
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMussel-inspired immobilization of Au on bare and graphene-wrapped Ni nanoparticles toward highly efficient and easily recyclable catalysts†
Fatima
Mahnaz
ab,
Mohammad
Mostafa-Al-Momin
a,
Md.
Rubel
a,
Md.
Ferdous
a and
Md. Shafiul
Azam
 *a
*a
aDepartment of Chemistry, Bangladesh University of Engineering and Technology (BUET), Dhaka 1000, Bangladesh. E-mail: mdshafiulazam@chem.buet.ac.bd
bDepartment of Chemical Engineering, Bangladesh University of Engineering and Technology (BUET), Dhaka 1000, Bangladesh
First published on 25th September 2019
Abstract
Bimetallic nanocatalysts have been gaining huge research attention in the heterogeneous catalysis community recently owing to their tunable properties and multifunctional characteristics. In this work, we fabricated a bimetallic core–shell nanocomposite catalyst by employing a mussel-inspired strategy for immobilizing gold nanoparticles (AuNP) on the surface of nickel nanoparticles (NiNP). NiNPs obtained from the reduction of Ni(II) were first coated with polydopamine to provide the anchoring sites towards the robust immobilization of AuNPs. The as-synthesized nanocomposite (Ni–PD–Au) exhibited outstanding catalytic activity while reducing methylene blue (MB) and 4-nitrophenol (4-NP) yielding rate constants 13.11 min−1 and 4.21 min−1, respectively, outperforming the catalytic efficiency of its monometallic counterparts and other similar reported catalysts by large margins. The superior catalytic efficiency of the Ni–PD–Au was attributed to the well-known synergistic effect, which was experimentally investigated and compared with prior reports. Similar bio-inspired immobilization of AuNPs was also applied on graphene-wrapped NiNPs (Ni-G) instead of bare NiNPs to synthesize another composite catalyst (Ni-G–PD–Au), which yet again exhibited synergistic catalytic activity. A comparative study between the two nanocomposites suggested that Ni–PD–Au excelled in catalytic activity but Ni-G–PD–Au provided noteworthy stability showing ∼100% efficiency over 17 repeated cycles. However, along with excellent synergistic performance, both nanocomposites demonstrated high magnetization and thermal stability up to 350 °C ascertaining their easy separation and sustainability for high-temperature applications, respectively.
Introduction
With the advancement of nanotechnology, multifunctional nanomaterials have received great attention in recent years owing to their unique ability to combine inorganic and organic moieties in a single assembly. Tuning and choosing the right combination of materials can significantly improve the already existing properties as well as introduce new functionality into the system. Among these materials, bimetallic nanocatalysts particularly draw greater attention due to their technological and scientific features for improving catalytic properties.1,2 For conventional heterogeneous catalysis, bimetallic catalysts often show synergistic effects with greater efficiency compared to their monometallic counterparts. Synergistic catalysis can also trigger chemical transformations that were otherwise unachievable, increase the effectiveness of existing transformations by causing a greater decrease in the activation energy, and create or improve catalytic enantioselectivity where stereocontrol was previously challenging.3,4 Consequently, various bimetallic nanomaterials consisting of noble metals such as Au, Ag, Pd, and Pt have been reported focusing on the control achieved over the catalytic properties, composition, and morphology.Numerous investigations were performed earlier for the synthesis of different bimetallic nanoparticles. For example, reduced graphene oxide (rGO) nanosheets decorated with Au–Pd bimetallic nanoparticles were synthesized and applied for photocatalytic degradation of phenolic compounds where the presence of Pd synergistically enhanced the catalytic ability of Au.5 Bimetallic Pt@Ag and Pd@Ag core–shell nanoparticles were also reported for exhibiting more catalytic activity than their corresponding monometallic nanoparticles while reducing Congo red.6 Again, Au@Ag core–shell bimetallic nanoparticles immobilized on electrospun TiO2 nanofiber was reported to be more efficient photocatalyst for the degradation of organic compounds compared to Au itself.7 Although bimetallic nanostructures in these cases provided better performance, separation and reuse of these expensive bimetallic catalysts have been a great concern for their effectiveness in industrial applications. Time-consuming separation processes such as filtration or centrifugation are usually performed to separate these catalysts from the reaction systems which often fail to isolate such small nanoparticles. Consequently, there has been an unfortunate declining trend towards the usage of these noble metals due to their inadequacy and high cost. The use of magnetic NPs as support materials in nanocomposites has particularly drawn attention as they can solve the separation problem as well as can combine the advantages of high dispersion in a liquid reaction system.
Fe3O4 has been used as the supporting material as well as the magnetic component in many adsorbent and catalytic nanomaterials.8–11 Various studies corroborated the potential of Fe3O4 to produce magnetically recoverable bimetallic nanoparticles. For instance, Fe3O4@Au–Pd hybrid nanoparticles exhibited good reusability towards the reduction of nitrophenol and potassium hexacyanoferrate.9 Magnetically responsive Fe3O4@polyaniline@Au hybrid nanocomposites were used for Congo red reduction up to five consecutive cycles.10 However, the issue with F3O4 is that they possess either no contribution or very little contribution to the overall catalytic activity of the composite materials. On the contrary, nickel nanoparticles have great potential to be used as the magnetic component in nanocomposites due to their ferromagnetic nature as well as they can enhance their catalytic activity at the same time. Copious studies evinced the improved catalytic activity of Ni-based bimetallic nanoparticles in different heterogeneous catalysis application.12–14
The aim of our work was to synthesize a bimetallic multifunctional nanocomposite incorporating gold and nickel nanoparticles in a single construct. AuNP was chosen as it has remarkable catalytic efficiency and has been used profusely in prior studies.15–19 Using NiNP in the nanocomposite provide means for easy magnetic separation and might also enhance the catalytic activity. For the fabrication of these nanocomposites, two core–shell structures were synthesized using bare NiNPs and graphene-coated NiNPs (Ni-G) as the core and polydopamine (PD) as the shell. Core–shell nanoparticles have been drawing noteworthy attention in recent years, as the shell material coating can modify the properties of the core particle such as decreasing reactivity or increasing stability, thus particles altogether exhibit distinctive properties.20 Polydopamine was used as the shell material as it has the ability to adhere to almost all material surfaces and provides a biocompatible and facile mean for the on-surface synthesis of metallic NPs. The presence of numerous amine and catechol functional groups in PD enables it to reduce the noble metallic salts and successively stabilize the produced metallic NPs.21–23 Herein, AuNPs were deposited on the surface of pre-synthesized core–shell structured Ni–PD and Ni-G–PD composite using PD as the organic linker. In the case of Ni-G–PD–Au nanocomposite, multilayered graphene was created on the surface of NiNP to separate the reactive NiNP from the outer system in order to render better stability and adsorption capacity. In this article, the fabrication method, catalytic activity, thermal stability, magnetic property, and recyclability were studied for both Ni–PD–Au and Ni-G–PD–Au nanocomposites. Additionally, the effect of introducing a graphene layer in the nanocomposite was studied in terms of catalytic activity, stability, and recyclability. Dramatic enhancement in the catalytic activity of the as-synthesized catalysts compared to their monometallic counterparts was explained in terms of synergistic effects.
Experimental
Materials
Hydrazine monohydrate (N2H4·H2O) (98%), nickel chloride hexahydrate (NiCl2·6H2O) (99.99%), methylene blue (95%), tri-ethylene glycol (TEG) (99%) and dopamine hydrochloride (98%) were purchased from Sigma-Aldrich. Sodium borohydride (98%) was obtained from Daejung Chemical & Metals. 4-Nitrophenol (98%) and sodium hydroxide (98%) were purchased from Loba Chemie, chloroauric acid (>47.8% gold basis) was bought from Shanghai Bojing Chemical. The chemicals and reagents were analytical grade and used without further purification. Milli-Q water was used as a solvent for preparing the solutions unless otherwise notified.Synthesis of nickel nanoparticles (NiNPs)
For synthesizing NiNPs, nickel chloride, hydrazine, and sodium hydroxide were used as the source materials. To control the size of the nanoparticles, a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :12:12 (NiCl2:N2H4:NaOH) was taken for the reaction. In the first step of synthesis, NiCl2·6H2O (2.4 g) was dissolved in water (3 mL) and added to 80% N2H4·H2O (7.6 g) in a 100 cm3 round bottom flask with vigorous stirring. A pale violet precipitate formed on the wall of the round bottom flask, which was washed with an additional 0.75 mL of distilled water for about 60 s. The solution temperature increased up to 65 °C due to the formation of a Ni complex between NiCl2 and N2H4. The solution was then allowed to cool down to 50 °C at an ambient atmosphere. An aqueous solution of NaOH (50%, 9.75 mL) was added instantaneously into the stock solution. The temperature of the solution increased spontaneously up to the second maximum of 54 °C due to the formation of Ni(OH)2. Finally, after 1 h, a black precipitate of NiNPs formed which was washed thoroughly several times with distilled water and dried at room temperature.24
:12:12 (NiCl2:N2H4:NaOH) was taken for the reaction. In the first step of synthesis, NiCl2·6H2O (2.4 g) was dissolved in water (3 mL) and added to 80% N2H4·H2O (7.6 g) in a 100 cm3 round bottom flask with vigorous stirring. A pale violet precipitate formed on the wall of the round bottom flask, which was washed with an additional 0.75 mL of distilled water for about 60 s. The solution temperature increased up to 65 °C due to the formation of a Ni complex between NiCl2 and N2H4. The solution was then allowed to cool down to 50 °C at an ambient atmosphere. An aqueous solution of NaOH (50%, 9.75 mL) was added instantaneously into the stock solution. The temperature of the solution increased spontaneously up to the second maximum of 54 °C due to the formation of Ni(OH)2. Finally, after 1 h, a black precipitate of NiNPs formed which was washed thoroughly several times with distilled water and dried at room temperature.24
Synthesis of Ni–PD–Au
To coat the surface of NiNPs with polydopamine, dopamine hydrochloride (0.05 g) and the prepared NiNPs (0.15 g) were dispersed in 70 mL of Tris buffer (pH = 8.5). The mixture was stirred at 60 °C for 24 hours.25 The prepared polydopamine coated NiNPs (Ni–PD) were washed with distilled water thoroughly. Next, gold nanoparticles (AuNPs) were deposited on the as-synthesized Ni–PD. Firstly, an aqueous solution of HAuCl4 (0.022 mol dm−3) was prepared by dissolving the salt in a certain volume of Milli-Q water. HAuCl4 (4.2 mL) solution was then added to water (120 mL) in a round bottom flask and charged with Ni–PD (150 mg). The reaction mixture was then placed in a dark place and stirred for 24 hours to allow the deposition of AuNPs onto the surface of Ni–PD.26Synthesis of Ni-G–PD–Au
NiNPs (1 g) were added to 80 mL solution of triethylene glycol (TEG). After that, 4 mL of aqueous NaOH solution (2 M) was added to the mixture. The solution with NiNPs was transferred into a Teflon lined autoclave and was allowed to keep at a temperature of 250 °C for 12 hours that finally led to the formation of carbon-incorporated NiNPs.27 The carbon-incorporated NiNPs were then annealed in a furnace under nitrogen atmosphere at 500 °C resulting in multilayer graphene (MLG)-encapsulated NiNPs.27 Dopamine (0.05 g) and Ni-G (0.15 g) were dispersed in 70 mL of Tris buffer (pH = 8.5). The mixture was stirred at 60 °C for 24 hours.25 The prepared Ni-G–PD was washed with distilled water for several times. Gold nanoparticles (AuNPs) were deposited on the as-synthesized Ni-G–PD by following a procedure published elsewhere. Briefly, an aqueous solution of HAuCl4 (0.022 mol dm−3) was first prepared by dissolving the salt in a certain volume of Milli-Q water. HAuCl4 solution (4.2 mL) was then added to 120 mL of water in a round bottom flask and charged with 150 mg of Ni-G–PD. The reaction mixture was then stirred for 24 hours in the absence of light to achieve uniform deposition of AuNPs on Ni-G–PD.26,28Synthesis of PD–Au
PD–Au was obtained by treating Ni–PD–Au (12 mg) with hydrochloric acid (5 mL, 0.25 M) in a 100 mL round bottom flask. The mixture was shaken for 24 hours using an orbital shaker at 100 rpm. The solution on the round bottom flask became pale green as the NiNPs got dissolved in the solution.27 The loss of magnetic property of the composite at the end suggested the complete etching of nickel. The nickel etching was repeated with another solution of hydrochloric acid (5 mL, 0.1 M) to confirm the complete removal of Ni from the Ni–PD–Au. The black precipitate of PD–Au nanocomposite was washed with distilled water (6 × 20 mL).Catalytic activity test
The catalytic activity of the Ni–PD, PD–Au, Ni–PD–Au and Ni-G–PD–Au nanocomposites was investigated for the reduction of two most commonly studied model organic pollutants, MB and 4-NP by a strong reducing agent, sodium borohydride (NaBH4). For the reduction of MB, a freshly prepared solution of NaBH4 (0.5 mL, 0.1 M) was added to an aqueous solution of MB (4.5 mL, 30 ppm). An accurately weighed amount of the nanocomposite (3.0 mg) was immediately transferred to the reaction mixture and the absorption spectra were recorded employing a UV-visible spectrophotometer (Shimadzu UV-1800). The absorbance of the principal absorption band of MB appeared at 664 nm was monitored as a function of time on the recorded UV-visible spectra. The reduction of 4-NP was studied similarly. Briefly, NaBH4 solution (2 mL, 0.1 M) was first mixed with a 4-NP solution (3 mL, 0.1 mM). The addition of NaBH4 immediately caused the formation of 4-nitrophenolate ions (4-NPL) resulting in a shift in the characteristic absorption peak from 317 nm (for 4-NP) to 400 nm (for 4-NPL). The resultant solution was then charged with the nanocomposites (3 mg) and the absorption spectra were recorded at different time intervals. However, for determining the rate constants for fast reactions, we allowed the UV-vis spectrophotometer to measure the absorbance only at the respective λmax (400 nm for 4-NP, and 664 nm for MB) at short intervals.Results and discussion
Fig. 1(a) shows the schematic illustration of the steps involved in synthesizing Ni–PD–Au nanocomposite catalyst. Spherical metallic NiNPs were first synthesized by the reduction of nickel(II) chloride with hydrazine hydroxide in the presence of sodium hydroxide. A Ni complex formed by the reaction between nickel(II) chloride and hydrazine solution and the addition of sodium hydroxide solution triggered series of reactions from the decomposition of the Ni complex via the formation of Ni(OH)2 and finally reducing the Ni(OH)2 gel into spherical NiNPs.24 To employ AuNPs on the surface of NiNPs, a metal–organic framework was produced in the form of polydopamine via the self-polymerization of dopamine.25 The gold nanoparticles were then loaded on the composite by the reduction of Au(III) solution and in situ deposition of AuNPs on the surface of Ni–PD. In this reaction, Au3+ ions are believed to diffuse into the PD layer from HAuCl4 solution and then are reduced to Au0 atoms by the catechol groups of PD causing in situ deposition of AuNPs.26 The catechol groups in PD are oxidized to corresponding quinones simultaneously. PD serves both as a reducer and a stabilizer which can help to prevent the AuNPs from agglomeration.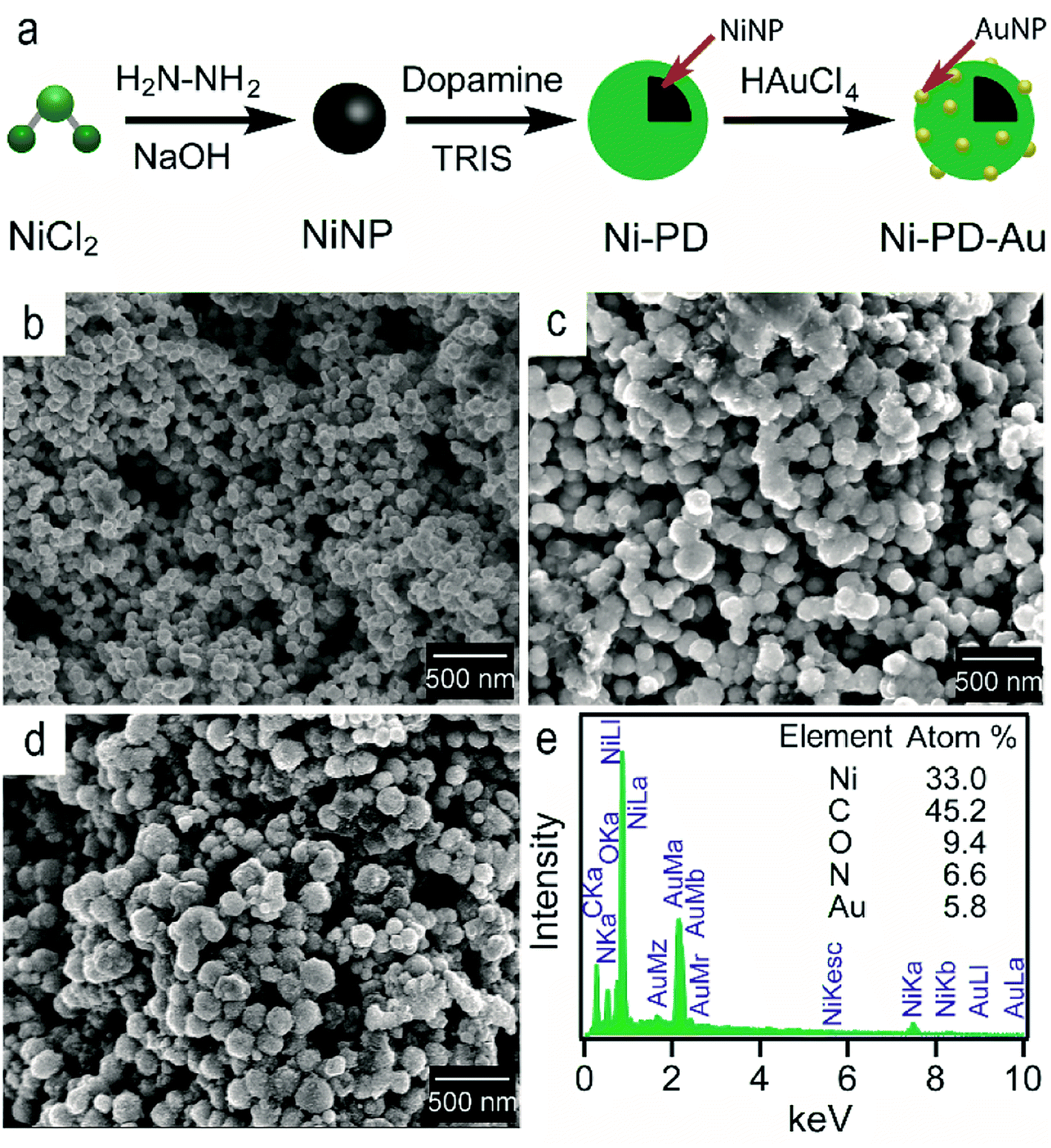 | ||
| Fig. 1 (a) The schematic representation of the synthesis of Ni–PD–Au nanocomposite from NiCl2 (b) SEM image of NiNPs (c) SEM image for Ni–PD (d) SEM image for Ni–PD–Au (e) EDS analysis for Ni–PD–Au. | ||
The surface morphologies of the as-synthesized NiNP, Ni–PD and Ni–PD–Au nanocomposites were monitored by scanning electron micrographs. Fig. 1(b) shows spherical and monodispersed NiNPs having the average diameter of ∼90 nm, and Fig. 1(c) shows NiNPs coated with polydopamine (Ni–PD) having the average size of ∼170 nm. The increase in diameter (∼80 nm) of the particles suggested the formation of ∼40 nm thick PD coating on the NiNPs, which is consistent with the previous reports.28,29 Ni–PD–Au was obtained by mixing the particles with HAuCl4 solution where Au3+ ions from the solution diffused into the PD layer and were reduced to AuNPs by the catechol groups present in the surface of Ni–PD. Fig. 1(d) shows the SEM image of Ni–PD–Au having the average size of ∼200 nm. Uniformly distributed spherical nanoparticles with the smaller size (∼20 nm) as seen from the image confirm the presence of immobilized AuNPs on the surface (Fig. S1†).30,31 Elemental analysis of Ni–PD–Au was done by energy-dispersive X-ray spectroscopy (EDS) as shown in Fig. 1(e). The EDS pattern of Ni–PD–Au indicated the peaks at 0.277, 0.392, and 0.525 keV for K cell of C, N, and O, correspondingly. It also confirmed the presence of Ni and Au by showing peaks at 0.851 keV for L shell of Ni and 2.121 keV for M shell of Au. The elemental atomic percentage is shown in the inset table where the atomic percentage of C, N, and O attributes to the PD layer present in the nanocomposite. From the atomic percentage of Ni and Au it can be concluded that the atomic proportion of AuNPs is lesser than NiNPs (∼1:6) which was expected as very low concentrated (6 × 10−4 M) HAuCl4 was used in gold deposition reaction to produce very small AuNPs on the PD surface.
Fig. 2(a) shows the FTIR spectra of Ni–PD and Ni–PD–Au nanocomposites attributing the functional groups present as well as the binding interactions between the PD and AuNPs. In the spectrum of Ni–PD, the absorption band near 3450 cm−1 corresponds to the stretching vibrations of –OH and N–H bonds of catechol and indole groups present in PD. The strong peak at 1624 cm−1 belongs to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibration of the aromatic ring while the peaks at 1477 cm−1 and 1101 cm−1 correspond to CN and C–O bonds, respectively, confirming the presence of PD in the nanocomposite. The spectrum of Ni–PD–Au shows distinct characteristic bonds of PD similar to that appear in the spectrum of Ni–PD indicating that the reaction process of gold deposition did not change the chemical structure of PD. However, the intensity of the peaks decreases in the spectrum of Ni–PD–Au as the catechol groups present in PD were oxidized to quinone group to reduce Au3+ ion to metallic Au0.28,32
C stretching vibration of the aromatic ring while the peaks at 1477 cm−1 and 1101 cm−1 correspond to CN and C–O bonds, respectively, confirming the presence of PD in the nanocomposite. The spectrum of Ni–PD–Au shows distinct characteristic bonds of PD similar to that appear in the spectrum of Ni–PD indicating that the reaction process of gold deposition did not change the chemical structure of PD. However, the intensity of the peaks decreases in the spectrum of Ni–PD–Au as the catechol groups present in PD were oxidized to quinone group to reduce Au3+ ion to metallic Au0.28,32
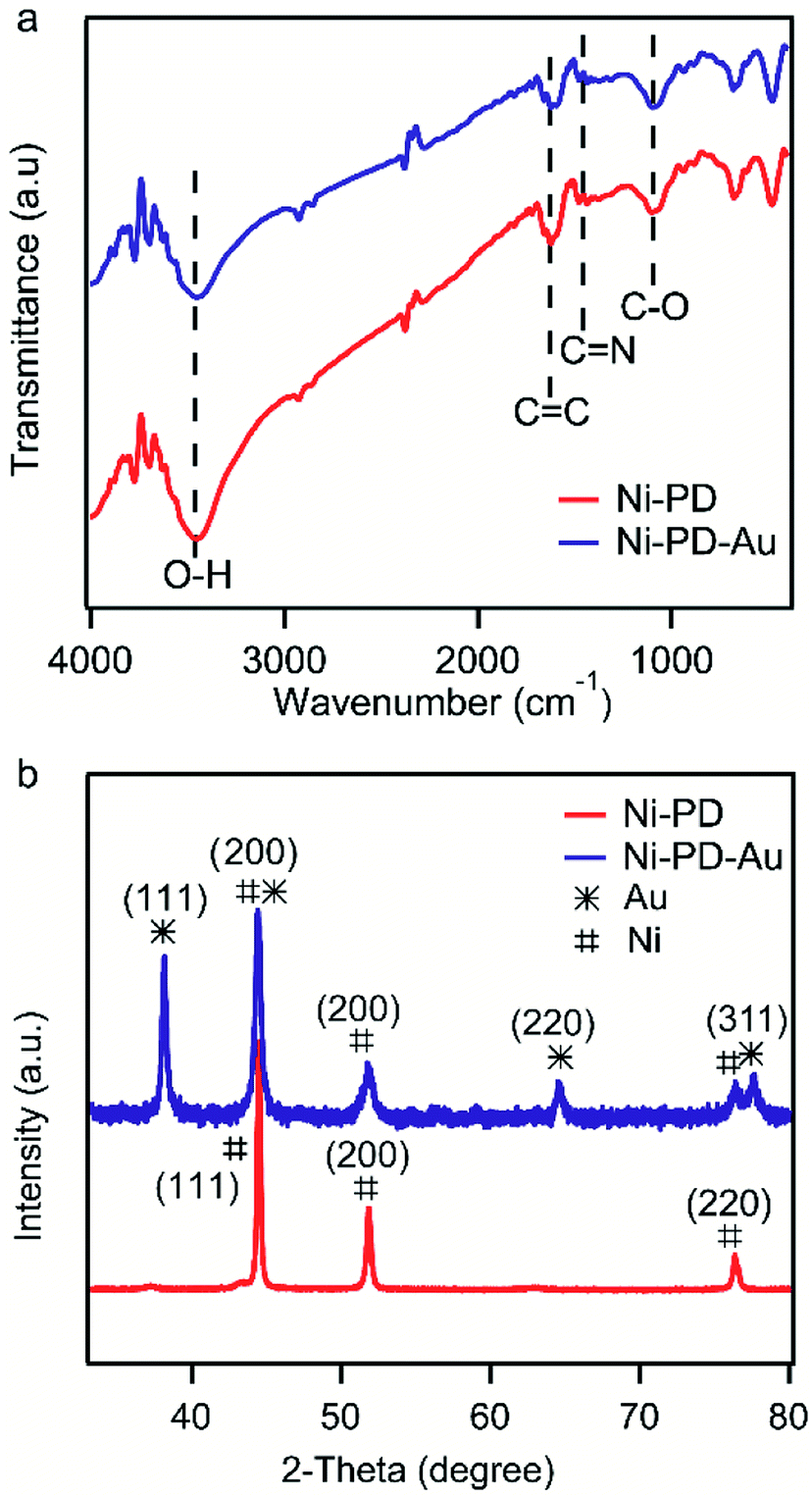 | ||
| Fig. 2 (a) FTIR spectra of Ni–PD and Ni–PD–Au (b) XRD pattern for Ni–PD and Ni–PD–Au. | ||
XPS measurements of Ni–PD–Au were carried out to further investigate the elemental composition, valence state of carbon as well as the oxidation state of the AuNPs. Fig. 3(a) exhibits the wide scan XPS spectra of Ni–PD–Au, which clearly shows the presence of C, O, Au, Ni, and N elements in the catalyst. The spectra were referenced first to the binding energy of the adventitious C 1s at 284.4 eV. The high-resolution C 1s spectra of Ni–PD–Au is shown in Fig. 3(b). Deconvolution of the C 1s spectra for PD-MCNT exhibited three peaks presenting CC/C–C (284.4 eV), C–N/C–OH (285.0 eV), and CO (288.1 eV). The appearance of C–N component in the C 1s spectra yet again confirms the formation of the PD layer on the surface of NiNPs.28 The presence of the CO component (288.1 eV) along with C–O (285.9 eV) indicates that a significant portion of the C–OH bonds of the catechol moieties of PD layer were oxidized to CO of quinone while reducing the Au3+ ions to AuNPs.28Fig. 3(c) presents the high-resolution spectrum of Au 4f. The binding energies at 84.0 and 87.6 eV are attributed to Au 4f7/2 and Au 4f5/2 of the metallic gold, confirming the zero oxidation state of the AuNPs deposited on the surface of the Ni–PD.28
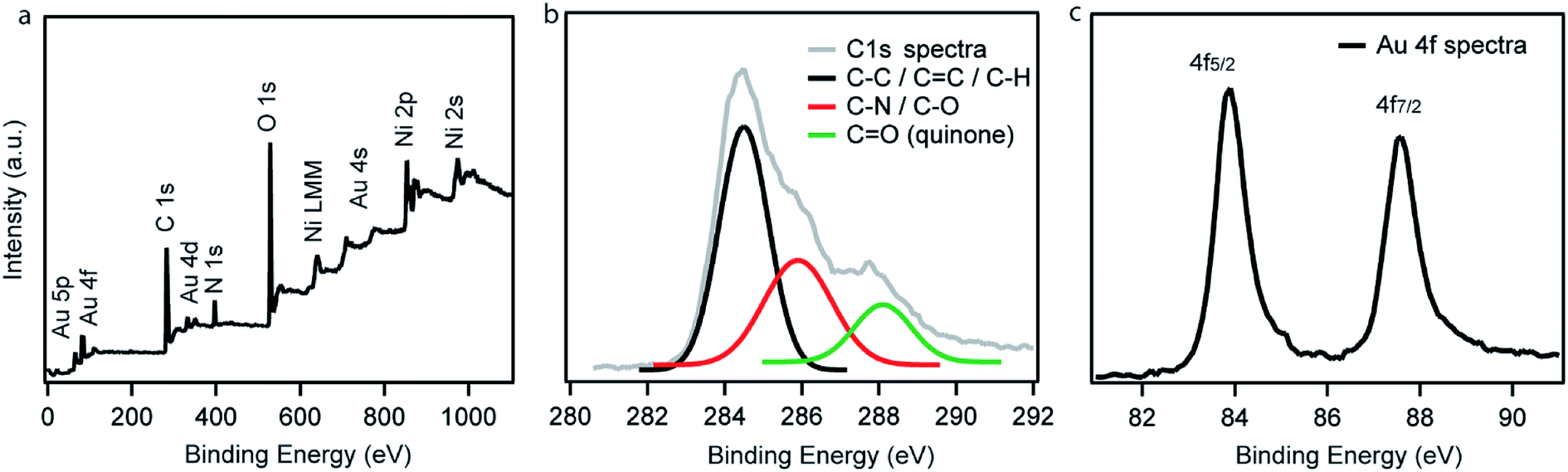 | ||
| Fig. 3 (a) Wide scan XPS spectrum, (b) high-resolution C 1s spectrum, and (c) high-resolution Au 4f spectrum of as-prepared Ni–PD–Au. | ||
The crystalline properties and phase identification of Ni–PD and Ni–PD–Au nanocomposites were then obtained by X-ray diffraction analysis. The red curve of Fig. 2(b) shows the characteristic three peaks found in Ni–PD sample. The peaks found for nickel have Bragg reflections at around 44.5°, 51.8° and 76.4° corresponding to (111), (200) and (220) lattice planes respectively which confirm the face-centered cubic phase of nickel in Ni–PD sample (JCPDS # 04-0850).33 The blue curve of Fig. 2 shows all the peaks found in Ni–PD–Au sample in which the characteristic peaks for gold are found at 38°, 43°, 63° and 77.7° corresponding to (111), (200), (220) and (311) lattice planes. These peaks are similar to face-centered cubic (FCC) phase of gold (JCPDS # 04-0783). The other peaks presented by the blue curve are due to the presence of nickel in the nanocomposite which infers that there was no change in the crystalline structure of the nickel in the core of the nanocomposite due to the in situ deposition reaction of AuNPs on the surface. XRD data were also used to estimate the size of AuNPs using Scherrer's equation and the average diameter of AuNPs was found to be ∼22 nm (see Table S1†), demonstrating a good agreement with the size determined from the SEM image.
The catalytic performances of the as-synthesized Ni–PD, PD–Au and Ni–PD–Au nanocomposites were evaluated by monitoring the reduction reactions of MB and 4-NP by NaBH4. In the presence of excess NaBH4, these reactions follow pseudo first order reaction kinetics. The reduction of organic dyes using aqueous NaBH4 is thermodynamically favorable, but kinetically not feasible due to the large potential difference between donor and acceptor molecules. Presence of metal NPs accelerates the reaction by facilitating electron relay from the donor BH4− to acceptor dye molecules and thereby overcome the kinetic barrier. Fig. 4 shows the UV-visible spectra for catalytic reduction of MB in the presence of Ni–PD, PD–Au, and Ni–PD–Au nanocomposites. The absorbance bands show that the reaction was very sluggish in case of Ni–PD and a substantial amount of MB (∼61%) retained in the solution even after 10 minutes of reaction (Fig. 4(a)). The catalytic performance of Ni–PD found in this work is similar to the activity of Ni catalyst found in prior investigations. Previously, Deka et al. showed that hetero-nanostructured Ni/α-Mn2O3 catalyst took about 15 to 60 minutes to catalytically degrade different dyes such as, MB, rhodamine B, rose bengal, Congo red and methyl orange where reduction of MB took 60 minutes with ∼12% MB retained in the solution.34 Greater catalytic activity was shown by PD–Au (Fig. 4(b)) compared to Ni–PD as 24% MB retained in solution after an equal length of time. The catalytic performance of AuNP found here is analogous to prior investigations.15,35 Earlier, Li et al. conducted an experiment on catalytic reduction of 4-NP and MB using Au/graphene hydrogel where reduction of MB took 14 minutes with ∼12% MB remaining in the solution. However, we observed a dramatic enhancement in the catalytic performance of Ni–PD–Au where we combine NiNPs and AuNPs in core–shell nanostructures. In this case, a complete reduction of MB was achieved within only 20 s demonstrating the synergistic effects of Ni and Au present in Ni–PD–Au.
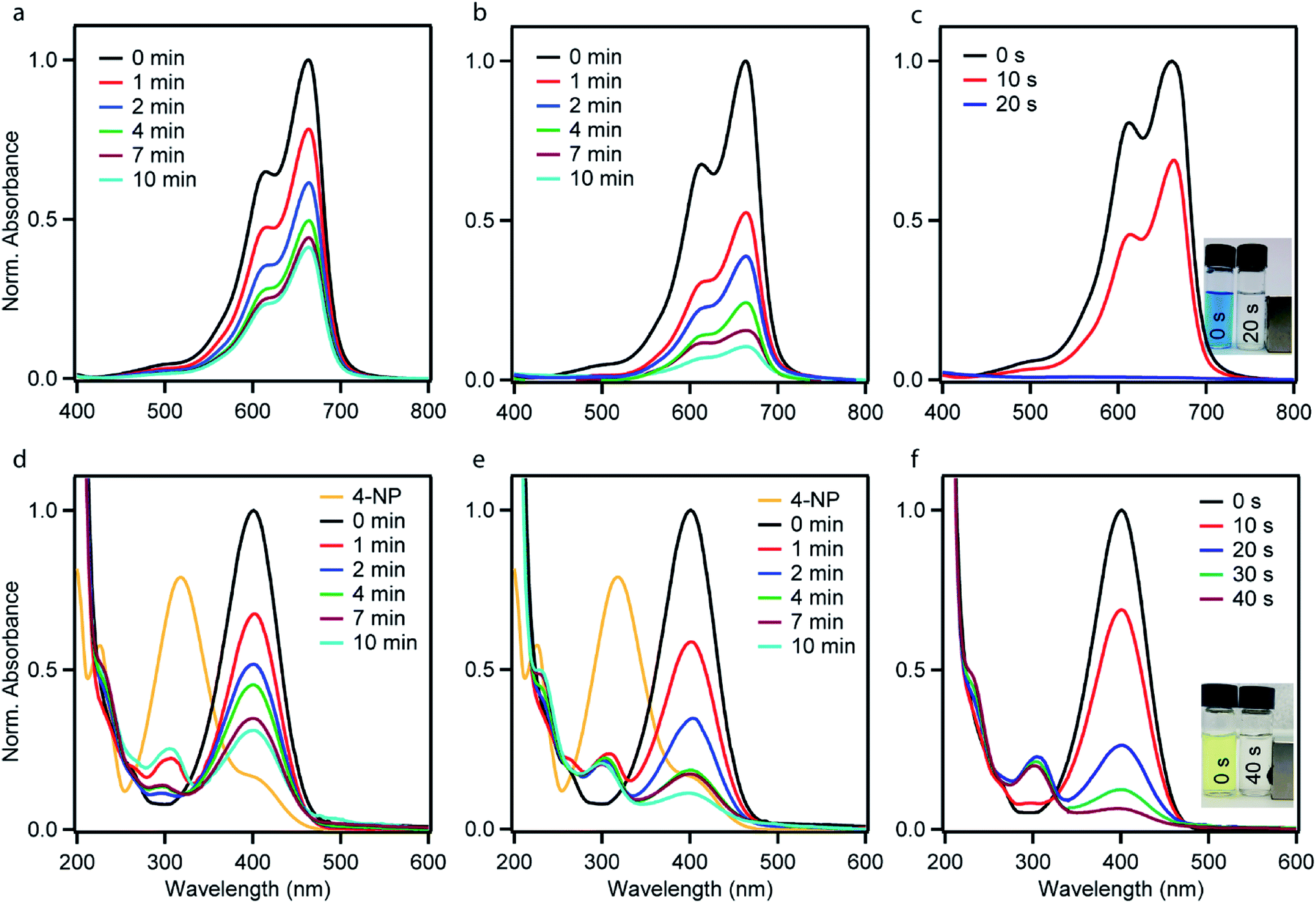 | ||
| Fig. 4 The representative UV-vis absorption spectra of an aqueous MB solution in the presence of NaBH4 using (a) Ni–PD (b) PD–Au (c) Ni–PD–Au as catalysts, and UV-vis absorption spectra of an aqueous 4-NP solution in the presence of NaBH4 using (d) Ni–PD (e) PD–Au (f) Ni–PD–Au as catalysts monitored as a function of time. | ||
In the catalytic reduction of 4-NP, the conversion of 4-NP into 4-AP occurs via the formation of an intermediate 4-nitrophenolate ion. 4-NP shows an absorbance peak at 317 nm which shifts to 400 nm due to the formation of 4-nitrophenolate ion (4-NPL) in the alkaline medium caused by NaBH4. Therefore, the progress of the reaction can be observed by monitoring the changes in the absorption spectra of 4-NPL at 400 nm. Fig. 4(d)–(f) depict successive absorption spectra of 4-NPL for Ni–PD, PD–Au, and Ni–PD–Au nanocomposites respectively. In each case, due to the addition of nanocomposite, a rapid decrease in the intensity of the absorption peak at 400 nm can be seen while there is a simultaneous appearance of a new peak at 300 nm due to the formation 4-AP. The reduction of 4-NP in presence of PD–Au (Fig. 4(e)) is faster than that with Ni–PD (Fig. 4(d)) indicating the higher catalytic activity of AuNP than NiNP, similar to what we observed for the reduction of MB. The Ni–PD–Au nanocomposite yet again exhibited the fastest catalytic activity by completely reducing 4-NP within 40 s as shown in Fig. 4(f), whereas the other two nanocomposites with either Ni or Au took more than 10 min for complete reduction. Thus the trend of catalytic performance of nanocomposites for reduction of 4-NP is similar to that found for MB as presumed.
To compare the rate constants for the nanocomposites, the linear relationships of absorbance vs. reaction time (t) are shown in Fig. 5(a) and (b). According to pseudo-first-order reaction kinetics:
| r = ln(ct/co) = kt, |
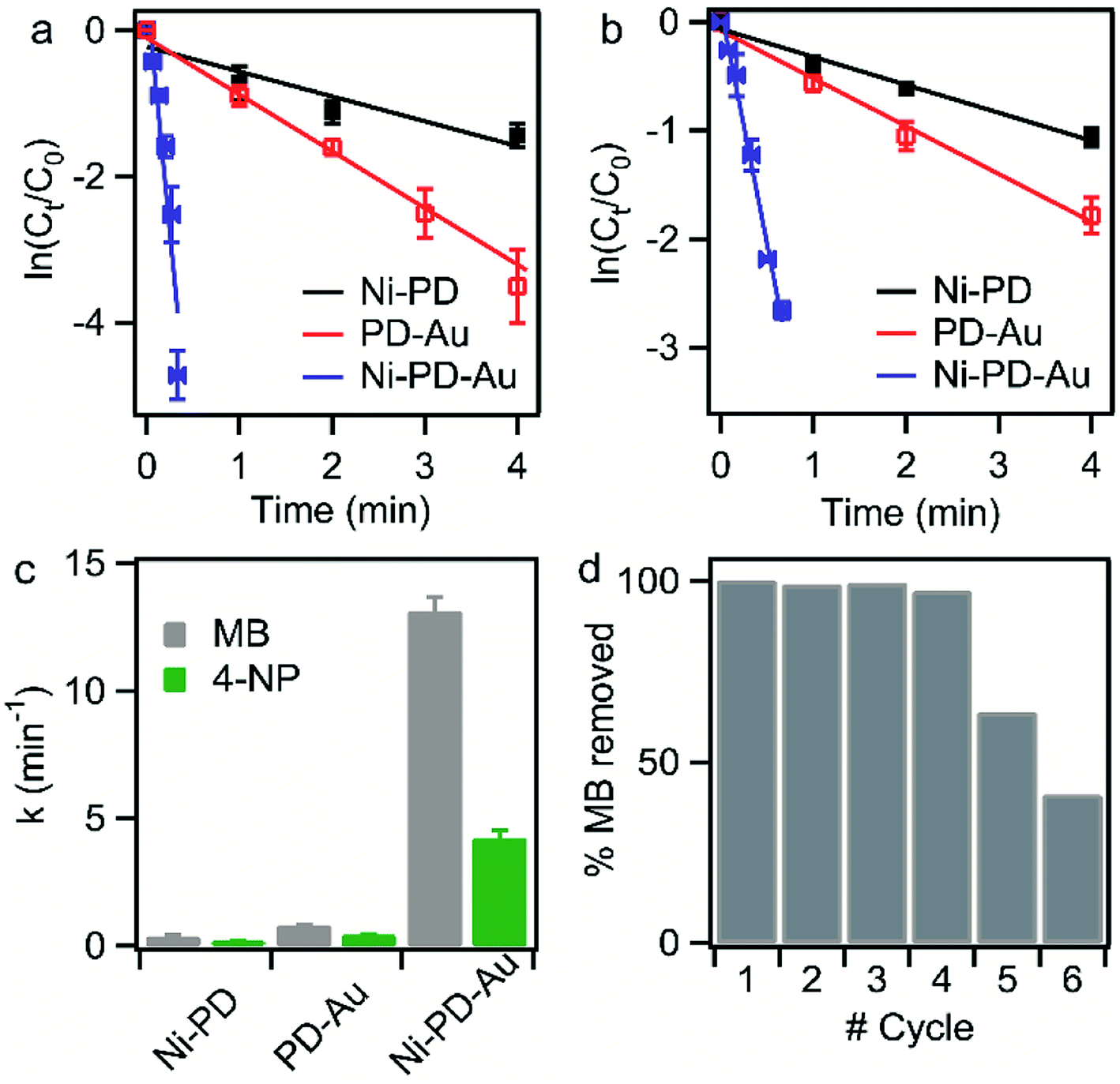 | ||
| Fig. 5 Pseudo-first order kinetic plot (ln(ct/c0) versus time, t) for the catalytic reduction of (a) MB and (b) 4-NP in presence of Ni–PD, PD–Au, and Ni–PD–Au nanocatalysts. The rate constants are determined from the line fit of the experimental data points. (c) Bar plot for the rate constants (d) plot of % removal of MB in catalytic reduction reaction using Ni–PD–Au as a catalyst over six successive cycles to represent the cyclic utilization of the nanocomposite. | ||
| Sample | Dyes | k (min−1) | R 2 | Reference |
|---|---|---|---|---|
| Ni–PD | MB | 0.33 | 0.955 | This work |
| 4-NP | 0.19 | 0.984 | This work | |
| PD–Au | MB | 0.76 | 0.999 | This work |
| 4-NP | 0.40 | 0.957 | This work | |
| Ni–PD–Au | MB | 13.11 | 0.945 | This work |
| 4-NP | 4.21 | 0.994 | This work | |
| Ni-G–PD–Au | MB | 1.76 | 0.997 | This work |
| 4-NP | 0.35 | 0.953 | This work | |
| Chitosan-Au hydrogel | 4-NP | 0.15 | — | 16 |
| Au/graphene hydrogel | 4-NP | 0.19 | — | 15 |
| MB | 0.23 | — | ||
| Metallic NiNP | 4-NP | 0.003 | — | 40 |
| Cu–Ni bimetallic NP (3:1) |
4-NP | 1.35 | — | 40 |
| NiNPs in p(AMPS) hydrogel | 4-NP | 0.06 | 0.999 | 12 |
| Ni/α-Mn2O3 | MB | 0.029 | 0.974 | 34 |
| 4-NP | 0.13 | — | ||
| Au@DNA hydrogel | 4-NP | 0.09 | — | 17 |
| AuNPs | 4-NP | 0.20 | 0.96 | 38 |
| AuNPs | MB | 0.24 | — | 35 |
| AuNPs | 4-NP | 0.09 | 0.982 | 18 |
| Au@Ni (bimetallic) | ||||
| 1:0 |
4-NP | 0.48 | — | 36 |
| 1:0.5 |
0.63 | |||
| 1:1 |
0.06 | |||
| 1:2 |
0.036 | |||
| Core–shell Au@Ni | 4-NP | 0.55 | — | 37 |
The reduction reaction of organic dyes by NaBH4 in the presence of metallic nanoparticles follows Langmuir–Hinshelwood mechanism as depicted in Fig. 6 where both BH4− ions and dye molecules are first adsorbed on the surface of the catalysts. The adsorption of BH4− ion on the surface triggers the formation of active hydrogen species that initiates the hydrogenation reaction. The adsorbed NaBH4 transfer hydride ions to the inner Ni and outer Au nanoparticles forming Ni–H and Au–H bonds, respectively. Next, MB molecules capture two hydride ions from the active metal surface, which is essentially the rate-determining step of the catalytic reduction. LMB is thereby formed and desorbed from the surface reactivating the catalyst again. In our investigation, the reaction rate constants for PD–Au and Ni–PD were measured 0.71 min−1 and 0.33 min−1, respectively. Recent reports demonstrated that BH4− ions create a negatively charged layer around the AuNPs in an aqueous environment facilitating the adsorption of the cationic dye molecules through electrostatic interaction and making the electron transfer faster.41,42 Also, the catalytic activity of noble metal nanoparticles improves when they are small-sized and highly dispersed enabling easy access of the reactant molecules to reach the surface. Thus the catalytic activity of AuNP is expected to be much higher than NiNP as evident in our case. Surprisingly, the rate constant for Ni–PD–Au catalyst was 13.11 min−1 which is distinctly higher than the corresponding monometallic counterparts. In this case, AuNPs along with NiNPs synergistically contributed in catalysis process improving the overall catalytic performance of the nanocomposite. The presence of AuNPs on the catalyst surface increases the number of active sites contributing to faster electron relay. The higher rate constant might be also attributed to the stabilizing desorption characteristic of AuNPs. Being a noble metal, desorption on AuNP surface is stabilizing and hydrogen is less strongly chemisorbed on AuNP surface making the hydrogenation reaction faster.43,44 Moreover, Au atoms were reported to induce electronic effects on d electrons of Ni similar to the Au@Ni core–shell alloys14,36 where catalytic reactivity depends on the combined electronic effect of two metal components. It was further concluded that Au atoms in the core of the Au@NiNPs induced an electronic effect on the local density of Ni d states via the presence of core Au atoms. As Au atoms have a high electron density, the neighboring Ni atoms experienced an enhanced electron density and a higher effective coordination number simultaneously similar to that proposed for Au–Ni surface alloy. d-Band theory has often been applied to understand this combined effect of two metal species.45 The primary principle underlying the d-band theory is that the binding energy of an adsorbate to a metal surface is largely dependent on the electronic structure of the surface itself. Earlier, bimetallic core–shell Au–NiNPs supported on CeO2 support were studied for CO oxidation and compared with their monometallic counterparts. As anticipated, Au/CeO2 catalysts exhibited better catalytic performance than the Ni/CeO2 sample. However, Au–Ni/CeO2 was remarkably more active than the Au/CeO2 catalyst, even though the characterization data corroborated that only nickel atoms were exposed on the surface during the reaction.41 In our case of Ni–PD–Au nanocatalyst, NiNP is separated from AuNP by PD layer and these nanoparticles are not as closely packed in the crystal structure as in alloys. But according to past investigations, PD promotes electron transfer processes due to its organic semiconductor nature.21,46 It was also proven from previous studies that the presence of quinone moieties in the PD rapidly channeled and accelerated the electrons derived from the electron donor (NaBH4) and acted as redox mediators for catalytic reduction of MB and rhodamine B.47 The contribution of PD in the catalytic reduction mechanism was also evidenced previously for Fe3O4@PD nanoparticles in reduction reaction where a synergistic effect between Fe(III) and the quinone moieties of PD contribute to the enhanced electron transfer rate from NaBH4 to methyl orange.21,47 On these bases, we hypothesize that in the Ni–PD–Au nanocomposite, AuNPs might have induced some electronic effect on NiNPs via the PD layer and enhanced the overall catalytic activity with the help of PD layer acting as redox mediators.
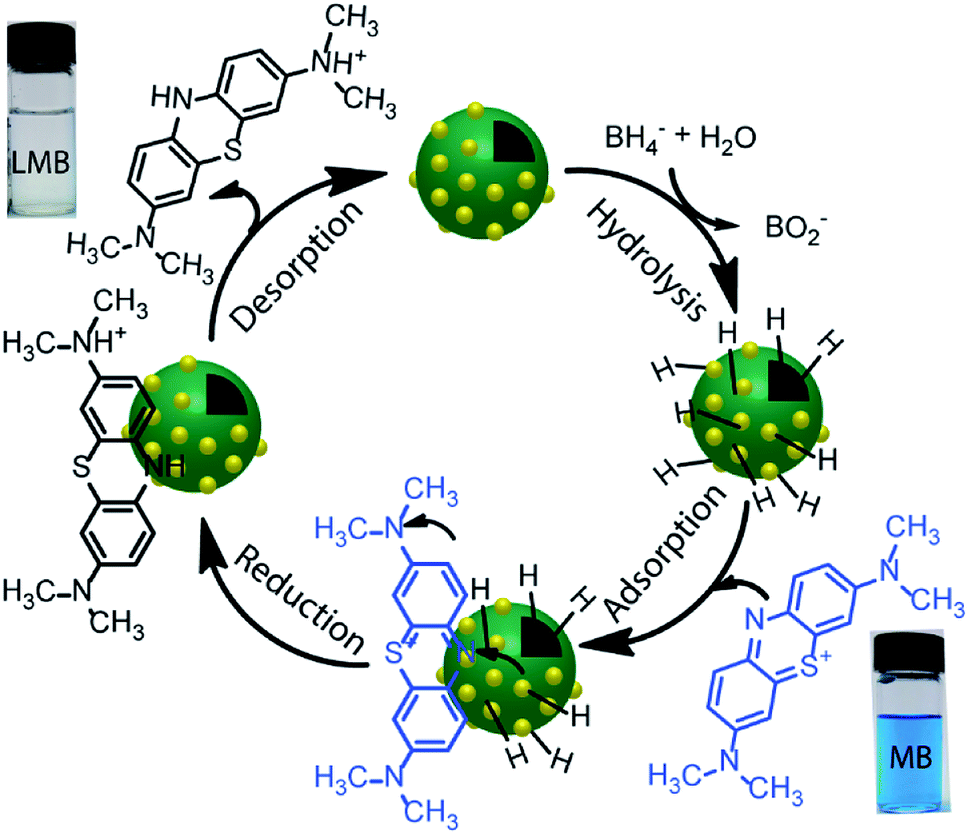 | ||
| Fig. 6 Scheme for a possible mechanism of catalytic reduction of MB by NaBH4. | ||
Reusability of the catalysts is another important property that is often required, particularly for the expensive catalysts, in addition to the high catalytic efficiency. Therefore, recyclability tests were performed to investigate the feasibility of the long-term use of our synthesized catalyst. We conducted the catalytic reduction reaction of MB six times employing repeatedly the same sample of Ni–PD–Au nanocomposite with NaBH4 while monitoring with the help of UV-visible spectroscopy. After each test, the particles were thoroughly washed with an aqueous solution of ethanol so that the reaction products were removed from the surface of the catalyst. From the absorbance spectrum monitored, the particles were found to be highly active for first four repeated runs with conversion efficiency around 100%. Nevertheless, we observed a substantial decrease in the catalytic activity after the fourth run as shown in Fig. 5(d). Being a transition metal, the reactivity of nickel can decrease over subsequent use due to oxidation or catalyst inactivation.12,48 We attributed this decrease in the synergistic catalytic activity to the fact that the NiNPs in the Ni–PD–Au were not completely protected by the PD shell and hence got oxidized to Ni2+ when exposed to aqueous solution for longer time. Thus to increase the stability of the Ni–PD–Au nanocomposite, another core–shell structure was fabricated using graphene-wrapped NiNPs as core instead of bare NiNPs. We hypothesized that the graphene wrapping of the NiNPs would provide additional stability in the harsh aqueous environment.48,49
Fig. 7(a) shows the scheme for synthesizing Ni-G–PD–Au nanocomposite. Spherical metallic NiNPs were synthesized in the first step by the reduction of nickel(II) chloride by hydrazine hydroxide in the presence of sodium hydroxide as done previously.24 Multilayered graphene over NiNPs was produced by solid carbon segregation method.27 In this process, NiNPs were carburized using TEG in the presence of NaOH. As a result, carbon diffused into NiNPs from TEG solution aided by the catalytic reaction of Ni and formed hexagonal nickel carbide (Ni3C). As the Ni3C phase is unstable at temperatures above 325 °C, during the annealing process, it decomposed into a mixture of phase-separated graphene layer and FCC phase nickel again.27 AuNPs were deposited over the surface of graphene wrapped NiNPs using polydopamine as organic linker similarly as done for Ni–PD–Au. The surface morphology of Ni-G–PD–Au is shown in Fig. 7(b) where spherical Ni-G–PD–Au nanocomposites seemingly have an average diameter of ∼254 nm. The difference in diameter between Ni-G–PD–Au and Ni–PD–Au is approximately 54 nm. The additional thickness (27 nm) corresponds to the multilayered graphene coating over NiNPs, which is consistent with previously reported investigations.27 It can be noticed that no deformation occurred in the spherical shape of NiNPs due to the carburization and annealing process done for producing graphene layer. Fig. 7(c) shows the XRD pattern for Ni-G–PD–Au which confirms the presence of both NiNP and AuNP in the sample. The peaks found at 44.5°, 51.8° and 76.4° corresponding to (111), (200) and (220) lattice planes of FCC Ni, which ascertained that hexagonal Ni3C of carburization step decomposed to phase-separated nickel and graphene layer after annealing at 500 °C. Peaks for gold are found at 38°, 43°, 63° and 77.7°, corresponding to (111), (200), (220) and (311) lattice planes. Using Scherrer's equation and XRD measurements, the average diameter of AuNPs was determined to be ∼24 nm, which are roughly the same size obtained for AuNPs on Ni–PD–Au, as described in Tables S1 and S2.† At this condition, graphene is not expected to show a distinct peak on XRD due to less crystallinity.50–52
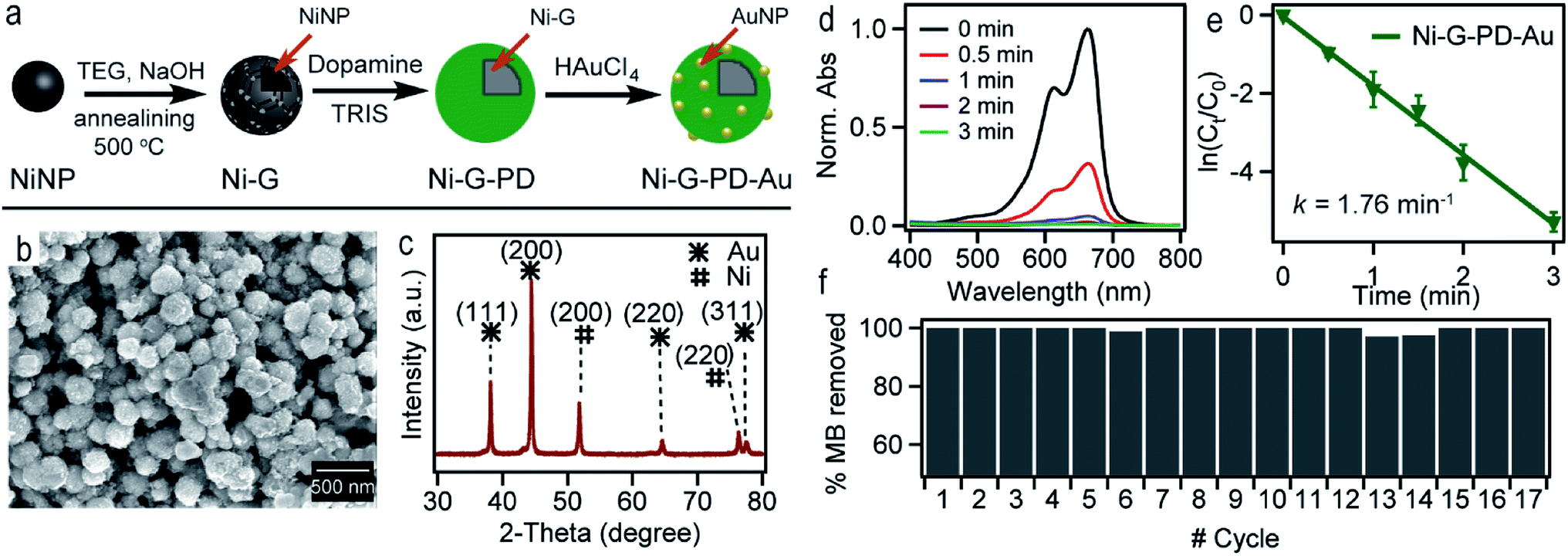 | ||
| Fig. 7 (a) The schematic representation for synthesizing Ni-G–PD–Au (b) SEM image for Ni-G–PD–Au (c) XRD pattern for Ni-G–PD–Au (d) UV-vis absorbance spectra for catalytic reduction of MB in presence of Ni-G–PD–Au monitored as a function of time (e) fitted curve plot for the kinetic estimation of catalytic reduction of MB Ni-G–PD–Au (f) plot of (%) removal of MB in catalytic reduction reaction using Ni-G–PD–Au as catalyst over 17 successive cycles. | ||
Fig. 7(d) shows the UV-spectra for reduction of MB by NaBH4 using Ni-G–PD–Au as catalyst where complete reduction of MB was achieved within 3 minutes. The reaction took over 10 minutes for completion in case of Ni–PD and PD–Au separately as mentioned earlier. Fig. 7(e) further indicates that the reaction rate constant for Ni-G–PD–Au is 1.76 min−1 which is higher than Ni–PD (0.33 min−1) and PD–Au (0.76 min−1). Thus the synergistic effect is also observed for Ni-G–PD–Au composite though not as prominent as observed for Ni–PD–Au. However, the rate constant for Ni-G–PD–Au is higher than individual Ni and Au based catalysts as mentioned in the literature (Table 1). It can also be concluded that synergistic catalytic effect is less for Ni-G–PD–Au than Ni–PD–Au due to the multilayered graphene coating over the core NiNPs. Although multilayered graphene coating decreased the catalytic activity of the nanocomposite to some extent, it provided great stability as evident by the recyclability test (Fig. 7(f)). As shown in the bar plot, recyclability test was done by conducting the catalytic reduction reaction of MB and NaBH4 for 17 times with the same sample of Ni-G–PD–Au nanocomposite. From the absorbance spectrum monitored, the nanoparticles were found to be highly active over 17 repeated runs having a conversion efficiency of around 100% with no substantial decrease in percentage removal of MB. On the contrary, the catalytic activity of Ni–PD–Au catalyst decreased after four repeated cycles. The recyclability test thus signifies the importance of graphene layer over the NiNPs to increase the stability of hybrid nanocomposite as well as vividly ascertains the long term use of Ni-G–PD–Au nanocomposites for catalysis application.
The aim of using NiNP as the core of our nanocomposites was to provide magnetic property for easy separation. Thus it was essential for this study to verify that the magnetic property and phase purity of NiNP was not altered in Ni–PD–Au and Ni-G–PD–Au nanocomposites by the shell materials or during fabrication steps. The magnetic properties of NiNPs, Ni–PD–Au and, Ni-G–PD–Au nanocomposites were evaluated by M–H hysteresis loops using vibrating sample magnetometer (Fig. 8(a)) where the magnetic saturation values are measured to be 61, 35 and 22 emu g−1 for NiNPs, Ni–PD–Au and Ni-G–PD–Au respectively. These magnetization curves show coercivity and remanence, confirming the ferromagnetic behavior of FCC phase Ni in both composites.53 It can be observed in the plot that the magnetic saturation value of Ni–PD–Au is lower than NiNPs due to the PD coating and deposition of AuNPs over the surface. In the case of Ni-G–PD–Au, the magnetic saturation value decreases further due to the addition of multilayered graphene over NiNP. However, the magnetic sensitivity of Ni-G–PD–Au is strong enough to easily separate the catalyst from the reaction system. The inset photograph shows the magnetic behavior of Ni-G–PD–Au composite and the separation process using an external magnet. Fig. 8(b) presents the thermogravimetric analysis (TGA) curves for Ni–PD–Au and Ni-G–PD–Au composites. The TGA curves depict similar decomposition patterns for both Ni–PD–Au and Ni-G–PD–Au. In both cases, the slight decomposition started at 70 °C, presumably because of physically adsorbed water in the material. After heating to about 300 °C, the adsorbed water seems to be driven off and the mass loss is due to moisture loss from the samples. The second step of decomposition for both composites started at 377 °C due to the oxidation of organic components present in the nanocomposites. Greater mass loss was observed between 384 °C to 410 °C due to the pyrolysis of oxygen-containing functional group. The decomposition pattern shows that the Ni-G–PD–Au is thermally more stable than the Ni–PD–Au due to the addition of multilayered graphene and thus making it more preferable to be used for high-temperature catalytic applications.
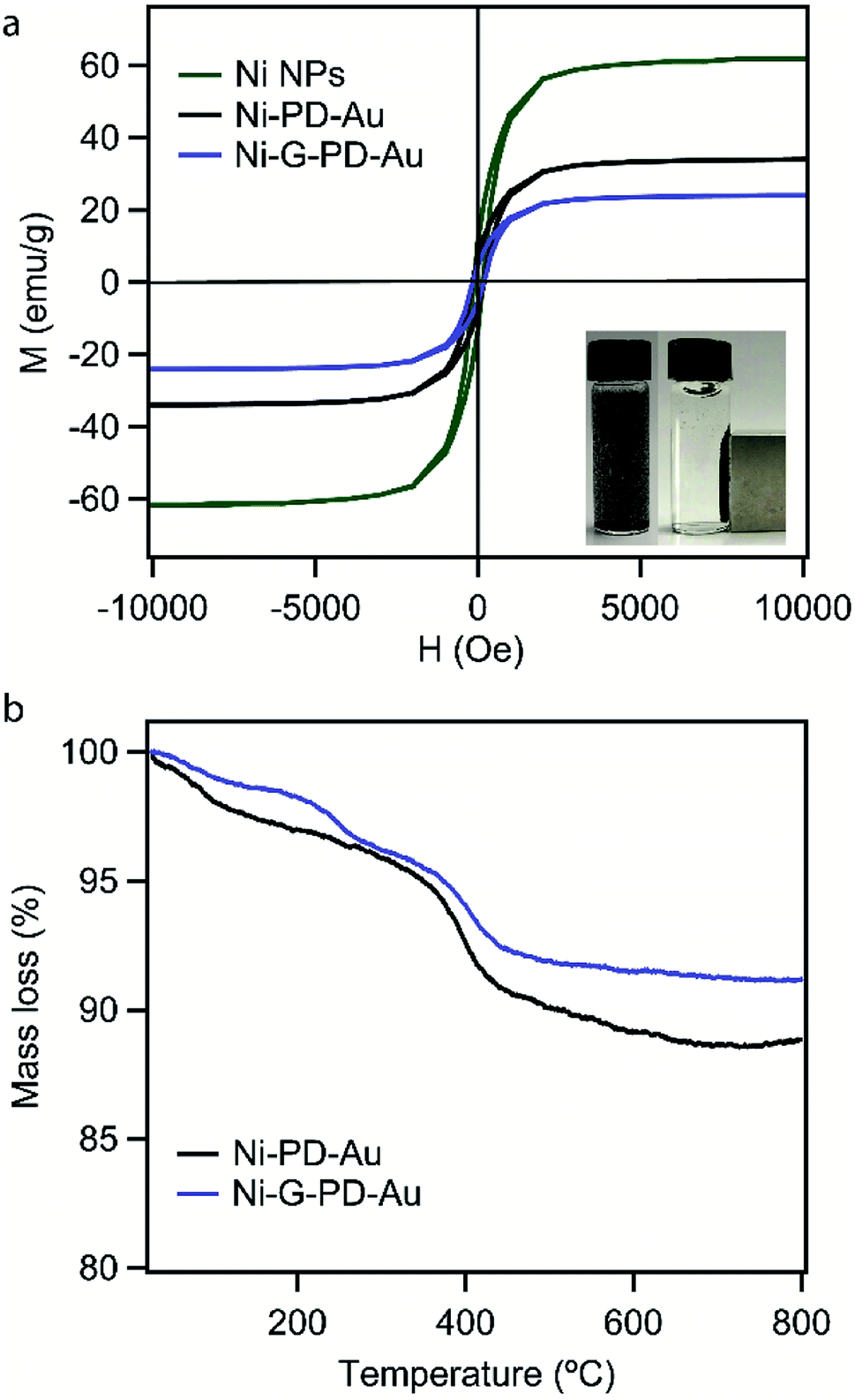 | ||
| Fig. 8 (a) Magnetization curves for NiNPs, Ni–PD–Au, and Ni-G–PD–Au (b) thermogravimetric analysis for Ni–PD–Au and Ni-G–PD–Au. | ||
Conclusions
We introduced two novel multifunctional nanocomposites comprised of Ni and AuNPs that exhibited remarkably enhanced catalytic activity for the reduction of MB and 4-NP demonstrating incredible synergistic effects. The synergistic catalytic activity of the as-synthesized nanocomposites dramatically surpassed the catalytic performance of similar Ni and Au catalysts reported in the literature by large margins suggesting the value of this report. A bio-inspired strategy was followed to immobilize Au nanoparticles on bare and graphene-wrapped Ni nanoparticles using polydopamine, which are believed to contribute to the catalytic action via transferring the electrons from Ni core to the AuNPs on the shell. Albeit Ni–PD–Au exhibited distinctly higher synergistic effect than Ni-G–PD–Au, its catalytic performance started degrading after four cycles. Whereas Ni-G–PD–Au showed excellent reusability without any substantial decrease in efficiency over 17 cycles. Use of graphene in Ni-G–PD–Au nanocomposite vividly provided better stability to the NiNPs in the aqueous solution, specifically when long term industrial applications are concerned. The thermogravimetric analysis showed that both of the nanocomposites were thermally stable approximately up to 350 °C which may be conducive to use the catalysts for the reaction at elevated temperatures. The mussel-inspired core–shell Ni–PD–Au and Ni-G–PD–Au catalysts synthesized in this work can also be extended to explore a broad range of industrially and environmentally important reactions including hydrodechlorination, hydrogenation, and steam reforming.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful to BUET for providing the funds for purchasing the chemicals used in this research. We also thank Prof. Julianne M. Gibbs and Benjamin Rehl (University of Alberta) for the help with XPS measurements.References
- K. McNamara and S. A. Tofail, Phys. Chem. Chem. Phys., 2015, 17, 27981–27995 RSC
.
- K. D. Gilroy, A. Ruditskiy, H.-C. Peng, D. Qin and Y. Xia, Chemical reviews, 2016, 116, 10414–10472 CrossRef CAS
- A. E. Allen and D. W. MacMillan, Chem. Sci., 2012, 3, 633–658 RSC
- S. Afewerki and A. Cordova, Chemical reviews, 2016, 116, 13512–13570 CrossRef CAS
- G. Darabdhara, P. K. Boruah, P. Borthakur, N. Hussain, M. R. Das, T. Ahamad, S. M. Alshehri, V. Malgras, K. C.-W. Wu and Y. Yamauchi, Nanoscale, 2016, 8, 8276–8287 RSC
- M. A. Salem, E. A. Bakr and H. G. El-Attar, Spectrochim. Acta, Part A, 2018, 188, 155–163 CrossRef CAS
- M. Misra, N. Singh and R. K. Gupta, Catal. Sci. Technol., 2017, 7, 570–580 RSC
- Y. Xie, B. Yan, H. Xu, J. Chen, Q. Liu, Y. Deng and H. Zeng, ACS Appl. Mater. Interfaces, 2014, 6, 8845–8852 CrossRef CAS
- Q. Xia, S. Fu, G. Ren, F. Chai, J. Jiang and F. Qu, RSC Adv., 2016, 6, 55248–55256 RSC
- M. Chen, P. Liu, C. Wang, W. Ren and G. Diao, New J. Chem., 2014, 38, 4566–4573 RSC
- J. Zhang, M. S. Azam, C. Shi, J. Huang, B. Yan, Q. Liu and H. Zeng, RSC Adv., 2015, 5, 32272–32282 RSC
- N. Sahiner, H. Ozay, O. Ozay and N. Aktas, Appl. Catal., A, 2010, 385, 201–207 CrossRef CAS
- P. Basnet, S. KC, R. K. Dhungana, B. Shrestha, T. J. Boyle and R. Giri, J. Am. Chem. Soc., 2018, 140, 15586–15590 CrossRef CAS
- S. De, J. Zhang, R. Luque and N. Yan, Energy Environ. Sci., 2016, 9, 3314–3347 RSC
- J. Li, C.-y. Liu and Y. Liu, J. Mater. Chem., 2012, 22, 8426–8430 RSC
- X.-Q. Wu, X.-W. Wu, Q. Huang, J.-S. Shen and H.-W. Zhang, Appl. Surf. Sci., 2015, 331, 210–218 CrossRef CAS
- A. Zinchenko, Y. Miwa, L. I. Lopatina, V. G. Sergeyev and S. Murata, ACS Appl. Mater. Interfaces, 2014, 6, 3226–3232 CrossRef CAS
- N. Bogireddy, U. Pal, L. M. Gomez and V. Agarwal, RSC Adv., 2018, 8, 24819–24826 RSC
- A. L. Chibac, T. Buruiana, V. Melinte, I. Mangalagiu and E. C. Buruiana, RSC Adv., 2015, 5, 90922–90931 RSC
- R. Ghosh Chaudhuri and S. Paria, Chemical reviews, 2011, 112, 2373–2433 CrossRef
- J. r. Liebscher, R. Mrówczyński, H. A. Scheidt, C. Filip, N. D. Hădade, R. Turcu, A. Bende and S. Beck, Langmuir, 2013, 29, 10539–10548 CrossRef CAS
- L. Guo, Q. Liu, G. Li, J. Shi, J. Liu, T. Wang and G. Jiang, Nanoscale, 2012, 4, 5864–5867 RSC
- M. S. Islam, N. Akter, M. M. Rahman, C. Shi, M. T. Islam, H. Zeng and M. S. Azam, ACS Sustainable Chem. Eng., 2018, 6, 9178–9188 CrossRef CAS
- J. Y. Choi, Y. K. Lee, S. M. Yoon, H. C. Lee, B. K. Kim, J. M. Kim, K. M. Kim and J. H. Lee, J. Am. Ceram. Soc., 2005, 88, 3020–3023 CrossRef CAS
- J. Miao, H. Liu, W. Li and X. Zhang, Langmuir, 2016, 32, 5365–5372 CrossRef CAS
- A. Ma, Y. Xie, J. Xu, H. Zeng and H. Xu, Chem. Commun., 2015, 51, 1469–1471 RSC
- S.-M. Yoon, W. M. Choi, H. Baik, H.-J. Shin, I. Song, M.-S. Kwon, J. J. Bae, H. Kim, Y. H. Lee and J.-Y. Choi, ACS Nano, 2012, 6, 6803–6811 CrossRef CAS
- T. Zeng, H.-y. Niu, Y.-r. Ma, W.-h. Li and Y.-q. Cai, Appl. Catal., B, 2013, 134, 26–33 CrossRef
- J. Jiang, L. Zhu, L. Zhu, B. Zhu and Y. Xu, Langmuir, 2011, 27, 14180–14187 CrossRef CAS
- X. Han, X. Chen, M. Yan and H. Liu, Particuology, 2019, 44, 63–70 CrossRef CAS
- L. Qin, D. Huang, P. Xu, G. Zeng, C. Lai, Y. Fu, H. Yi, B. Li, C. Zhang and M. Cheng, J. Colloid Interface Sci., 2019, 534, 357–369 CrossRef CAS
- W. Zheng, H. Fan, L. Wang and Z. Jin, Langmuir, 2015, 31, 11671–11677 CrossRef CAS
- M. Tian, C. Dong, X. Cui and Z. Dong, RSC Adv., 2016, 6, 99114–99119 RSC
- P. Deka, P. Sarmah, R. C. Deka and P. Bharali, ChemistrySelect, 2016, 1, 4726–4735 CrossRef CAS
- B. R. Ganapuram, M. Alle, R. Dadigala, A. Dasari, V. Maragoni and V. Guttena, Int. Nano Lett., 2015, 5, 215–222 CrossRef CAS
- A. Vysakh, C. L. Babu and C. Vinod, J. Phys. Chem. C, 2015, 119, 8138–8146 CrossRef CAS
- V. A. Bharathan, V. Yadukiran, A. Lazar, A. P. Singh and C. P. Vinod, ChemistrySelect, 2016, 1, 140–146 CrossRef CAS
- A. Ahmad, Y. Wei, F. Syed, M. Imran, Z. U. H. Khan, K. Tahir, A. U. Khan, M. Raza, Q. Khan and Q. Yuan, RSC Adv., 2015, 5, 99364–99377 RSC
- S. K. Maji and A. Jana, New J. Chem., 2017, 41, 3326–3332 RSC
- V. Seethapathy, P. Sudarsan, A. K. Pandey, A. Pandiyan, T. V. Kumar, K. Sanjeevi, A. K. Sundramoorthy and S. B. K. Moorthy, New J. Chem., 2019, 43, 3180–3187 RSC
- P. Saikia, A. T. Miah and P. P. Das, J. Chem. Sci., 2017, 129, 81–93 CrossRef CAS
- X. Bi, H. Ma and P. Westerhoff, Environ. Sci. Technol., 2018, 52, 13289–13297 CrossRef CAS
- A. M. Ruppert, M. Jędrzejczyk, N. Potrzebowska, K. Kaźmierczak, M. Brzezińska, O. Sneka-Płatek, P. Sautet, N. Keller, C. Michel and J. Grams, Catal. Sci. Technol., 2018, 8, 4318–4331 RSC
- C. Deraedt, L. Salmon, S. Gatard, R. Ciganda, R. Hernandez, J. Ruiz and D. Astruc, Chem. Commun., 2014, 50, 14194–14196 RSC
- P. Kratzer, B. Hammer and J. No/rskov, J. Chem. Phys., 1996, 105, 5595–5604 CrossRef CAS
- J. H. Kim, M. Lee and C. B. Park, Angew. Chem., Int. Ed., 2014, 53, 6364–6368 CrossRef CAS
- S. Du, Z. Liao, Z. Qin, F. Zuo and X. Li, Catal. Commun., 2015, 72, 86–90 CrossRef CAS
- W. Lin, H. Cheng, J. Ming, Y. Yu and F. Zhao, J. Catal., 2012, 291, 149–154 CrossRef CAS
- S. Chen, L. Brown, M. Levendorf, W. Cai, S.-Y. Ju, J. Edgeworth, X. Li, C. W. Magnuson, A. Velamakanni and R. D. Piner, ACS Nano, 2011, 5, 1321–1327 CrossRef CAS
- B. C. Bayer, D. A. Bosworth, F. B. Michaelis, R. Blume, G. Habler, R. Abart, R. S. Weatherup, P. R. Kidambi, J. J. Baumberg and A. Knop-Gericke, J. Phys. Chem. C, 2016, 120, 22571–22584 CrossRef CAS
- H. Song, H. Cui and C. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 13765–13769 CrossRef CAS
- D. Wu, C. Zhang, C. Liang, Y. Zhu, S. Xu, D. Xiong, S. Xue, L. Wang and P. K. Chu, J. Mater. Chem. C, 2016, 4, 2079–2087 RSC
- Y. Chen, D.-L. Peng, D. Lin and X. Luo, Nanotechnology, 2007, 18, 505703 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra05736f |
| This journal is © The Royal Society of Chemistry 2019 |