
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHeterogeneous photocatalytic performances of CO2 reduction based on the [Emim]BF4 + TEOA + H2O system
Jinliang Lin *,
Youfeng Li and
Bo Xie
*,
Youfeng Li and
Bo Xie
Department of Chemical and Engineering, Zunyi Normal College, Zunyi, 563000, P. R. China. E-mail: jinliang_lin@163.com; Fax: +86-851-28927159; Tel: +86-851-28927159
First published on 4th November 2019
Abstract
The photochemical reduction of CO2 was studied in a 1-ethyl-3-methylimidazolium tetrafluoroborate, triethanolamine and water ([Emim]BF4 + TEOA + H2O) system under visible light irradiation. The integration of CdS and the Co–bpy complex, which acted as a photocatalyst and cocatalyst, respectively, was employed as an efficient catalytic system for the CO2-to-CO conversion. The utilization of [Emim]BF4 and water took advantage of their green properties. The amount of CO production showed that the test medium containing 10 vol% H2O was favourable for the catalytic performance of the CO2 reduction. In order to further study the factors that influenced the current system, the physical and spectroscopy properties were characterized by altering the composition ratio of the ingredients. Relevant parameters, including the viscosity, conductivity, solubility and coordination, were adjusted using the ratio of the H2O/[Emim]BF4 addition, resulting in a different catalytic performance. All of these attempts led to an optimal reaction condition for the CO2 reduction process.
1 Introduction
In recent decades, the massive consumption of fossil fuels by human beings has caused both energy shortages and excessive CO2 emission, which have been accompanied by serious environmental changes.1,2 Meanwhile, CO2 is also an abundant and sustainable carbon resource that can be used as an industrial feedstock to produce value-added chemicals.3–5 More specifically, the photocatalytic process addresses the tough challenges and provides a feasible way to use CO2 as a resource. Therefore, it was simultaneously developed as an approach to store renewable solar energy in the form of chemical energy. In other words, solar energy along with CO2 can be converted into a high-energy density fuel, which is convenient for transportation and storage. There are a number of works that focus on the photocatalytic CO2 conversion. To date, these reports are generally classified into following categories: (1) exploiting highly efficient and stable photocatalysts, especially for visible light responding materials;6,7 (2) the development of reaction types, including the reduction of CO2 into Cl chemicals and liquid fuels8 as well as the synthesis of organic compounds such as poly/carbonates, and aromatic carboxylic acids;9 (3) designing gimmickry or a reaction system for various purposes, especially for green processes and air resistance systems.10,11 (4) The mechanism investigation is also a promising approach to yield a high efficiency CO2 conversion.More specifically, the perspective of green chemistry plays an important role in the design of chemical products and catalytic processes. Water is considered to be a green solvent. Therefore, many CO2 reduction experiments have been conducted in an aqueous solution over the past few decades. Indeed, the use of water as a reductant and reaction medium provides an ideal approach for the CO2 conversion with the concept of green chemistry. However, inevitable obstacles exist that hinder the CO2 reduction reaction. The primary factors include a very low CO2 solubility (0.033 mol L−1 at 25 °C) and hydrogen generation.12 The poor solubility leads to slow kinetics during the reaction. A competing reaction of the H2 evolution results in a large consumption of photogenerated electrons.
On the other hand, ionic liquids (ILs) were also reported as green solvents and are widely investigated to address the issues with CO2 capture and activation.13 ILs possess many desirable and unique properties for chemical reactions such as wide electrochemical potential window, high ionic conductivity, good thermal and chemical stability as well as low vapour pressure. In particular, they exhibited a high CO2 solubility.14 For example, recent research studies proved that an imidazolium-based IL exhibited an excellent performance for the catalytic CO2 reduction.15–17 Grills et al. revealed that imidazolium cations, a component of IL molecules, can significantly promote the CO2-to-CO conversion by stabilizing carbonate intermediates.18 Thus, ILs were widely employed as a reaction medium and catalysts during the electro/chemical reduction of CO2 in other works.19–23 However, some inherent drawbacks of ILs hinder their catalytic efficiency such as high viscosity and binding energy. Rosen et al. demonstrated the production of CO from a CO2-saturated IL/H2O solvent with a high faradaic yield and overpotentials below 0.2 V in earlier years.24 Therefore, to overcome the drawbacks, a study on the CO2 reduction conducted in an IL/organic or IL/water solution proved ILs to be an outlet in electrochemistry.25,26 A number of parameters, including the density, viscosity and gas capture behaviour were evaluated in mixtures involving ionic liquids, water and amines.27 The benefits of the mixture suggested that the strategy of solvent engineering was a feasible way to reduce costs and increase the efficiency towards CO2 conversion.
With respect to the photocatalytic CO2 activation, we previously demonstrated a photocatalytic system that exhibited a promotion effect of the ILs for the reduction of CO2 into CO at 1 atm. In our previous work, the reaction system was tested in a homogenous system and Ru(bipy)3Cl2 was used as a light photosensitizer.13 Surely, both ILs and H2O exhibited potential in the performances for CO2 reduction. However, the heterogeneous system for the photocatalytic CO2 conversion in the ILs/H2O mixture was barely covered. Based on the photocatalytic CO2 reduction platform that used CdS as a photocatalyst and Co–bpy complex as cocatalyst, the system with volatile organic compounds was expected to be a green and functional solvent. Herein, we reported that the visible light driven catalytic reduction of the CO2 system was assessed in an ionic liquid medium as well as in the presence of water and triethanolamine (denoted as ([Emim]BF4 + H2O + TEOA)) with CdS being employed as a photocatalyst.13 An innovation in this work was addressing the extension of the green system from a homogenous to heterogeneous process, which allowed for an easier separation and additional recycling. In addition, the relevant physical properties, coordination capability and electrochemical behaviours of the system were evaluated in different conditions. Detailed information on the mechanism of the CO2 reduction and the design of an environmentally friendly system for the CO2 reaction was provided.
2 Experimental
2.1 Chemicals
All of the reagents were commercially obtained and used without further purification. 2,2′-Bipyridine (bpy, Alfa), cobalt chloride hexahydrate (CoCl2·6H2O, Sigma), cadmium sulfide (CdS, Sigma, 99.9%) and 1-ethyl-3-methylimidazolium tetrafluoroborate ([Emim]BF4, ≥98% Shyfhx Co.) were used as received. Triethanolamine (TEOA) was of reagent grade and was purchased from China Sinopharm Chemical Reagent Co. Ltd. The used water was ultrapure with a resistivity of ca. 18 mΩ cm−1.2.2 Viscosity measurement
The viscosity of the reaction medium was measured by an Ubbelohde viscometer (0.47 mm). The testing temperature was maintained at 20 °C. The mixture (10 mL) was prepared in a beaker at different composition ratios by volume (V([EMIM]BF4)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :V(H2O) = 2:8, 4:6, 6:4 and 8:2). Each sample was determined three times in parallel and the average value was taken.
:V(H2O) = 2:8, 4:6, 6:4 and 8:2). Each sample was determined three times in parallel and the average value was taken.
2.3 Spectrographic characterization
Absorption spectra were obtained on a UV-Vis spectrophotometer (Varian Cary 500). The samples were prepared in 5 mL solvents ([Emim]BF4, H2O or TEOA) with different solutes (CoCl2 or Co(bpy)3Cl2). To determine the Co(bpy)+ transition, the experiment was conducted in a sealed container adaptable for the recording equipment and the compound was exposed to light irradiation for 30 min before the test.2.4 Conductivity test
The electrical conductivity was measured on a conductivity meter (DDSJ-318, INESA Scientific Instrument Co. Ltd). The mixture (10 mL) was added in the electrolysis cell at different composition ratios by volume, which was the same as the viscosity measurement. The electrolysis cell was placed in a constant temperature water bath at 20 °C. Each sample was analyzed three times in parallel and the average value was taken.2.5 Photocatalysis activity test
All of the experiments were performed in a Schlenk flask (80 mL) under an atmosphere of CO2. In the Schlenk flask, CdS (50 mg) and CoCl2·6H2O (10 μmol) were added to a 5 mL mixture of (solvent + water)/TEOA (4:1 by volume). This mixture system was subjected to vacuum degassing and backfilling with pure CO2 gas. This process was repeated (3 times) and after the last cycle, the flask was back-filled with CO2. Then, the system was irradiated for 2 hours with four 300 W LED light sources under vigorous stirring at 20 °C with a controlled water-cooling system. The produced gases (CO and H2) were detected using a gas chromatography system (Agilent 7890B, Agilent Technologies) equipped with a packed molecular sieve column (TDX-1 mesh 42/10). Ar was used as the carrier gas for the GC.
3 Results and discussion
The photocatalytic performance was tested by fixing the total volume at 5 mL and the volume of TEOA at 1 mL, while altering the addition of H2O and [Emim]BF4. As illustrated in Fig. 1, the amount of gaseous production (CO and H2) was closely related to the ratio of [Emim]BF4/water in the reaction medium. Under anhydrous conditions, the evolution of CO and H2 was moderate (11.2 and 5.6 μmol) under visible light illumination. An increasing evolution of CO was observed after adjusting the H2O content from 0 mL to 10 vol%. 31.8 μmol of CO was obtained in the reaction medium containing 10% H2O by volume. Clearly, the reaction was more feasible to start with H2O, as both electrons and holes preferred charged reaction partners. Afterwards, the addition of H2O resulted in decrements towards the production of CO. The activity for the CO production (3.1 μmol) was also sluggish when [Emim]BF4 was removed, which was lower than that without H2O (11.0 μmol). However, a gradually increased yield of H2 was generated when the system increased the amount of H2O. Obviously, the production of both CO and H2 were closely related to the different ratios of [Emim]BF4/H2O. The result suggested that there was a synergistic effect for [Emim]BF4 and H2O, which played a vital role in the enhancement of the photocatalytic performance of the CO2 reduction. It is well known that [Emim]BF4 possesses a high viscosity, which affects the mass transfer. We can acknowledge from the above result that the role of water in the adjustment of the catalytic performance was related to the dilution effect and proton source supplier.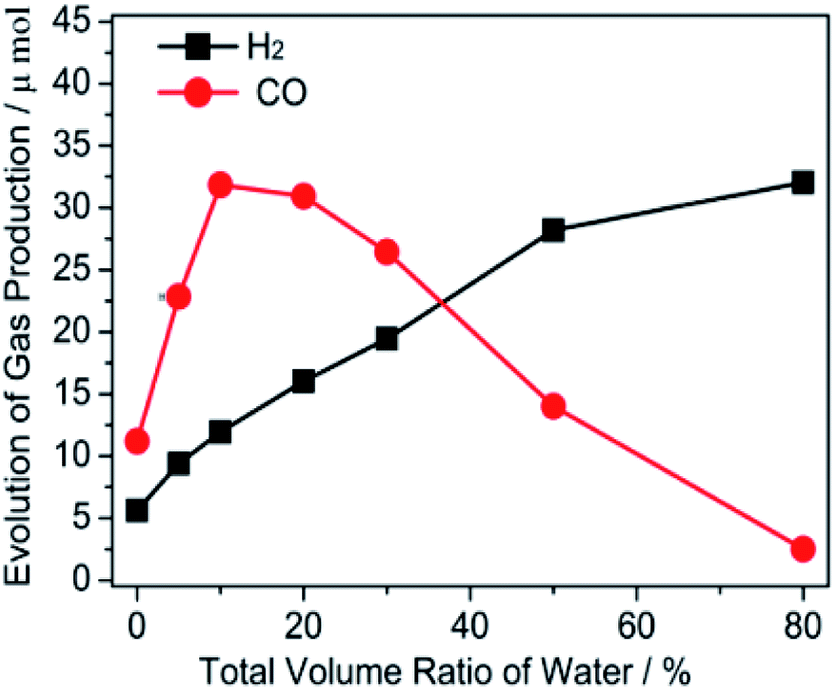 | ||
| Fig. 1 The effect of the different volume ratios of the [Emim]BF4/H2O/TEOA medium on the gas evolution of CO and H2. | ||
The hypothesis for the relationship between the catalytic performance and viscosity was still unclear when H2O was used as a conditioning agent because H2O also acted as a hydrogen source that affected the catalytic performance. It is well known that the viscosity can be easily adjusted by temperature. Thus, a further reaction was conducted at a different temperature. It should be mentioned that the influence of the molecular activation by the finite-amplitude temperature was negligible in this study. Based on above results, we fixed a 10 vol% H2O solution as the reaction medium for this study. As shown in Fig. 2, a low catalytic performance for both, the H2 and CO evolution, was obtained at 5 °C, which was attributed to the viscous solution impeding the mass transfer. Generally, a steadily increasing evolution rate for H2 was obtained as the temperature increased. An increasing amount of CO was generated when the temperature rose from 5 °C to 20 °C. This meant that the mobility of the reaction medium promoted both the CO2 and H2O reduction. Afterwards, the increase in the temperature resulted in a negative effect on CO evolution. A gradual decrease in the CO selectivity was therefore exhibited in this figure when the temperature was increased. This phenomenon was mainly caused by the high temperature leading to a low CO2 solubility. The formation of the CO2− intermediate transition was an adverse process when CO2 escaped from the solution.
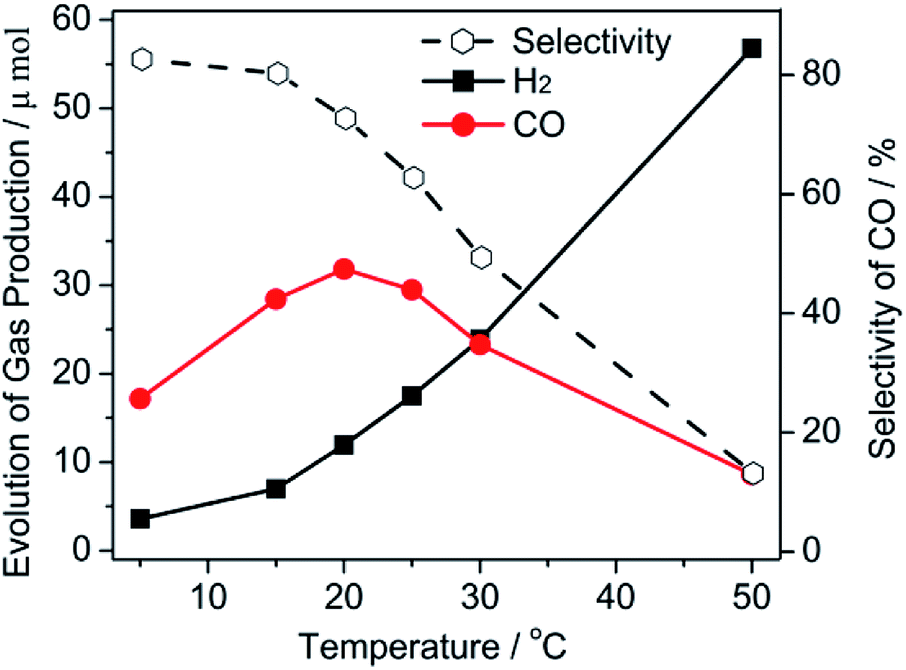 | ||
| Fig. 2 The gas evolution of CO and H2 and the selectivity of CO as a function of the temperature (selectivity of CO = mol CO/mol (H2 + CO) × 100). | ||
As shown in Fig. 3, the viscosity of the [Emim]BF4/H2O/TEOA solution continuously decreased with the increase in the H2O volume fractions. Meanwhile, the conductivity of the [Emim]BF4/H2O/TEOA solution increased as the H2O volume fractions increased in the low concentration range. After reaching a maximum value of 32.5 mS cm−1 at 50 vol%, the conductivity of the solution started to decrease with an increasing H2O concentration. At the initial stage, the change in the conductivity was consistent with the catalytic performance when the water content was within 10 vol%. The viscosity of the solution was 7.8 mPa s. The reaction medium with a low concentration of water provided a proper viscosity and proton supply. The lower viscosity resulted in a high efficiency by accelerating the mass transfer, which was evidently reflected by increasing the conductivity. Another reason to boost the CO evolution originated from the water addition, providing a typical hydrogen source for the dispersed surrounding CO2 molecules, which were readily available for the proton coupling process. Both promoted the photocatalytic CO2 reduction. However, the maximum conductivity for the water content at 50 vol% was different from that of the catalytic performance at 10 vol%. The decreasing CO evolution suggested a mass-transfer and solubility process that had a negative effect on CO2 transfer, while promoting the competitive reaction of hydrogen (H2) evolution.
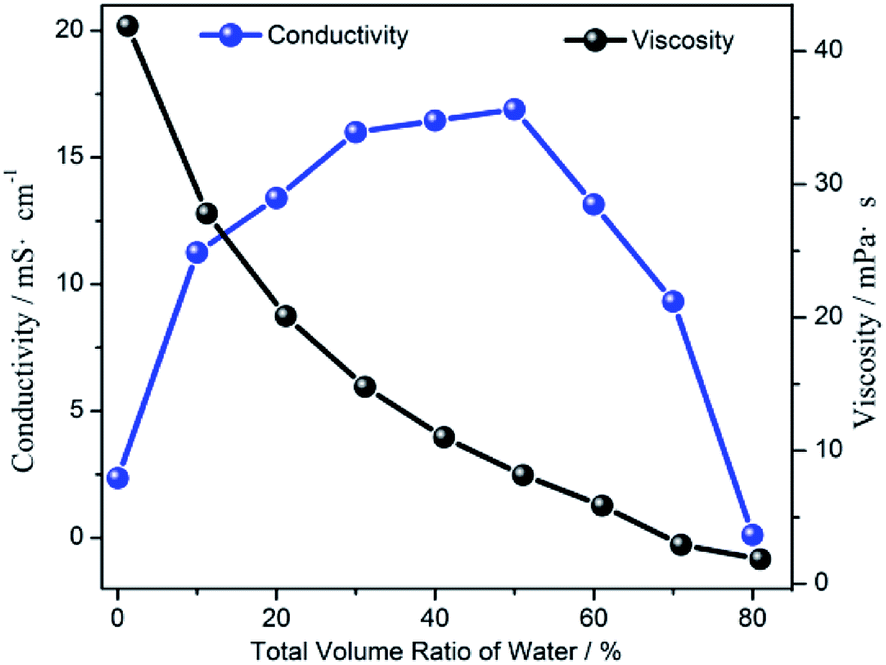 | ||
| Fig. 3 The viscosity and the electrical conductivity of the [Emim]BF4/H2O solution as a function of the water volume fraction. | ||
The spectrochemical method revealed a definite catalytic mechanism that may provide important insights for the further rational design of the catalytic system. Thus, a UV-Vis absorption measurement was conducted to collect the coordination information for the selected solvents. Initially, in order to evaluate the coordination capabilities of the metal center with solvent molecules, the experiments were tested in solutions by directly dissolving Co2+ (CoCl2) in an aqueous solution and [Emim]BF4, respectively.28 The UV-Vis absorption spectra (Fig. 4) obtained in both solutions generally exhibited two absorption bands. The absorption bands located within the ultraviolet region were attributed to the ligand π–π* transition and a red-shift likely from a metal-to-ligand charge transition (MLCT) band (Fig. 4A and B, black line).29 The absorption bands positioned in the visible region were due to the d–d transition (Fig. 4A and B, black line).30 A different absorption region was also presented in the spectra of an aqueous solution (410–570 nm, Fig. 4A, red line) and [Emim]BF4 solution (500–715 nm, Fig. 4B, red line). This observation indicated a different interaction between the ligand part and metal center in the current system when water and [Emim]BF4 were used as the reaction medium.
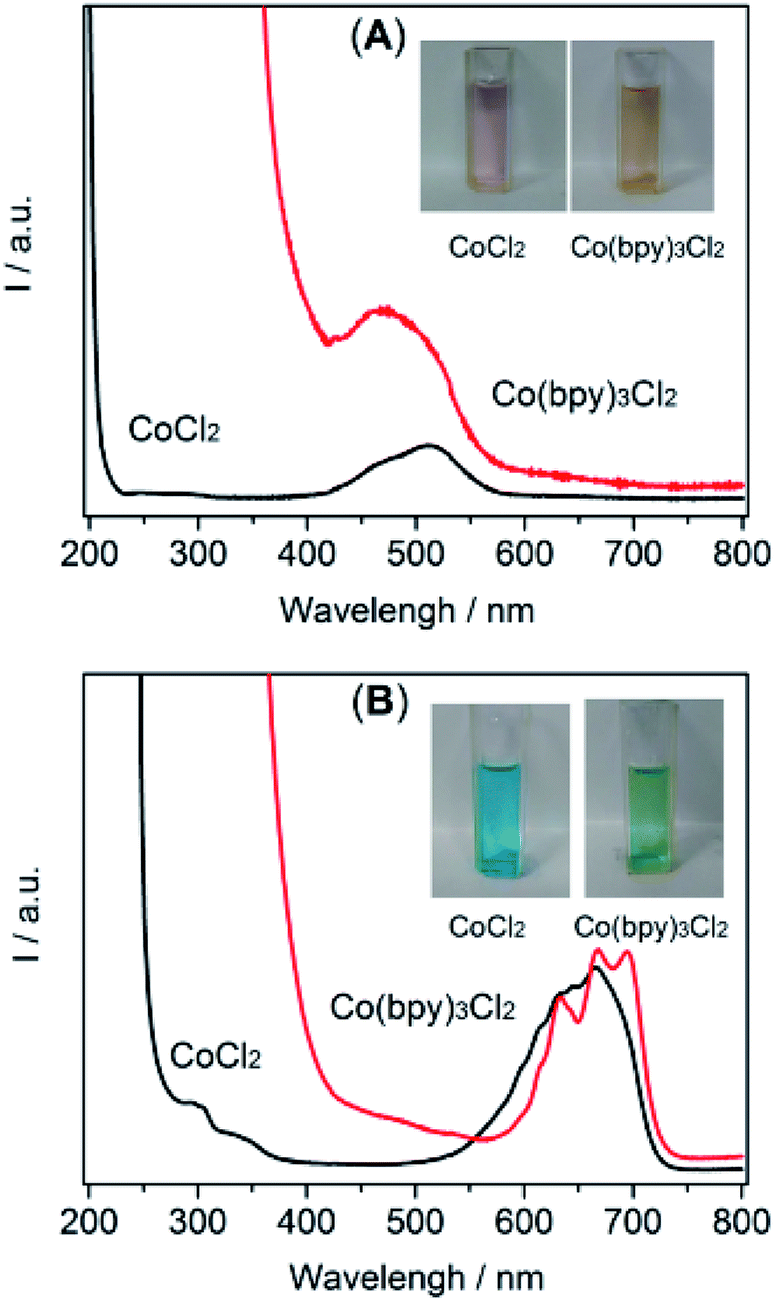 | ||
| Fig. 4 UV-Visible absorption spectra of CoCl2/Co(bpy)3Cl2 recorded in water (A) and [Emim]BF4 (B) solutions. (Inset shows the corresponding photograph of each sample). | ||
Fig. 4B displays the UV-Vis absorption spectra of the solution in the presence of bpy. As shown, after the introduction of bpy, the absorption position extending to the near-ultraviolet region experienced an obvious red-shift, highlighting the enhanced MLCT process. A new absorption (peaks at 450 nm) appeared in the CoCl2/bpy containing aqueous solution, while the absorption intensity at 410–570 nm was weakened. This change made the solution turn from red to an orange colour. In the [Emim]BF4 solution, a red-shift near 400 nm and an alternation in 550–725 nm appeared, which resulted in a colour change from blue to green. Compared with the solution without bpy, these results indicated that the cobalt center was favored to coordinate with bpy and form Co(bpy)3Cl2 in these two solvents. It also should be noted that there were different absorptions between the solutions of [Emim]BF4 and H2O. These changes were mainly induced by the alternation in the chemical nature of the solvent molecules such as the charge transfers between the solvent and metal ions, solvent dependent aggregation and complexation.31 In addition, the tertiary amine, TEOA, may have also contributed to the solvent effect in the photocatalytic CO2 reduction systems by surviving as a ligand to coordinate with the cobalt center. The results of the UV-Vis absorption characterizations demonstrated that the ligand (e.g., bpy) in the CO2 reduction system altered the charge distribution of the metal center and eventually affected the CO2 adsorption and the catalytic performance of the CO2 reduction system. In addition, the spectra also presented solid evidence of a coordination effect between the cobalt center and [Emim]BF4 or H2O.
Interestingly, before the photocatalytic reaction, all of the coordination sites for cobalt were occupied by three bipyridine molecules (six N atoms) and should not have been affected by the local environments. Therefore, a confirmation in the vacancy of the Co complex was further investigated. As demonstrated in Fig. 5, a new absorption range from 500–750 nm was observed during light irradiation, which suggested that Co(bpy)+ was formed.32 The orange suspension turned dark blue during irradiation. The color shift was probably related to Co(I) πd to bpy π* back-bonding in the Co(bpy)n+ series. In addition, the occurrence of a partial dissociation between cobalt and bpy generated the possibility of anchoring again with the surrounding solvent molecules. This spectroscopic result provided evidence in support of the speculates that coordinated between the Co(bpy)+ complexes and solvents.
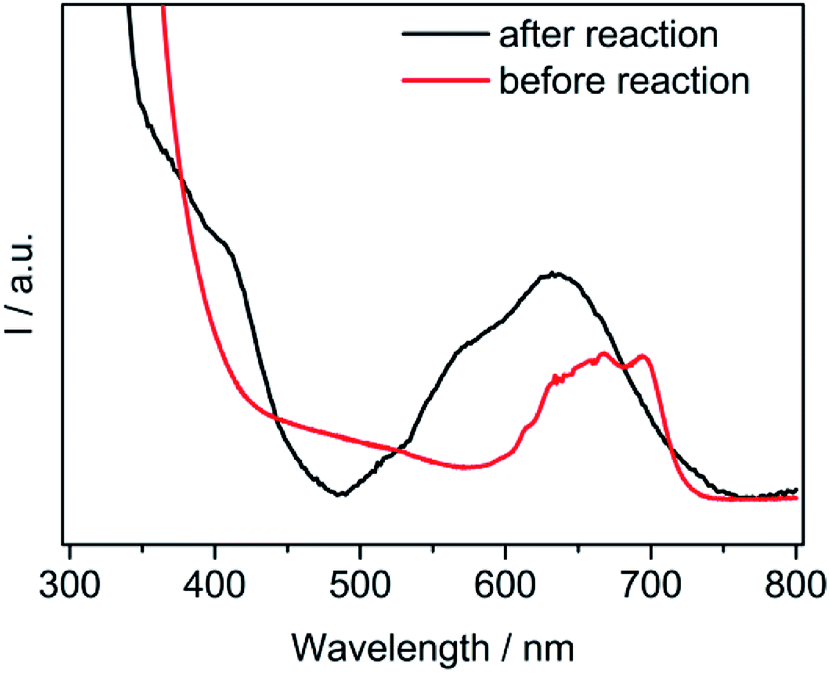 | ||
| Fig. 5 Absorption spectra of the Co(bpy)2+ complexes (before reaction) and Co(bpy)+ complexes (after reaction). The ingredients in the solution were the same as the reaction with 10 vol% H2O. | ||
In fact, the redox process for the Co-complex was previously inspected by electrochemical experiments.33 Those reports indicated an existence of CoII/I and CoI/0 redox pairs during cyclic voltammetry (CV) scans. The generation of Co(I) species subsequently served as an important activated catalyst for CO2. Further observations verified the binding of CO2 with the Co(I) species to give a [Co–CO2]− adduct. It is well known that the electrochemical behaviors are strongly influenced by surrounding factors. Ionic liquids such as [Emim]BF4 can greatly lower the over-potentials of the CO2 reduction.24 It happened that there was a similar case. The electrocatalysts were able to facilitate the proton-coupled multi-electron reactions, which required lower potentials than that for the single-electron reaction occurring at −1.9 V. These results indicated that the ingredients water and [Emim]BF4 in a proper proportion simultaneously exhibited a synergistic effect for the catalytic CO2 reduction.
Hence, the mechanism for the catalytic CO2 reduction by a CdS/Co(bpy)32+ hybrid catalytic system was described in previous reports.34 CdS liberated the electrons upon light irradiation. The Co(II) complex with bipyridinium, an electron-withdrawing anchoring group, easily grasped the electrons and then turned into an active Co(I) intermediate.35 At this stage, one of the functions of water was that of a diluting agent, which lowered the viscosity of the reaction medium. It promoted the mass transfer process by enhancing the mobility, which was directly recorded by the high conductivity (Fig. 2).
Next, as confirmed in Fig. 5, one bipyrimidine ligand left the metal center. Therefore, the molecular compounds ([Emim]+, H2O, TEOA) around the complex may have come into contact with the metal centers and functioned as a stabilizer for a highly unstable intermediate (Fig. 4). A reasonable example in a cobaltic system was reported that involved an ionic liquid with an imidazole-group function, serving as a stabilizer of the CoI species.36 The reductive cobalt intermediate allowed for a strong interaction with CO2 to form a metal carbonate. It should be noted that [Emim]BF4 with a high CO2 solubility assisted the combination rate for CO2 and cobalt. On the other hand, many proton sources dissociated from water. Therefore, the protonation promoted the cleavage of the C–O bond in the [Co–CO2]− adduct and resulted in the yield of CO.37 The H2 evolution with a combination of a Co(I) intermediate with protons to form the Co(III)-hydride was probably similar to the CO2 conversion.38 During the catalytic processes, the generation and stability of the active species was greatly enhanced by the favorable chemical environments of the reaction medium.39 More specifically, the coordination ability of [Emim]BF4 contributed to this process, and thereafter promoted the final CO2 conversion. As expected, the process for the charge transfer between the cobalt ion and coordinated CO2 was inevitably influenced by the nature of [Emim]BF4.
4 Conclusions
In summary, a heterocatalytic system was mediated by [Emim]BF4/H2O/TEOA for the selective reduction of CO2. The influence factors for the catalytic performance were characterized in detail. (i) The role of water was that of a dilution agent to adjust the viscosity of the current system and that of a proton source to assist the proton-coupling process, which was responsible for the CO2 conversion. (ii) The activity and selectivity were closely related to the viscosity and conductivity. (iii) The coordination between the metal center and [Emim]+ may have promoted the stability of the CO2 intermediates. Therefore, all of these results were helpful for studying a broader range of the green system, developing a new, efficient, artificial photosynthesis system, better understanding composition–activity relationships and optimizing this system.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was sponsored by the Natural Science Foundation of Guizhou Province Education Department ([2018]311) and the Natural Science Foundation of Guizhou Province ([2016]7011 & [2017]5727-11).Notes and references
- M. He, Y. Sun and B. Han, Angew. Chem., Int. Ed., 2013, 52, 9620–9633 CrossRef CAS.
- I. Yasuo, Coord. Chem. Rev., 2013, 257, 171–186 CrossRef.
- W. L. Dai, L. Chen, S. F. Yin, W. H. Li, Y. Y. Zhang, S. L. Luo and C. T. Au, Catal. Lett., 2010, 137, 74–80 CrossRef CAS.
- X. Shi, Z. Q. Han, X. L. Peng, P. Richard, T. Qian, X. X. Wu, M. W. Qiu, S. C. Wang, J. P. Hu, Y. J. Sun and H. Ding, Nat. Chem., 2017, 9, 1019 CrossRef.
- L. Q. He, H. Yang, J. J. Huang, X. H. Lu, G. R. Li, X. Q. Liu, P. P. Fang and Y. X. Tong, RSC Adv., 2019, 9, 10168–10173 RSC.
- J. L. Lin, Z. M. Pan and X. C. Wang, ACS Sustainable Chem. Eng., 2013, 2(3), 353–358 CrossRef.
- L. Yuan and Y. J. Xu, Appl. Surf. Sci., 2015, 342, 154–167 CrossRef CAS.
- J. Wei, Q. Ge, R. Yao, Z. Wen, C. Fang, L. Guo, H. Xu and J. Sun, Nat. Commun., 2017, 8, 15174 CrossRef PubMed.
- H. Kawanami, A. Sasaki, K. Matsui and Y. Ikushima, Chem. Commun., 2003, 34(31), 896–897 RSC.
- C. Guan, Y. P. Pan, E. P. L. Ang, J. S. Hu, C. G. Yao, M. H. Huang, H. F. Li, Z. P. Lai and K. W. Huang, Green Chem., 2018, 20, 4201–4205 RSC.
- R. Snoeckx and A. Bogaerts, Chem. Soc. Rev., 2017, 46, 5805–5863 RSC.
- M. Jitaru, J. Univ. Chem. Technol. Metall., 2007, 42(4), 333–344 CAS.
- J. L. Lin, Z. X. Ding, Y. D. Hou and X. C. Wang, Sci. Rep., 2013, 3, 1056 CrossRef.
- S. Bazhenov, M. Ramdin, A. Volkov, V. Volkov, T. J. H. Vlugt and T. W. D. Loos, J. Chem. Eng. Data, 2014, 59(3), 702–708 CrossRef CAS.
- J. P. Feng, S. J. Zeng, J. Q. Feng, H. F. Dong and X. P. Zhang, Chin. J. Chem., 2018, 36(10), 961–970 CrossRef CAS.
- J. P. Feng, S. J. Zeng, H. Z. Liu, J. Q. Feng, H. S. Gao, L. Bai, H. F. Dong, S. J. Zhang and X. P. Zhang, ChemSusChem, 2018, 11(18), 3191–3197 CrossRef CAS.
- Y. L. Gu, Q. H. Zhang, Z. Y. Duan, J. Zhang, S. G. Zhang and Y. Q. Deng, J. Org. Chem., 2005, 70(18), 7376–7380 CrossRef CAS.
- D. C. Grills, Y. Matsubara, Y. Kuwahara, S. R. Golisz, D. A. Kurtz and B. A. Mello, J. Phys. Chem. Lett., 2014, 5, 2033–2038 CrossRef CAS.
- Y. Oh and X. L. Hu, Chem. Soc. Rev., 2013, 42, 2253–2261 RSC.
- B. C. M. Martindale and R. G. Compton, Chem. Commun., 2012, 48, 6487–6489 RSC.
- D. Zhao, M. Wu, Y. Kou and E. Min, Catal. Today, 2002, 74, 157–189 CrossRef CAS.
- M. Galinski, A. Lewandowski and I. Stepniak, Electrochim. Acta, 2006, 51, 5567–5580 CrossRef CAS.
- S. M. Xia, K. L. Chen, C. H. Fu and L. N. He, Front. Chem., 2018, 6, 468 CrossRef.
- B. A. Rosen, A. S. Khojin, M. R. Thorson, W. Zhu, D. T. Whipple, P. A. Kenis and R. I. Masel, Science, 2011, 334(6056), 643–644 CrossRef CAS.
- D. W. Yang, Q. Y. Li, F. X. Shen, Q. Wang, L. Li, N. Song, Y. N. Dai and J. Shi, Electrochim. Acta, 2016, 189, 32–37 CrossRef CAS.
- T. N. Huan, P. Simon, G. Rousse, I. Génois and V. A. M. Fontecave, Chem. Sci., 2017, 8(1), 742–747 RSC.
- Y. S. Zhao, X. P. Zhang, S. J. Zeng, Q. Zhou, H. F. Dong, X. Tian and S. J. Zhang, J. Chem. Eng. Data, 2010, 55(9), 3513–3519 CrossRef CAS.
- P. F. Zhang, Y. T. Gong, Y. Q. Lv, Y. Guo, Y. Wang, C. M. Wang and H. R. Li, Chem. Commun., 2012, 48, 2334–2336 RSC.
- T. Shimoda, T. Morishima, K. Kodama, T. Hirose, D. E. Polyansky, G. F. Manbeck, J. T. Muckerman and E. Fujita, Inorg. Chem., 2018, 57, 5486–5498 CrossRef CAS.
- K. Sivaraj and K. P. Elango, J. Solution Chem., 2010, 39, 1681–1697 CrossRef CAS.
- G. Goutam and G. Suhrit, Chem. Commun., 2018, 54, 5720–5723 RSC.
- H. A. Schwarz, C. Creutz and N. Sutin, Inorg. Chem., 1985, 24(3), 433–439 CrossRef CAS.
- N. V. Rees and R. G. Compton, Energy Environ. Sci., 2011, 4, 403–408 RSC.
- Z. G. Chai, Q. Li and D. S. Xu, RSC Adv., 2014, 4, 44991–44995 RSC.
- M. Cheng, X. Yang, J. Li, C. Chen, J. Zhao, Y. Wang and L. Sun, Chem.–Eur. J., 2012, 18(50), 16196–16202 CrossRef CAS.
- Y. Hori, B. H. Wake, T. Tsukamoto and O. Koga, Electrochim. Acta, 1994, 39, 1833–1839 CrossRef CAS.
- Y. A. Daza and J. N. Kuhn, RSC Adv., 2016, 6(55), 49675–49691 RSC.
- S. C. Marinescu, J. R. Winkler and H. B. Gray, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15127–15131 CrossRef CAS.
- J. L. Lin, B. Qin and G. L. Zhao, J. Photochem. Photobiol., A, 2018, 354, 181–186 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |