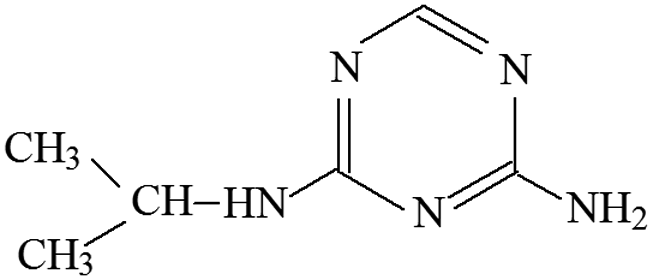 |
4-Isopropylamino-6-amino-s-triazine |
| P2 | ESI+ | 156, 142, 114, 97, 71, 69 | 184 | 183 | C7H13N5O | 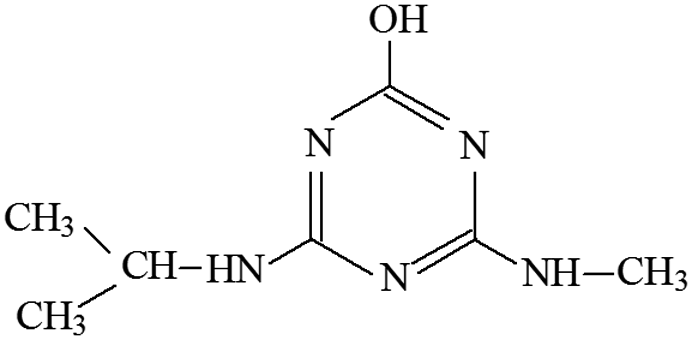 |
2-Methoxy-4-methylamino-6-isopropylamino-s-triazine |
| P3 | ESI+ | 145, 97, 89, 71, 65 | 196 | 196 | C8H14N5O | 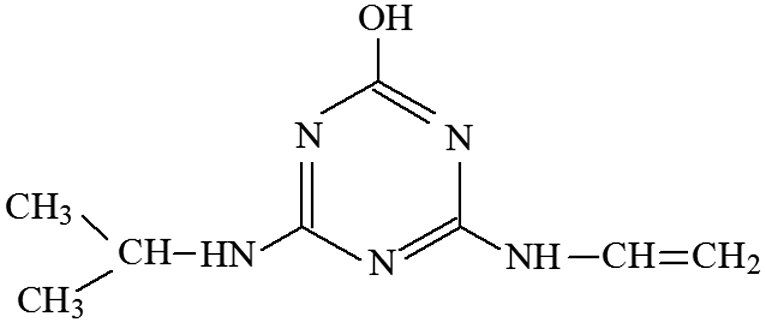 |
2-Hydroxy-4-isopropylamino-6-vinylamino-s-triazine |
| P4 | ESI+ | 156, 128, 114, 97, 86, 69 | 198 | 197 | C8H15N5O | 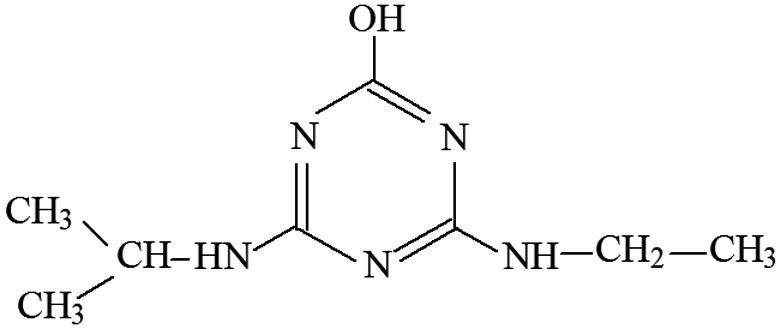 |
2-Hydroxy-4-ethylamino-6-isopropylamines-s-triazine |
| ESI− | 168, 154, 125, 111, 83, 69 | 196 | |||||
| P5 | ESI+ | 156, 139, 113, 96, 85, 71 | 198 | 197 | C7H11N5O2 | 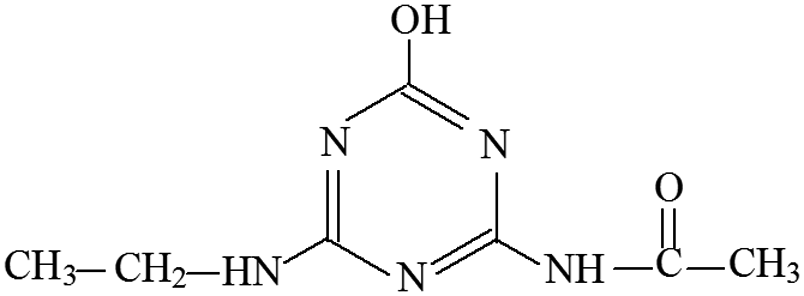 |
2-Hydroxy-4-acetamido-6-ethylamino-s-triazine |
| ESI− | 137, 111, 83, 69 | 196 | |||||
| P6 | ESI+ | 156, 153, 127, 113, 85, 71 | 198 | 197 | C7H11N5O2 | 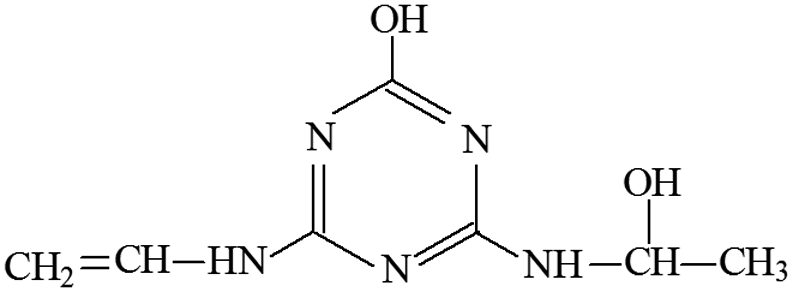 |
2-Hydroxy-4-(2-hydroxy-ethylamino)-6-vinylamino-s-triazine |
| ESI− | 151, 125, 111, 83 | 196 | |||||
| P7 | ESI+ | 170, 128, 86, 68 | 212 | 211 | C8H13N5O2 | 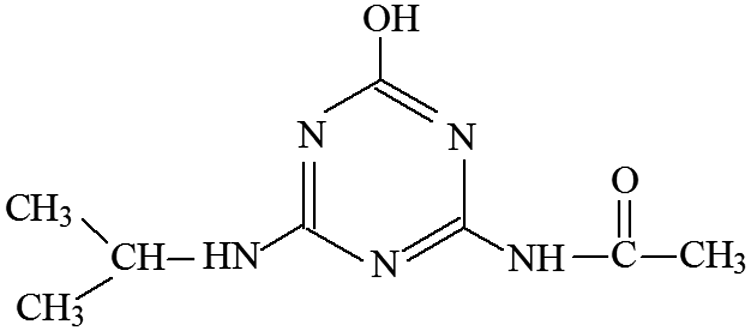 |
2-Hydroxy-4-acetamido-6-isopropylamino-s-triazine |
| P8 | ESI+ | 182, 170, 142, 128, 114, 97 | 212 | 211 | C9H17N5O | 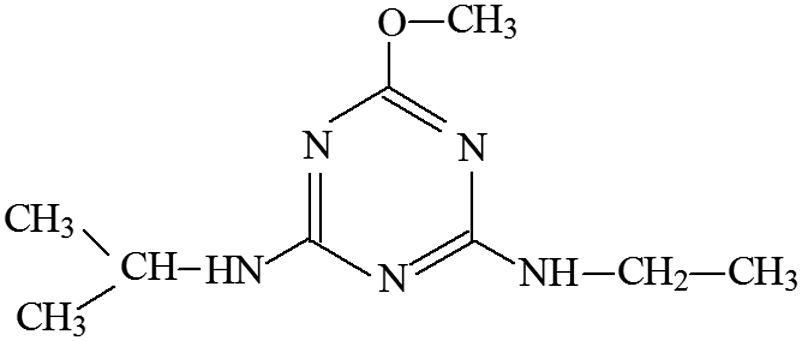 |
2-Methoxy-4-isopropylamino-6-ethylamino-s-triazine |
| P9 | ESI+ | 174, 146, 96, 71, 68 | 216 | 215.7 | C8H14N5Cl | 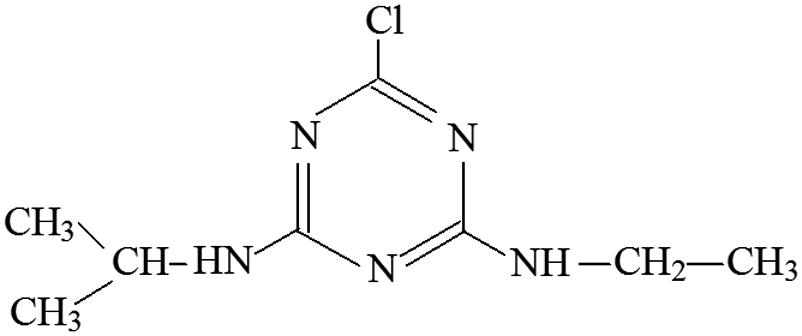 |
2-Chloro-4-ethylamino-6-isopropylamino-s-triazine |
| P10 | ESI+ | 81, 72 | 139 | 138 | C5H6N4O | 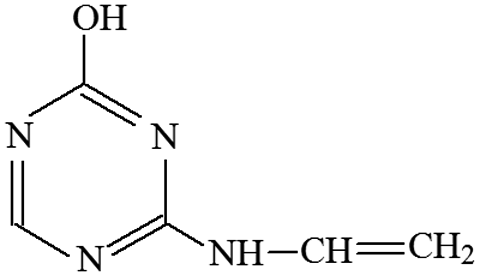 |
2-Hydroxy-4-vinylamino-s-triazine |
| P11 | ESI+ | 198, 220 | 198 | 197 | C8H15N5O | 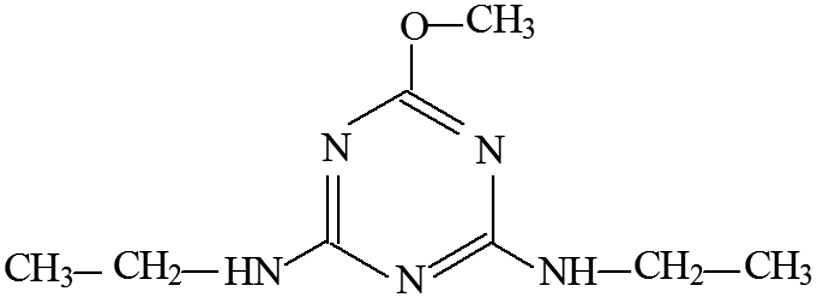 |
2-Methoxy-4,6-diethylamino-s-triazine |
| ESI− | 196 | 196 |
Molecular structures of all these intermediates were derived based on MS spectra and MS/MS spectra.
P1 was only detected in ESI+ mode, and m/z 154 was the main fragments in MS spectra. The positive ion was produced by the addition of H+ to the P1 molecule, inferring the MW of P1 is 153 Da. Moreover, fragment ions of m/z 112, 85, 70 and 68 in the MS/MS spectra were detected (Fig. S5†). The fragment ion of m/z 112 reduced 42 than m/z 154, losing –CH(CH3)2 on the precursor ion. The m/z 85 ion reduced 27 than m/z 112, indicating the s-triazine ring of the fragment was opened by CID. The fragment ion of m/z 85 might generate by losing one carbon atom and one –NH2 connected to s-triazine ring (see Fig. S5†). The fragment ion of m/z 85 loss –NH2, generating the m/z 70 ion. Fragment ions suggested that the molecular structure of P1 included –NHCH(CH3)2 and –NH2 attached to the s-triazine ring, as presented in Table 1.
P2 was only detected in ESI+ mode, and m/z 184 was the main fragments in MS spectra with MW of 183 Da. Fragment ions of m/z 156 (loss of two –CH3) and 142 (loss of –CH(CH3)2), and 97 (loss of –CH(CH3)2, –NHCH3 and –OH) in the MS/MS spectra (Fig. S6†) suggesting that the molecular structure of P2 included –OH, –NHCH3 and –NHCH(CH3)2 attached to the s-triazine ring (Table 1). The possible structures of m/z 114, 71 and 69 are shown in Fig. S2.† Based on above information, molecular structure of P2 was proposed, presented in Table 1 and Fig. S6.†
P3 was only detected in ESI+ mode, and m/z 196 was the main fragments in MS spectra with MW of 195 Da. Based on the mass spectra and chromatographic of P3 and results of previous studies,23,39 the possible molecular structure of P3 was deduced (Table 1 and Fig. S7†).
P4 generated molecular ion m/z 198 in ESI+ mode and m/z 196 in ESI− mode with MW of 197 Da. In addition, m/z 220 (M + Na), 395 (2M+) and 417 (2M + Na) in ESI+ full scan mode and 393 (2M−) in ESI− full scan mode was also detected. The fragments of m/z 198 (ESI+) and 196 (ESI−) were selected as the precursor ions to identify molecule structure of P4 by CID in the daughter scan mode. The fragment ions of m/z 156 (loss –CH(CH3)2), 128 (loss –CH(CH3)2 and –CH2CH3), 114 (same as m/z 114 of P2), 97 (loss –NHCH(CH3)2, –CH2CH3 and –OH) and 86 (the s-triazine ring was addition in) was detected in the MS/MS spectra (ESI+) (Fig. S8†). The fragment ions of m/z 168 (loss two –CH3), 154 (loss –CH(CH3)2), 125 (lost –NHCH(CH3)2 and –CH3), 111 (loss –NHCH(CH3)2 and –CH2CH3) and 83 was detected in the MS/MS spectra (ESI−) (Fig. S8†). Based on above information, molecular structure of P4 was proposed (Table 1).
P5 produced molecular ion m/z 198 in ESI+ mode and m/z 196 in ESI− mode with MW of 197 Da. The fragments of m/z 198 (ESI+) and 196 (ESI−) were selected as the precursor ions to identify its molecule structure by CID, the MS/MS spectra of P5 was obtained (Fig. S9†). The main fragment ions of m/z 156 (loss –COCH3), 139 (loss –NHCOCH3) and 113 (loss m/z –NHCOCH3 and –CH2CH3) were detected in the MS/MS spectra (ESI+), the structures of m/z 96, 85 and 71 were deduced based on its MS/MS spectra. Meanwhile, m/z 137, 111, 83 and 69 were detected in the MS/MS spectra (ESI−). The structure information of each fragment is shown in Fig. S9.† The possible molecular structure of P5 is shown in Table 1.
The MS spectra of P6 in the ESI+ and ESI− mode are shown in Table S2.† The fragments of m/z 198 (M+), 220 (M + Na), 395 (2M+), 417 (2M + Na) and 127 in ESI+ full scan mode and the m/z 196 (M−) and 125 in ESI− full scan mode were detected. Therefore, the MW of P6 was deduced to be 197 Da. The fragment ions of m/z 198 (ESI+) and 196 (ESI−) were selected as the precursor ions to identify its molecule structure by CID, and the MS/MS spectra of P6 was also obtained (Fig. S10†). In ESI+ mode, m/z 156 (loss –CHCH2 and –OH), 153 (loss –NHCHOHCH3 with –CH3 restructured at this position), 127 (loss –NHCHCH2, –CH3 and –OH), 113 (loss –NHCHCH2 and –CHOHCH3) and 85 was detected in the MS/MS spectra of P6. At the same time, in ESI− mode, m/z 151, 125, 111 and 85 was also detected. The structures information of each fragment ions are shown in Fig. S10,† and the possible molecular structure of P6 is shown in Table 1.
P7 generated molecular ion m/z 212 in ESI+ mode and m/z 210 in ESI− mode with MW of 211 Da. The fragment ions of m/z 212 (ESI+) and 210 (ESI−) were selected as the precursor ions to identify its product ions by CID. In ESI+ mode, the fragment ions of m/z 170 (loss –CH(CH3)2), 128 (loss –CH(CH3)2 and –COCH3), 86 and 68 (Fig. S11†) was detected. However, the MS/MS spectra of P7 in ESI− mode was not obtained. Based on above information, the structure of P7 and its daughter ions fragment were proposed (Fig. S11† and Table 1).
The MS spectra of P8 in ESI+ mode indicated its molecular ion was m/z 212. Furthermore, the fragment ions of m/z 182 (loss –OCH3), 170 (loss –CH(CH3)2), 142 (loss –CH(CH3)2 and –CH2CH3) and 128 (loss –CH(CH3)2, –CH2CH3 and –OH) were detected in the MS/MS spectra (Fig. S12†), suggesting the structure of P8 included –NHCH(CH3)2, –NHCH2CH3 and –OCH3 attached to the s-triazine ring (Table 1).
The fragment ion of m/z 216 was detected as P9 in MS spectra under ESI+ mode. The P9 was deduced to be ATZ by comparing the TIC of ATZ solution before and after UV irradiation treatment (Fig. S13†). P10 was only detected in ESI+ mode and fragment ion of m/z 139 was detected (Table S2†). The molecular structure of P10 was proposed in Fig. S14† and Table 1.
The MS spectra of P11 in the ESI+ and ESI− mode are shown in Table S2.† In ESI+ mode, m/z 198 (M+), 220 (M + Na), 395 (2M+) and 417 (2M + Na) were detected. Nevertheless, only one fragment ion of m/z 196 (M−) was detected in ESI− mode. Based on previous studies29 and the information of chromatography and mass spectra, molecular structures of P11 was proposed as presented in Table 1.
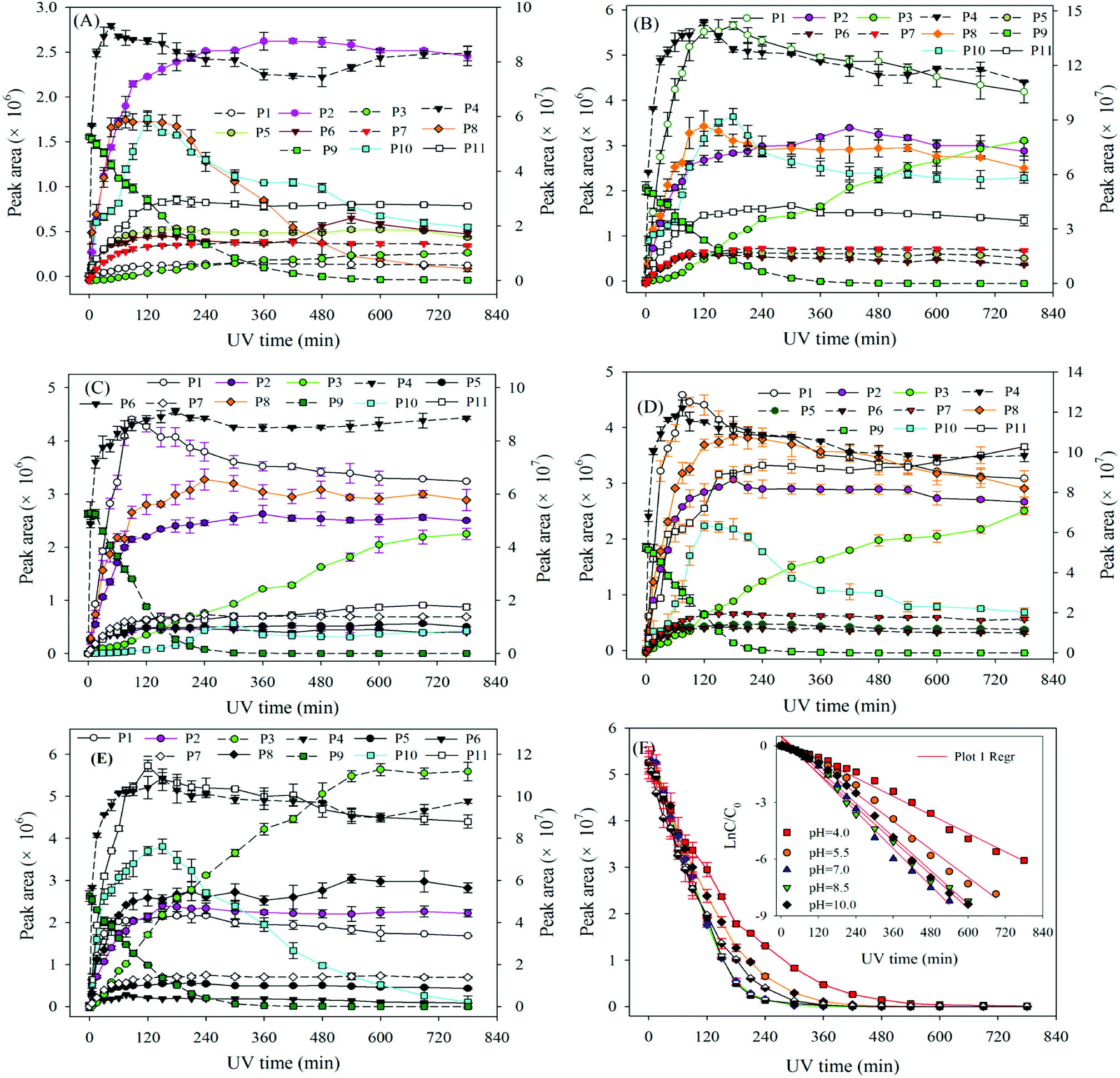 | ||
| Fig. 2 The degradation of ATZ and formation of intermediates versus irradiation time in sole-UV process: (A) pH = 4.0; (B) pH = 5.5; (C) pH = 7.0; (D) pH = 8.5; (E) pH = 10.0. Note: solid line corresponds left axis; dotted line corresponds right axis. | ||
The degradation pathways of ATZ and the formation intermediates exhibited significant difference under five solution pH conditions. The concentrations of P1, P2, P4 and P10 increased at the initial stage of reaction and then decreased with the gradually increase of irradiation time at initial pH values of 4.0, 5.5, 7.0, 8.5 and 10.0 (Fig. 2(A)–(E)). The formation of P1 gradually decrease with the increase of solution pH values (compared with pH = 4.0, maximum formation of P1 reduced by 11%, 31%, 39% and 68% at the pH value of 5.0, 7.0, 8.5 and 10.0, respectively). Apparently, the concentration of P4 was the largest among all degradation products, the concentration of P4 increased fast from 0 to 120 min at all the pH value conditions (Fig. 2(A)–(E)). This phenomenon suggested that dechlorination is the dominant reaction of ATZ in sole-UV process. Dechlorination could occurred either through homolytic cleavage of the C–Cl bond followed by an electron transfer from the carbon to the chlorine radicals or the heterolytic cleavage of the excited-ATZ molecule favored by polar solvents.43 The results of Fig. 2 indicated that dechlorination-hydroxylation product P4 can be further degraded. The formation of P10 first increased and then gradually decreased with UV irradiation time increase at all the five pHs as observed previously.30 The concentration of P10 at pH 7.0 was lower than those at the other pHs. In addition, the irradiation time at maximum formation of P10 varied from solution pH values.
The formation of P3 increased gradually with the increase of UV irradiation time from 0 to 780 min. However, under acidic conditions (pH 4.0) and alkaline (pH 10.0) conditions, the concentrations of P10 were significant greater than that at other pH (5.5, 7.0 and 8.5) conditions.
The concentrations of P5, P6, P7, P8 and P11 increased gradually at the initial stage of reaction and then remain unchanged at all five solution pHs. The maximum concentrations of P5 at pH 4.0, 7.0 and 10.0 was similar, whereas the maximum formation amount of P5 at pH 5.5 and 8.5 was about 3 times that of the above three pH conditions. The formation of P6 at pH 5.5 and 8.5 were significantly higher than those at the other three pH conditions. The effects of pH on the formation of P7 were evidently minimal. At pH 4.0, the formation of P8 reached maximum at 75 min and then decreased rapidly as the UV irradiation treatment continued. However, under other four pH conditions, the concentrations of P8 increase at the initial stage and then remained unchanged with the increase of UV irradiation time. The formation of P11 under alkaline conditions (pH 10.0 and 8.5) is significantly higher than those under neutral (pH 7.0) and acidic (pH 5.5 and 4.0) conditions, the results indicating that P11 may be a basic catalytic product.
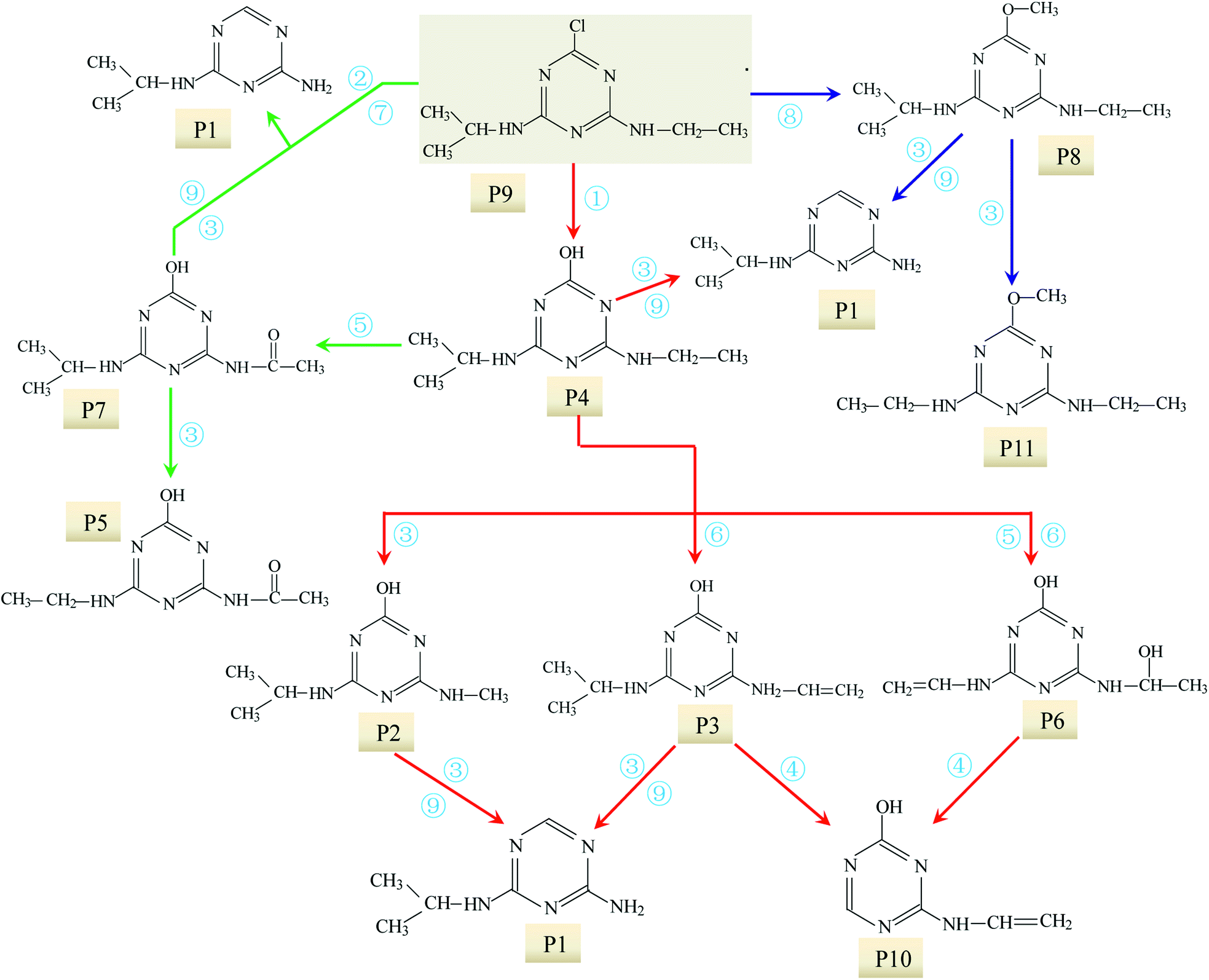 | ||
| Fig. 3 Degradation mechanism of ATZ in sole-UV process: ① dechlorination-hydroxylation reaction; ② dechlorination–dealkylation reaction; ③ dealkylation reactions; ④ deamination and lateral chains connected with the group of –NH2; ⑤ alkylic-oxidation of lateral chains; ⑥ dehydrogenation–olefination reaction; ⑦ dechlorination–hydrogenation reaction; ⑧ dechlorination–methoxylation reaction; ⑨ dehydroxylation reaction. | ||
The differences in ATZ degradation products under different pH conditions can be attributed to the difference of UV-vis absorption spectrum of ATZ under different pH conditions. UV-vis absorbance spectra of ATZ solutions after photo-degradation treatment (0–780 min, 21 sampling intervals) under different pH values were shown in Fig. S15.† Nice isosbestic points were observed, indicating clean interconversion to the primary products.44 According to the position and change of the UV-vis spectra of ATZ and its products, the absorption spectrum in Fig. S15† was divided into four regions, namely (I), (II), (III) and (IV). The arrow direction in the Fig. S15† was the change direction of the absorption spectrum with UV irradiation time increases. When the irradiation time was less than 120 min, the absorbance of the four regions in the spectrum all shows significant changes with the increase of irradiation time. However, the change range of the spectrum decrease with irradiation time further increased.23 The absorbances of the regions (i) and (iii) in Fig. S15† gradually increased with the increase of the irradiation time. However, the absorbance of the regions (ii) and (iv) gradually decreased with the increase of the irradiation time. The regions of (ii) and (iv) were the absorption regions of ATZ, and the wavelengths at the maximum absorbance were 220 nm and 265 nm, respectively.
As shown in Fig. S15,† there was an intense band around 220 nm and a rather weak one around 265 nm, the 220 nm band previously was assigned to a π to π* transition, the 265 nm was assigned to n to π*.23 The absorption value at the maximum absorption wavelength of 220 nm decreased gradually with irradiation treatment time increase, indicating that UV irradiation induced fast decomposition of ATZ. The slight blue shift was observed from Fig. S15,† which may be caused by theoretically partial or complete loss of lateral chains and substitution amino by hydroxyl group.23 Upon irradiation with 254 nm light, these bands were seen to decrease in intensity and new absorptions grew in at 210 and 240 nm. The peak at 210 nm might be caused by the formation of cyanuric acid, ammelide and ammeline.45 The chromophoric group of heteroatom ring, such as –Cl, was possibly replaced by –OH, which led to larger conjugative effect.
In addition, pH could significantly affect the UV-visible absorption spectrum of the ATZ solution after photo-oxidation. For example, there were large differences in the absorption spectra of ATZ solution at pH values of 4.0, 5.5, and 7.0 (Fig. S15(a)–(c)†), while the difference of ATZ solution absorption spectra at pH = 7.0, 8.5, and 10.0 is relatively small. (Fig. S15(c)–(e)†).
The above difference in absorption spectra of ATZ solution was caused by the difference of ATZ degradation rate, the formation and dissociation form of intermediates and/or by-products under different pH conditions.44
UV radiation would result in the decomposition of organic molecules by bond cleavage and free radical generated.46 The toxicity of ATZ was believed to be connected to the chlorine group.21 Thus de-chlorination through photo-oxidation could reduce the overall toxicity of ATZ and its degradation by-products.22 For ATZ, the bond length of C–Cl bond (1.734 Å) was the longest among all bonds and the bond polarity (0.293) was relatively lower, so C–Cl bond was the easiest scissile bond compared with other bonds.23 Previous studies have also shown that the chlorine (Cl) in ATZ can be replaced by hydroxyl group (–OH) during UV radiation or combined processes with oxidants or catalysts in water.23,24 Intense UV radiation could induce cleavage of C–Cl bond to form dechloro-hydrogenated products, one of typical pathway of photolysis of ATZ.23 In present study, all the identified intermediates in UV irradiation treatment process were dechlorination products. It is generally accepted that the first step of the degradation process is dechlorination–hydroxylation reaction, generated abundant P4, which appeared to be quite stable in direct photolysis.27
In addition, C–N bond and C–C bond in amino-alky groups might also cleave easily and induced dealkylation reaction. The bond polarity of C–C (0.343) in ethylamino group was smaller than that in isopropylamino group (0.366 and 0.365), inferring that deethylation reaction should be easier occurred than deisopropylation reaction.23 In ethylamino group, possible reaction was as follows eqn (3) and (4):
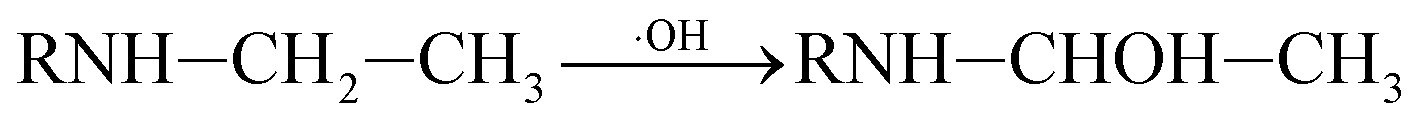 | (3) |
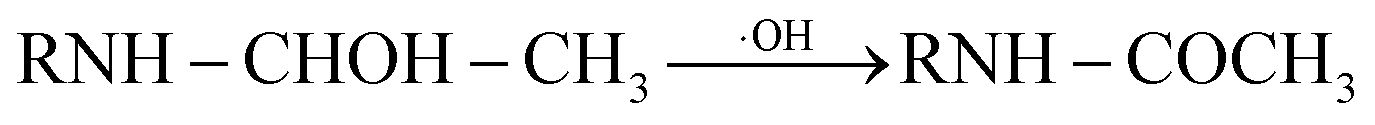 | (4) |
P5, P6 and P7 can be generated through these reactions (eqn (3) and (4)). While in the case of the isopropylamino group, the alkylic-oxidated products might be produced in parallel through different reaction mechanism:47
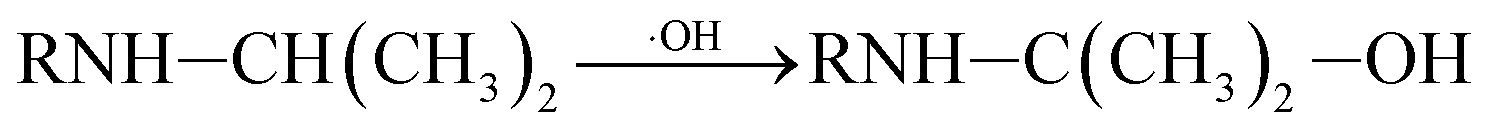 | (5) |
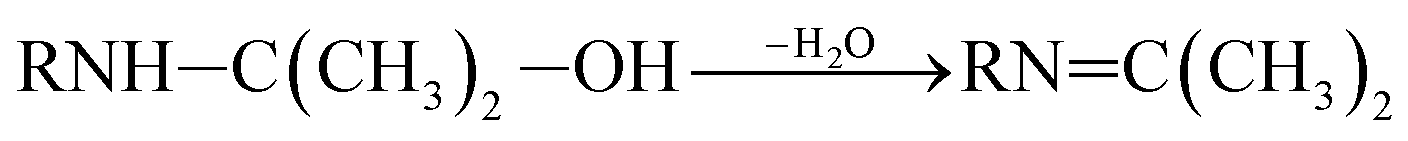 | (6) |
RN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(CH3)2–OH ⇌ RNH–C(CH3)2CH2 C(CH3)2–OH ⇌ RNH–C(CH3)2CH2
| (7) |
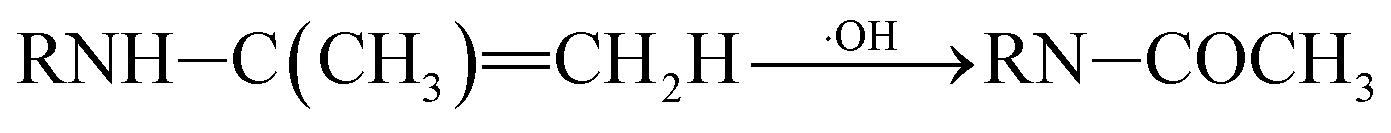 | (8) |
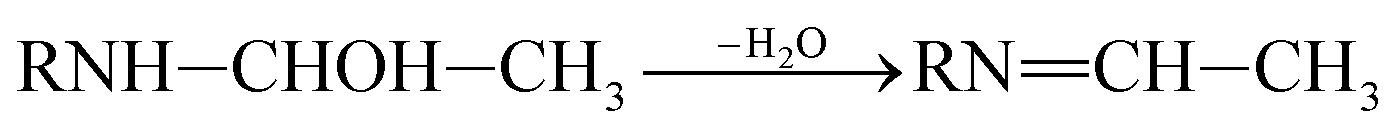 | (9) |
|
RNCH–CH3 ⇌ RNH–CHCH2
| (10) |
Based on the above formation of intermediates or by-products, the main reactions of ATZ in sole-UV process were proposed as follows:
① Dechlorination–hydroxylation reaction: replacement of Cl with an OH. It is notable that dechlorination–hydroxylation was the dominating reaction during direct UV photolysis.
② Dechlorination–dealkylation reaction: replacement of Cl with an OH, and the same time, the removal of alkyl groups from the lateral chains. The dealkylation reactions could also arise from the homolytic or heterolytic cleavage of the C–C and N–C bonds of ethylamino and/or isopropylamino of the excited ATZ molecules during UV irradiation. Intermediates P2 was obtained through dechlorination–dealkylation reaction during UV irradiation.
③ Dealkylation reactions: removal of alkyl groups from the lateral chains (methylamino, ethylamino and/or isopropylamino) of ATZ and its photo-degradation intermediates.
④ Deamination and lateral chains connected with the group of –NH2. In this study, the formation of P10 relates to the reaction.
⑤ Alkylic-oxidation of lateral chains: the reaction may be resulted from H-abstraction by ·OH/–SO4˙ leading to the formation of a carbon radical compound. In this study, P5 and P7 were obtained by the reaction of the alkylic-oxidation during UV irradiation.
⑥ Dehydrogenation–olefination reaction: olefination by-products can be resulted from the dehydration of alkylic-hydroxylation by-products. The H-abstraction by reactive radicals from lateral chains of ATZ can also lead to the formation of olefination by-products. P3 and P6 were obtained by the reaction of the dehydrogenation–olefination in sole-UV process.
⑦ Dechlorination–hydrogenation reaction: similar to dechlorination–hydroxylation mechanism except that chlorine is possibly replaced by H from water molecules. The degradation intermediates in UV irradiation through dechlorination–hydrogenation reaction include P1 and P10.
⑧ Dechlorination–methoxylation reaction: a series of dechlorination–methoxylation products (such as P8 and P11) were detected and these products might be caused by slight amount of methanol from reserving liquid.
⑨ Dehydroxylation reaction: the degradation products results in the removal of hydroxyl from s-triazine ring of ATZ and its photo-degradation intermediates. P1 can be formation through dehydroxylation reaction.
3.2 UV photo-oxidation intermediates and possible pathways of ATZ in UV/H2O2 process
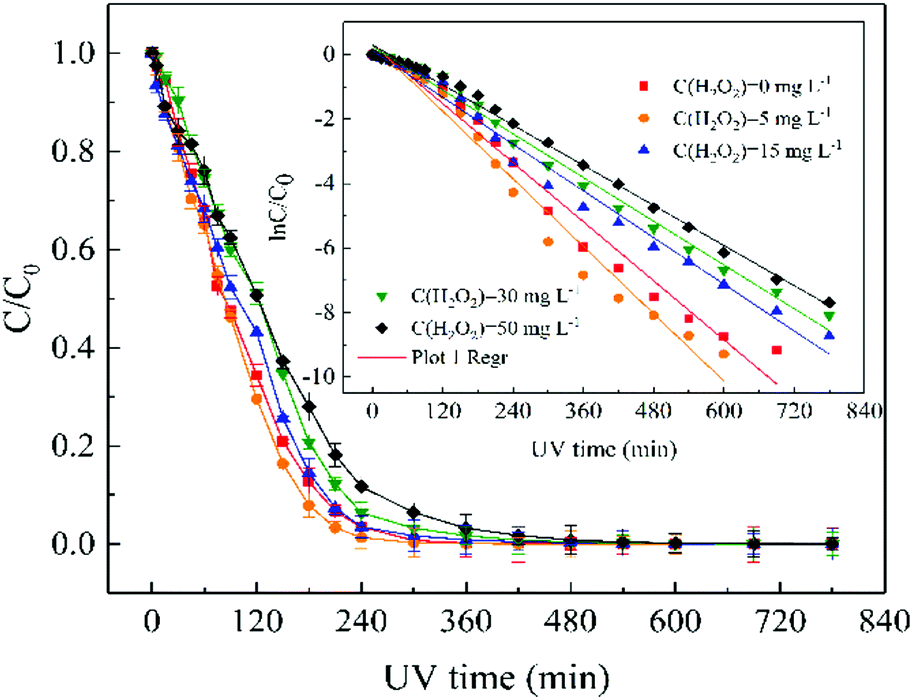 | ||
| Fig. 4 Effect of H2O2 does on removal of ATZ. Raw ATZ solution: 5 mg L−1, initial pH 7.0. | ||
Base on eqn (2), the values of kinetic parameters at different H2O2 dose were calculated, and the detailed information is shown in Table S3.† When the UV irradiation time was greater than 300 min, the data points have gradually to upward deviate from the fitted curve. The results indicated that the degradation rate of ATZ gradually decreased with irradiation time increase as have been reported previously.49 Two possible explanations for the retardation of reaction rate were proposed.
(i) The intermediates of ATZ formed in the solution would compete for the photons which retards the degradation of ATZ. This effect would become more and more dominant since the intermediates accumulate to higher levels in reaction solution.50
(ii) The presence of intermediates and/or by-products in solution might absorb light, thereby attenuating the incident UV intensity available for photo-degradation. The reduction of the ATZ reaction rate can be also explained from the point of view of the collision theory of reaction rates.49 Such an effect is believed to be applicable to all the degradable components in the solution.50
Besides, it is interesting to note that the solution pH would gradually increase or decrease as the initial pH level is in acidic or basic conditions during the photolysis process, respectively.50 The changes of pH between before and after reaction is relatively small (see Table S3†), thus the effect of pH changes on the degradation rate of ATZ can be ignored.
In this study, although the degradation rate of ATZ decreasing with the reaction time increases, but the reduction degree of ATZ degradation rate with irradiation time increase was significantly lower than that of previously reported, and it has an good linear correlation coefficient (r2 ≥ 0.979) of pseudo first-order reaction kinetics for ATZ in water. Therefore, the degradation kinetics curve of ATZ was not stage fitting under different reaction conditions in this study. UV-vis absorbance spectra of ATZ solutions at pH of 7.0 in UV/H2O2 process was shown in Fig. S16.†
No obvious changed of H2O2 (<3%) was observed in the dark controls. According to eqn (2), the data on the H2O2 concentration versus UV irradiation time was fitted using the pseudo-first-order kinetics model (Fig. S17†), showing good linear relationship (r2 ≥ 0.984).
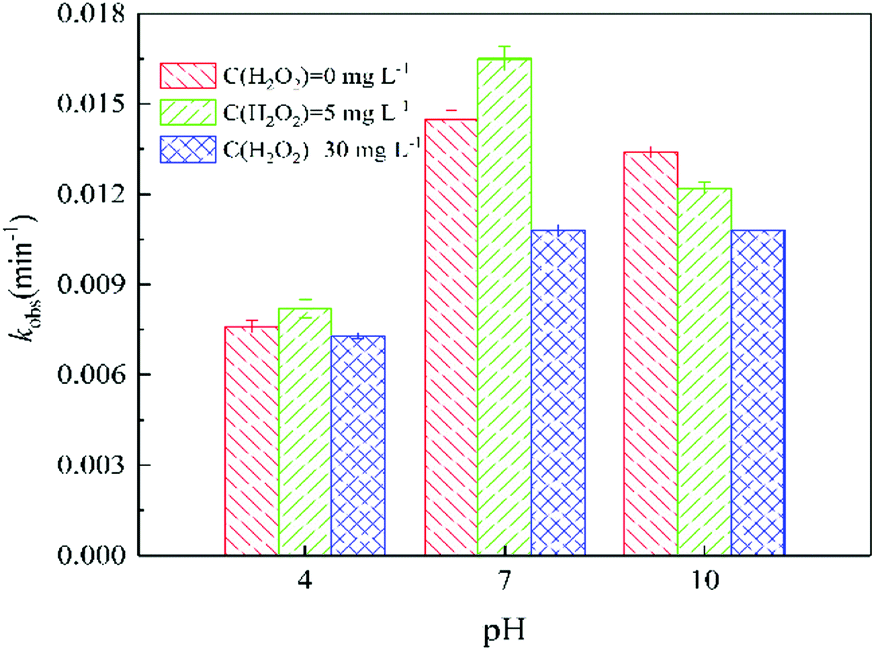 | ||
| Fig. 5 Effect of solution pH on the pseudo-first-order reaction rate constants (kobs) of ATZ during UV irradiation treatment under different H2O2 does. Raw ATZ solution: 5 mg L−1. | ||
At pH 10.0, the degradation rate of ATZ gradually decreases in UV/H2O2 process with the H2O2 dose increasing (Fig. S19†). UV-vis absorbance spectra of ATZ solutions at pH of 4.0 and 7.0 in UV/H2O2 process was shown in Fig. S20 and S21,† respectively. The results indicate that addition of H2O2 did not promote the formation of ·OH at pH 10.0. In addition, the degradation rate of H2O2 during UV irradiation treatment increased with the solution pH increasing (Fig. S17, S22 and S23†).
At pH 7.0, ATZ and its ten intermediates (P1, P2, P3, P4, P5, P6, P7, P8, P10 and P11) were detected in sole-UV process (Fig. S24(A)†). During UV/H2O2 process, supererogatory five intermediates was detected, marked as P12, P13, P14, P15 and P16.
The formation of these intermediates varies with solution pH during UV/H2O2 process. At pH 7.0, 15 intermediates (P1, P2, P3, P4, P5, P6, P7, P8, P10, P11, P12, P13, P14, P15 and P16) of ATZ were detected (Fig. S24(D)†). All these intermediates were detected at pH 4 except P13 (Fig. S24† (F)). However, at pH 10.0, another five new chromatographic peaks (P17, P18, P19, P20 and P21) were detected. Retention time (RT) and MS spectral information in full scan modes of the above 11 chromatographic peaks (P1–P11) are shown in Table S2,† and detailed information of all other chromatographic peaks are shown in Table S4.†
The molecular structures of P1, P2, P3, P4, P5, P6, P7, P8, P10 and P11 were proposed in above part. The precursor ions of P12, P13, P14, P15, P16, P17, P18, P19, P20 and P21 in daughter scan mode are shown in Table 2, and MS/MS spectra of these intermediates are shown in Fig. S25–S34.† Molecular structures of these intermediates or by-products were derived based on MS spectra and MS/MS spectra (Fig. S25–S34†).
| Name | ESI model | MS fragment ions (m/z) | Precursor ions (m/z) | MW (Da) | Formula | Proposed structure | Chemical name |
|---|---|---|---|---|---|---|---|
| P12 | ESI+ | 168, 152, 123, 115 | 210 | 209 | C8H11N5O | 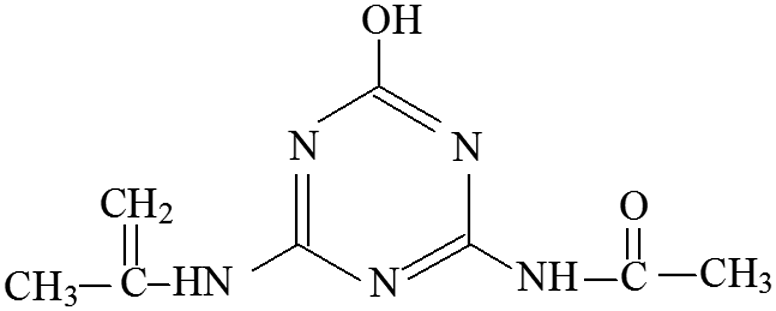 |
2-Hydroxy-4-acetamido-6-isopropenylenylamino-s-triazine |
| P13 | ESI+ | 97, 139 | 139 | 138 | C5H6N4O | 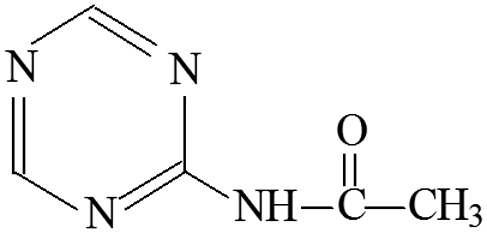 |
4-Acetamido-s-triazine |
| P14 | ESI+ | 184, 170, 142, 100, 85, 68 | 212 | 211 | C8H13N5O2 | 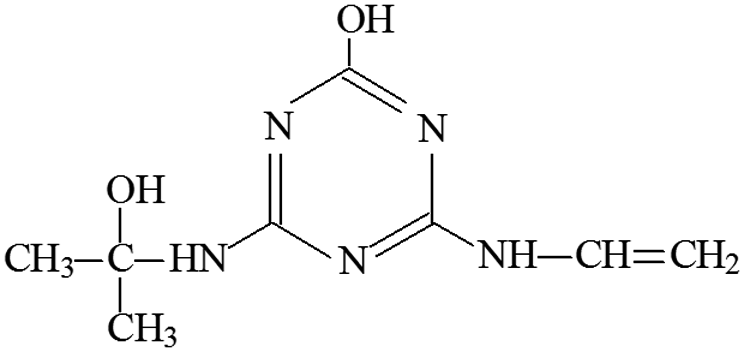 |
2-Hydroxy-4-acetamido-6-(2-hydroxyisopropylamino) -s-triazine |
| ESI− | 182, 166, 136, 115, 111, 98 | 210 | |||||
| P15 | ESI+ | 170, 103, 86 | 212 | 211 | C8H13N5O2 | 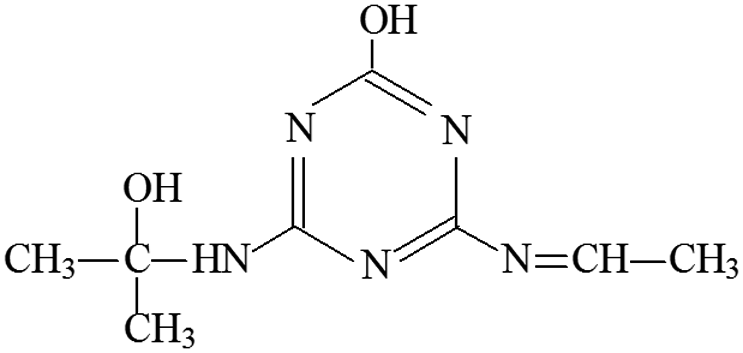 |
2-Hydroxy-4-ethylimine-6-(2-hydroxyisopropylamino) -s-triazine |
| ESI− | 167, 152, 123, 115, 101, 66 | 210 | |||||
| P16 | ESI+ | 173, 214 | 214 | 213 | C7H8N5OCl | 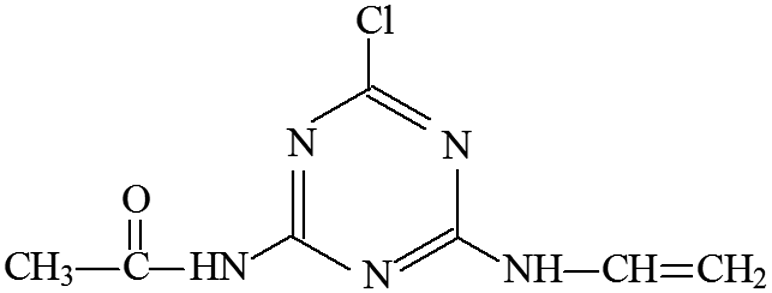 |
2-Chloro-4-vinylamino-6-acetamido-s-triazine |
| P17 | ESI+ | 196, 170, 143, 129 | 214 | 213 | C8H15N5O2 | 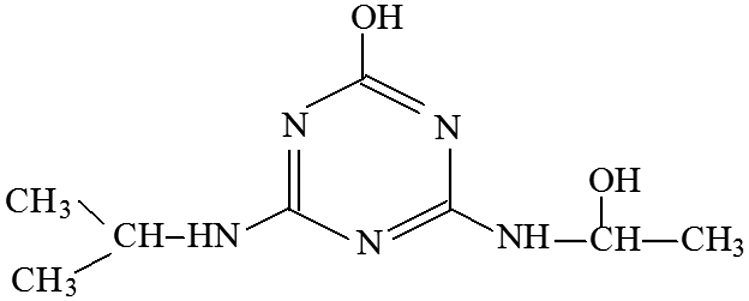 |
2-Hydroxy-4-(2-hydroxy-ethylamino)-6-isopropylamino-s-triazine |
| P18 | ESI+ | 214 | 214 | 213 | C8H15N5O2 | 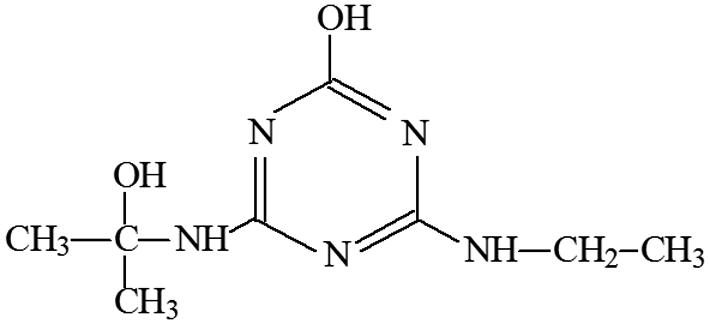 |
2-Hydroxy-4-ethylamino-6-(2-hydroxyisopropylamino) -s-triazine |
| P19 | ESI+ | 218, 154, 127 | 196 | 195 | C8H13N5O | 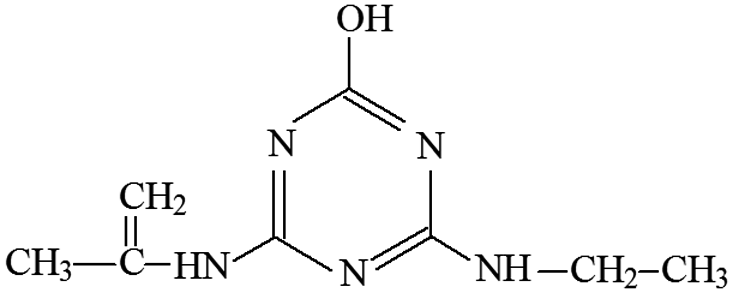 |
2-Hydroxy-4-ethylamino-6-isopropenylenylamino-s-triazine |
| P20 | ESI+ | 172, 194 | 172 | 171 | C5H9N5O2 | 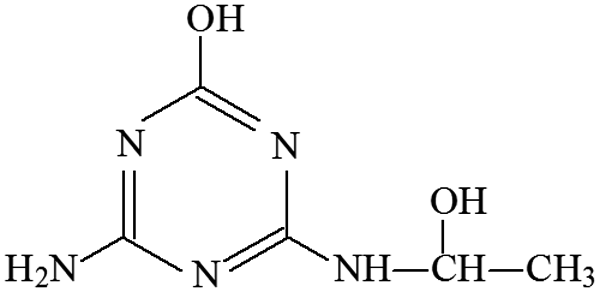 |
2-Hydroxy-4-(2-hydroxy-ethylamino)-6-amino-s-triazine |
| ESI− | 170 | 170 | |||||
| P21 | ESI+ | 172, 194 | 172 | 171 | C5H6N5Cl | 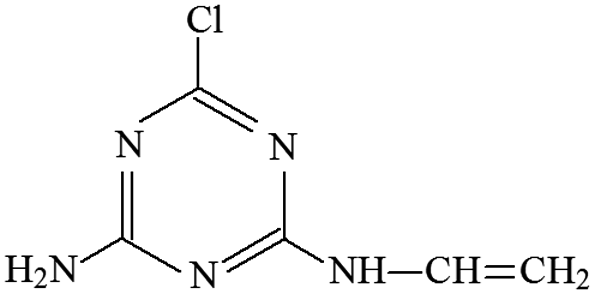 |
2-Chloro-4-vinylamino-6-amino-s-triazine |
| ESI− | 170 | 170 |
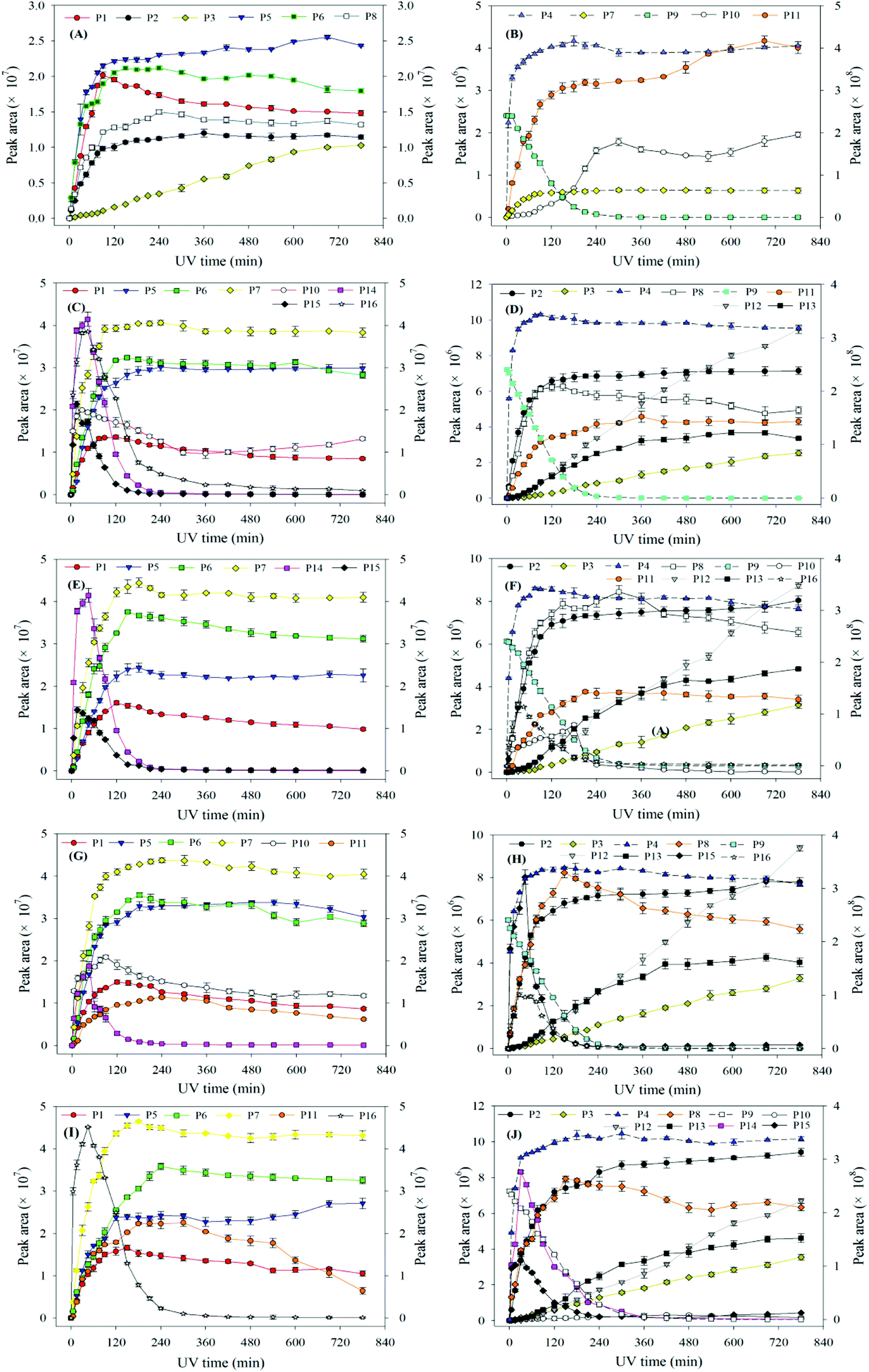 | ||
| Fig. 6 The degradation of ATZ and formation of identified intermediates versus UV irradiation time in UV/H2O2 process (pH = 7.0): (A) and (B) 0 mg L−1 H2O2; (C) and (D) 5 mg L−1 H2O2; (E) and (F) 15 mg L−1 H2O2; (G) and (H) 30 mg L−1 H2O2; (I) and (J) 50 mg L−1 H2O2. Note: solid line corresponds left axis; dotted line corresponds right axis. | ||
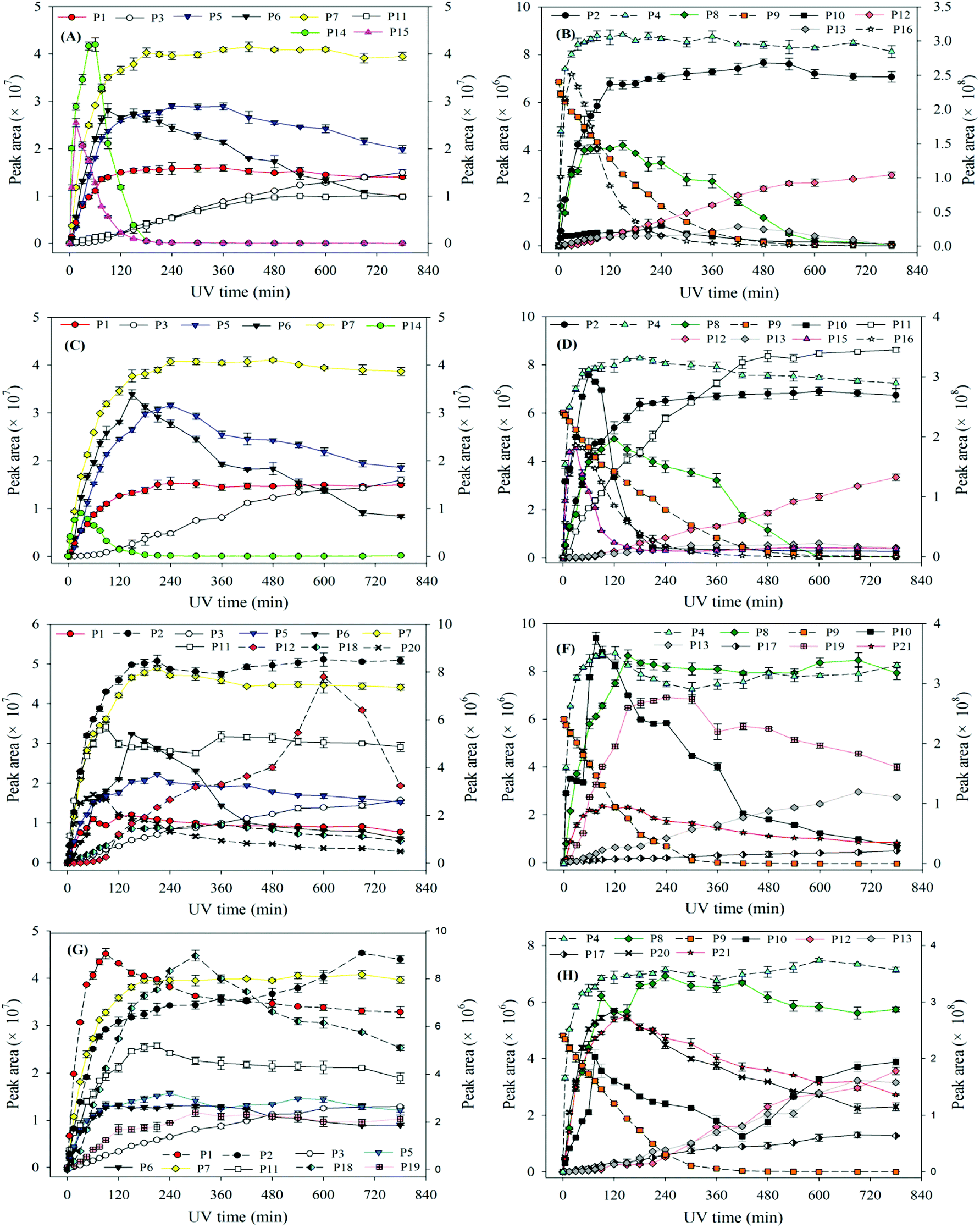 | ||
| Fig. 7 The degradation of ATZ and formation of identified intermediates versus UV irradiation time in UV/H2O2 process: (A) and (B) pH = 4.0, 5 mg L−1 H2O2; (C) and (D) pH = 4.0, 30 mg L−1 H2O2; (E) and (F) pH = 10.0, 5 mg L−1 H2O2; (G) and (H) pH = 10.0, 30 mg L−1 H2O2. Note: solid line corresponds left axis; dotted line corresponds right axis. | ||
Compared to the sole-UV process, the species and formation of intermediates of ATZ had significant changes in UV/H2O2 process. During sole-UV process, ten intermediates were detected as shown in Fig. S24(A).† Nevertheless, fifteen intermediates were detected during UV/H2O2 process as shown in Fig. S24(B).† As the H2O2 dose increased from 5 to 50 mg L−1, the speciation of the intermediates remained unchanged (Fig. S24(B)–(E)†). During sole-UV and UV/H2O2 processes, there was an obvious difference in the concentration of P1, P2, P3, P4, P5, P6, P7, P8, P10 and P11. In addition, H2O2 dose also has remarkable effect on the formation of the ten intermediates. During UV/H2O2 process, P12 increased gradually with the increase of UV irradiation time. However, the formation amount of the intermediate decreased slightly with the increase of H2O2 dose at the same irradiation time. The formations of P13, P14 and P15 firstly increased and then decreased with the increase of UV irradiation time. In addition, P14 was one of the major products at low H2O2 dose and irradiation time.
At the same H2O2 dose, the concentration of P16 first increased sharply and attained a maximum at the UV irradiation time of 30 min and then decreased rapidly with the increase of irradiation time. In addition, the maximum formation amount of P16 first increased and then decreased with the increase of H2O2 dose, and the maximum value was obtained at H2O2 of 15 mg L−1.
In UV/H2O2 process, the same intermediates were detected at the pH 4.0 and 7.0. The formations of these intermediates are shown in Fig. 7(A)–(D). Except for P14, P15 and P16, all the other intermediates at pH 10.0 were also detected. In addition, another five new intermediates (P17, P18, P19, P20 and P21) were also detected at pH 10.0. The main degradation products of ATZ at pH 10.0 recorded as a function of irradiation time were displayed in Fig. 7(E)–(H).
The results indicated that the intermediates P14, P15 and P16 were detected under acidic and neutral solution conditions, and P17, P18, P19, P20 and P21 were only detected under alkaline condition.
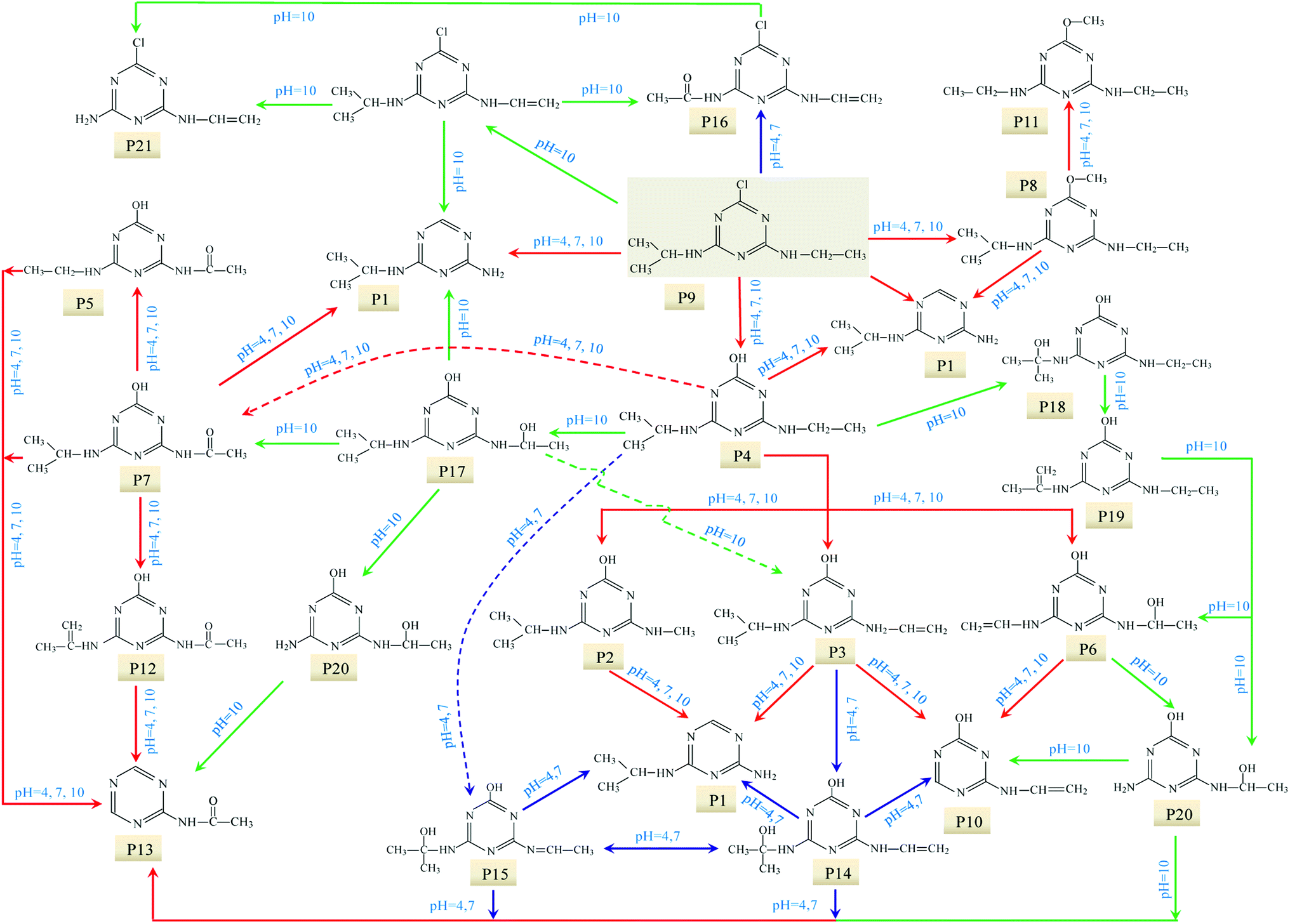 | ||
| Fig. 8 Proposed UV photo-degradation pathway of ATZ in UV/H2O2 process. | ||
4. Conclusions
This study explored the degradation of ATZ in sole-UV and UV/H2O2 process. The degradation of ATZ followed pseudo-first-order kinetics in the two processes. It was observed that ATZ could be removed from water by UV-254 irradiation treatment, and the combination of low H2O2 dose with UV radiation significantly accelerated the degradation efficiency.The structures of the main intermediates or by-products were identified based on MS and MS/MS spectra, which showed that the UV oxidation intermediates or by-products varied with reaction conditions. Ten degradation products were detected in sole-UV process, and there are significant differences for the formation of these products under different solution pH conditions. In UV/H2O2 process, fifteen intermediates were detected at pH 7.0 whereas the speciation of the intermediates remained unchanged as the H2O2 dose increased. The photo-oxidation intermediates of ATZ were found to vary with solution pH conditions in UV/H2O2 process. Except for P13, all the other intermediates at pH 4 were also detected; at pH 10.0, in addition of the above fifteen intermediates, another five new intermediates were also detected. Potential degradation mechanisms for ATZ were evaluated exhibiting nine different degradation pathways including dechlorination–hydroxylation, dechlorination–dealkylation, de-alkylation, deamination, alkylic-oxidation of lateral chains, dehydrogenation–olefination, dechlorination–hydrogenation, dechlorination–methoxylation and dehydroxylation. The plausible degradation pathways of ATZ in sole-UV and UV/H2O2 process were proposed.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Natural Science Foundation of Shandong Province (No. ZR2017BEE016), National Natural Science Foundation of China (No. 51609207), and Science Fund of Yantai University (No. TM17B19). The authors gratefully acknowledge the support of the three foundations.References
- Z. Zhao, Q. Wu, T. Nie and W. Zhou, RSC Adv., 2019, 9, 4162–4171 RSC.
- S. Singh, V. Kumar, A. Chauhan, S. Datta, A. B. Wani, N. Singh and J. Singh, Environ. Chem. Lett., 2018, 16, 211–237 CrossRef CAS.
- N. Chen, D. Valdes, C. Marlin, H. Blanchoud, R. Guerin, M. Rouelle and P. Ribstein, Sci. Total Environ., 2019, 652, 927–938 CrossRef.
- G. Sagratini, M. Ametisti, M. Canella, G. Cristalli, E. Francoletti, D. Giardina, M. C. Luminari, G. Paparelli, Y. Pico, R. Volpini and S. Vittori, Fresenius Environ. Bull., 2007, 16, 973–979 CAS.
- A. Pannier, T. Lehrer, M. Vogel, U. Soltmann, H. Boettcher, S. Tarre, M. Green, J. Raff and K. Pollmann, RSC Adv., 2014, 4, 19970–19979 RSC.
- P. F. Coldebella, M. R. Fag-Undes-Klen, D. Rezende, A. T. Alves Baptista, L. Nishi, Q. L. Shimabuku and R. Bergamasco, Desalin. Water Treat., 2018, 126, 248–258 CrossRef CAS.
- J. A. Godfrey and A. L. Rypstra, Chemosphere, 2018, 201, 459–465 CrossRef CAS.
- S. E. Oliveira, P. M. Costa, S. B. Nascimento, W. V. Castro, R. I. Maciel De Azambuja Ribeiro, H. B. Santos and R. G. Thome, Aquat. Toxicol., 2018, 202, 57–64 CrossRef CAS.
- R. S. D. Calder and K. A. Schmitt, Environ. Sci. Technol., 2010, 44, 8008–8014 CrossRef CAS.
- P. Westerhoff, Y. Yoon, S. Snyder and E. Wert, Environ. Sci. Technol., 2005, 39, 6649–6663 CrossRef CAS.
- T. C. Zhang and S. C. Emary, Environ. Eng. Sci., 1999, 16, 417–432 CrossRef CAS.
- I. Akpinar and A. O. Yazaydin, J. Chem. Eng. Data, 2018, 63, 2368–2375 CrossRef CAS.
- Y. Zhang, B. Cao, L. Zhao, L. Sun, Y. Gao, J. Li and F. Yang, Appl. Surf. Sci., 2018, 427, 147–155 CrossRef CAS.
- M. Saxena, H. Brahmbhatt, D. Anjali Devi and A. Bhattacharya, Desalin. Water Treat., 2015, 55, 575–586 CrossRef CAS.
- V. S. Babu, M. Padaki, L. P. D'Souza, S. Deon, R. G. Balakrishna and A. F. Ismail, Chem. Eng. J., 2018, 334, 2392–2400 CrossRef CAS.
- S. Wu, H. He, X. Li, C. Yang, G. Zeng, B. Wu, S. He and L. Lu, Chem. Eng. J., 2018, 341, 126–136 CrossRef CAS.
- D. Wang, H. Xu, J. Ma, X. Lu, J. Qi and S. Song, Chem. Eng. J., 2018, 354, 113–125 CrossRef CAS.
- K. Zhao, X. Quan, S. Chen, H. Yu, Y. Zhang and H. Zhao, Chem. Eng. J., 2018, 354, 606–615 CrossRef CAS.
- K. Zhu, X. Wang, X. Ma, Z. Sun and X. Hu, Electrocatalysis, 2019, 10, 35–44 CrossRef CAS.
- R. Hong, L. Zhang, W. Zhu and C. Gu, Sci. Total Environ., 2019, 652, 224–233 CrossRef.
- A. Beatriz Baranda, A. Barranco and I. Martinez De Maranon, Water Res., 2012, 46, 669–678 CrossRef.
- G. W. Stratton, Arch. Environ. Contam. Toxicol., 1984, 13, 35–42 CrossRef CAS.
- C. Chen, S. Yang, Y. Guo, C. Sun, C. Gu and B. Xu, J. Hazard. Mater., 2009, 172, 675–684 CrossRef CAS.
- H. Chen, E. Bramanti, I. Longo, M. Onor and C. Ferrari, J. Hazard. Mater., 2011, 186, 1808–1815 CrossRef CAS.
- L. Xu, H. Zang, Q. Zhang, Y. Chen, Y. Wei, J. Yan and Y. Zhao, Chem. Eng. J., 2013, 232, 174–182 CrossRef CAS.
- M. Gmurek, M. Olak-Kucharczyk and S. Ledakowicz, Chem. Eng. J., 2017, 310, 437–456 CrossRef CAS.
- K. Lekkerkerker-Teunissen, M. J. Benotti, S. A. Snyder and H. C. van Dijk, Sep. Purif. Technol., 2012, 96, 33–43 CrossRef CAS.
- J. De Laat, H. Gallard, S. Ancelin and B. Legube, Chemosphere, 1999, 39, 2693–2706 CrossRef CAS.
- Y. Tian, W. Shen, F. Jia, Z. Ai and L. Zhang, Chem. Eng. J., 2017, 330, 1075–1081 CrossRef CAS.
- K. H. Chan and W. Chu, Appl. Catal., B, 2005, 58, 165–174 CrossRef CAS.
- A. Torrents, B. G. Anderson, S. Bilboulian, W. E. Johnson and C. J. Hapeman, Environ. Sci. Technol., 1997, 31, 1476–1482 CrossRef CAS.
- N. Ta, J. Hong, T. Liu and C. Sun, J. Hazard. Mater., 2006, 138, 187–194 CrossRef PubMed.
- S. Trapp, A. Franco and D. Mackay, Environ. Sci. Technol., 2010, 44, 6123–6129 CrossRef CAS PubMed.
- L. K. Ge, J. W. Chen, X. X. Wei, S. Y. Zhang, X. L. Qiao, X. Y. Cai and Q. Xie, Environ. Sci. Technol., 2010, 44, 2400–2405 CrossRef CAS.
- X. X. Wei, J. W. Chen, Q. Xie, S. Y. Zhang, L. K. Ge and X. Qiao, Environ. Sci. Technol., 2013, 47, 4284–4290 CrossRef CAS.
- W. Li, Y. Liu, J. Duan, J. van Leeuwen and C. P. Saint, Chem. Eng. J., 2015, 274, 39–49 CrossRef CAS.
- R. O. Rahn, M. I. Stefan, J. R. Bolton, E. Goren, P. S. Shaw and K. R. Lykke, Photochem. Photobiol., 2003, 78, 146–152 CrossRef CAS.
- N. V. Klassen, D. Marchington and H. Mcgowan, Anal. Chem., 1994, 66, 2921–2925 CrossRef CAS.
- G. Imoberdorf and M. Mohseni, Chem. Eng. J., 2012, 187, 114–122 CrossRef CAS.
- J. A. Khan, X. He, N. S. Shah, H. M. Khan, E. Hapeshi, D. Fatta-Kassinos and D. D. Dionysiou, Chem. Eng. J., 2014, 252, 393–403 CrossRef CAS.
- Y. Ku, J. L. Chang and S. C. Cheng, Water, Air, Soil Pollut., 1998, 108, 445–456 CrossRef CAS.
- Y. Liu, J. Duan and W. Li, Acta Chim. Sin., 2015, 73, 1196–1202 CrossRef CAS.
- V. Hequet, C. Gonzalez and P. Le Cloirec, Water Res., 2001, 35, 4253–4260 CrossRef CAS.
- M. E. D. G. Azenha, H. D. Burrows, M. Canle, L. R. Coimbra, M. I. Fernández, M. V. García, M. A. Peiteado and J. A. Santaballa, J. Phys. Org. Chem., 2003, 16, 498–503 CrossRef CAS.
- M. E. Balmer and B. Sulzberger, Environ. Sci. Technol., 1999, 33, 2418–2424 CrossRef CAS.
- Z. Gao, S. Yang, T. Na and C. Sun, J. Hazard. Mater., 2007, 145, 424–430 CrossRef CAS.
- S. Nelieu, L. Kerhoas and J. Einhorn, Environ. Sci. Technol., 2000, 34, 430–437 CrossRef CAS.
- H. Shemer and K. G. Linden, J. Hazard. Mater., 2006, 136, 553–559 CrossRef CAS PubMed.
- W. Li, J. Duan and D. Mulcahy, J. Water Supply: Res. Technol.-AQUA, 2012, 61, 82–93 CrossRef CAS.
- T. K. Lau, W. Chu and N. Graham, Chemosphere, 2005, 60, 1045–1053 CrossRef CAS PubMed.
- M. Hincapie, M. I. Maldonado, I. Oller, W. Gernjak, J. A. Sanchez-Perez, M. M. Ballesteros and S. Malato, Catal. Today, 2005, 101, 203–210 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra05747a |
| This journal is © The Royal Society of Chemistry 2019 |