
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pyrrolizidine alkaloids quantified in soil and water using UPLC-MS/MS†
Jawameer R.
Hama
 * and
Bjarne W.
Strobel
* and
Bjarne W.
Strobel
Department of Plant and Environmental Sciences, University of Copenhagen, Thorvaldsensvej 40, 1871 Frederiksberg, Denmark. E-mail: jawameer@plen.ku.dk
First published on 25th September 2019
Abstract
Pyrrolizidine alkaloids (PAs) are produced in plants as defence compounds against insects. PAs present a serious health risk to humans and livestock; therefore it is necessary to have a validated analytical method to monitor PAs in the environment. The objective of this work is to present an UPLC-MS/MS method for quantification of PAs in environmental samples of both soil and water. A fast, reliable, and sensitive approach is developed to identify and quantify PAs in soil and water. Sample preparation was performed by clean-up and pre-concentration of the samples using MCX solid phase extraction cartridges with full optimization, and then PAs were determined by UPLC coupled with TQ-MS. In the liquid chromatography, most of the parameters were optimized and tested including gradient time, solvents, additives, and pH of the mobile phases and flow rate. In addition, the MS parameters of cone voltage, desolvation temperature, cone flows, and collision energy were optimized. The instrument limit of detection (2–7 μg L−1) and limit of quantification (5–9 μg L−1) were determined experimentally, and the method was linearity validated up to 1000 μg L−1. The method was applied to analyse soil and surface water samples collected in April and May 2018 in Vejle, Borup, and Holte, Denmark. In total, 15 PAs were quantified and reported for the first time in environmental samples, in a range of 3–1349 μg kg−1 in soil and 4–270 μg L−1 in surface water.
1. Introduction
Pyrrolizidine alkaloids (PAs) are secondary plant metabolites, consisting of nitrogen between two fused five-membered rings. PAs are esters of hydroxylated methyl pyrrolizidines, with a necine base and necic acid moiety that can be either saturated or unsaturated with a double bond between the 1 and 2 positions.1 PAs are classified into three classes: retronecine, otonecine and platynecine. They exist as either the free base (PA, tertiary amine) or N-oxide (PANO) form.2,3 So far, several hundred PAs have been found in nature and their structures determined.4 Retronecine and otonecine types have a 2-double bond in the necine bases, which are hepatotoxic and may lead to cancer in humans. However the platynecine type has a saturated necine base.5–7 Plants use PAs mostly as a defence mechanism against insects. It is estimated that approximately 3% of all flowering plants contain at least one PA. PAs are found in many plant species in the Asteraceae, Boraginaceae and Fabaceae families.8,9 PAs are among the most abundant group of natural toxins produced by plants.10,11Toxicity of individual PAs is related to the chemical structure and physical properties.7 Cases of human exposure to PAs originate from direct consumption of PA-containing teas, herbal dietary and supplements.9 Moreover, exposure of PAs is likely by consuming tea, honey and animal product s which are contaminated with PAs.7,12 Due to safety hazards, some guidelines and regulations were issued to limit daily intake. These regulation are for particular cases and cannot be used for all situations.9,12–14 German Federal Institute for Risk Assessment (BfR) recommended 0.007 μg PAs intake for humans per kg bodyweight.12 While EFSA chose 237 μg per kg bodyweight as a new reference point for MDL10 calculation from riddelliine's toxicity study, and corresponds to a maximum intake of 0.024 μg PA/PANO per kg bodyweight.9
Previous analytical methods are optimized to detect PAs in a certain commodities. In particular, LC-MS methods optimized to detect PAs in foods, is more for quality control of food and of foodstuffs.1,10,15–17 The UPLC coupled with MS has been used to detect a trace-level of organic pollutants with high selectivity and sensitivity, with shorter run time and sharper peaks, this may reduce or eliminate the matrix effects issue.18–20 High resolution MS can provide accurate mass information that resulted in enhanced application of the instrument.1,21 Quantification and target analysis can be processed using high MS resolution with multiple reaction monitoring (MRM) transitions, however the exact masses is required.22 MRM transition is good for quantification of PAs, as they have more common fragmentation patterns and produce similar product ions.17,21,23 Cases of PA poisoning cannot be eliminated due to the lack of accurate analytical methods for a wider range of sample types. Thus, a robust analytical method to determine PAs (Fig. 1) in complex matrices is still missing.24
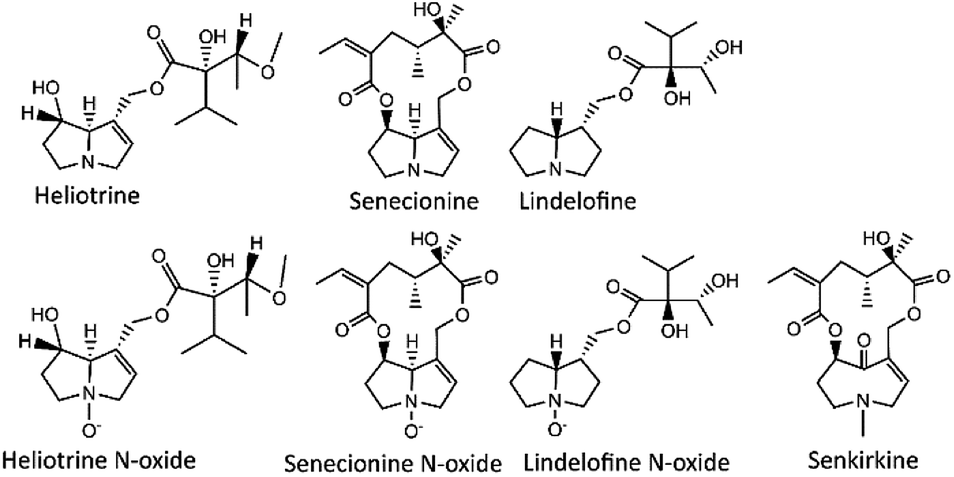 | ||
| Fig. 1 Structures of selected types of pyrrolizidine alkaloids. | ||
Here, we report a quick and robust UPLC-MS/MS method to monitor and quantify PAs in a wide range of environmental samples such as plant, soil and water. The method enables analysis of small and large volumes of environmental samples after fast SPE to pre-concentration and clean-up the samples.
2. Experimental
2.1 Chemicals and reagents
Methanol (MeOH) (MS grade), acetonitrile (MeCN) (MS grade), formic acid (FA) (MS grade), ammonium formate (MS grade), acetone (HPLC grade), sulfuric acid, hydrochloric acid (Analytical reagent grade) and caffeine (internal standard, for spiking experiments and method validation) were purchased from Sigma-Aldrich (Steinheim, Germany). Glass fibre (SiO2, particle size 0.2–0.8 mm) was purchased from Merck (Darmstadt, Germany). Certified reference materials of heliotrine, monocrotaline, monocrotaline N-oxide, senecionine, senecionine N-oxide, erucifoline, erucifoline-N-oxide, retrorsine, jacobine, jacobine N-oxide senkirkine, heliotrine N-oxide, echimidine, echimidine N-oxide, purchased from Phytolab (Vestenbergsgreuth, Germany), also lindelofine, lindelofine N-oxide purchased from AKos Consulting & Solutions (Steinen, Germany). Oasis MCX 6 cc, 150 mg Sorbent, 30 μm particle size purchased from Waters (Milford, USA). MilliQ water (resistivity 18.2 Mohm × cm, TOC less than 1 μg L−1) was produced in-house with a type I ultrapure water purification system from ELGA-Veolia LabWater (High Wycombe, UK).2.2 Soil and water samples
Two surface water, three topsoil and one subsoil samples were collected in amber glass bottles in April and May 2018 in Vejle, Borup and Holte, Denmark (coordinates: N 55°41′00.0, E 9°25′00.0: N 55°27′00.0, E 12°00′00.0; N 55°49′00.0, E 12°30′00.0, respectively). The areas had Ragwort (Senecio jacobaea L.) plants in densities 20–28 plants per square meter as a source of PAs. Water samples were filtered with filter paper (Whatman® quantitative-Grade 42) to remove any suspended particles and acidified to pH 3 with diluted FA. Soils were sieved on 0.2 mm to remove the coarse sand and gravel, and visible plant roots removed. All samples were stored at −20 °C prior to extraction, in most cases the extraction was performed within 48 h after sampling. Each time, 10 g soil or 500 mL water were collected in duplicate, and stored at −20 °C prior to the analysis.2.3 Sample preparation and extraction
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :acetone (85:15 v/v%) solvent used for a third and fourth extraction. The extraction efficiency was approved with 1 mL of the fourth aliquot of MeOH:acetone (85:15 v/v%) extract tested directly by UPLC-MS/MS method, and no PAs were detected. Finally, the three extracts were combined, centrifuged and filtered with a 0.45 μm PTFE Membrane filter, and then passed through MCX SPE.
:1, v/v). The eluates were combined and dried under gentle nitrogen flow in a heating block at 40 °C. The dried extract was dissolved in 1 mL 40% MeCN and filtered through 0.2 μm PTFE membrane filter prior to analysis.
:acetone (85:15 v/v%) solvent used for a third and fourth extraction. The extraction efficiency was approved with 1 mL of the fourth aliquot of MeOH:acetone (85:15 v/v%) extract tested directly by UPLC-MS/MS method, and no PAs were detected. Finally, the three extracts were combined, centrifuged and filtered with a 0.45 μm PTFE Membrane filter, and then passed through MCX SPE.
:1, v/v). The eluates were combined and dried under gentle nitrogen flow in a heating block at 40 °C. The dried extract was dissolved in 1 mL 40% MeCN and filtered through 0.2 μm PTFE membrane filter prior to analysis.
2.4 Instrumentation and analytical conditions
In the optimized method, the column temperature was set to 35 °C. LC mobile phase was composed of A (water + 0.01% FA) and B (acetonitrile + 0.01% FA). Gradient conditions were: 0–4 min 10% B, 7 min 20% B, 10 min 50% B, 15 min 90% B, 15–17 min 90% B. The column was equilibrated for 6 min before each run, the total run time was 23 minutes. Flow rate and injection volume were set to 0.450 mL min−1 and 2.5 μL, respectively. All solutions and standards were made in 40% MeCN to make injection of solvent as the same composition of the mobile phase in the LC system.
To verify the method development approach, individual standard solution of heliotrine, senecionine, jacobine and senkirkine with 1000 μg L−1 were injected directly into the MS, using the Intellistart (Waters, Milford) software. The approach worked and helped to protect the MS from direct injections of high concentration of standard solutions for many times.
After optimizing the MS parameters, MS/MS functions and one full scan were performed. In the MS/MS MRM mode, the ion traces was obtained for apex retention time (tR) ± 0.15 min. Corresponding CV and CE for PAs is listed in Table 1. The capillary voltage of 3.5 kV. The desolvation temperature 600 °C, desolvation gas flow 1000 L h−1, and cone gas flow 20 L h−1 was used. Data processing with MassLynx 4.0.2.3 (Waters, Milford, USA).
| PA | t R (min) | MS m/z | MS/MS m/z | CV (V) | CE (eV) |
|---|---|---|---|---|---|
| Monocrotaline | 4.59 | 326.15 | 120.08, 138.09 | 40 | 40 |
| Monocrotaline N-oxide | 5.42 | 342.15 | 120.08, 138.09 | 35 | 35 |
| Erucifoline | 5.66 | 350.15 | 120.08, 138.09 | 40 | 35 |
| Jacobine | 7.18 | 352.17 | 120.08, 138.09 | 40 | 40 |
| Jacobine N-oxide | 7.89 | 368.16 | 120.08, 138.09 | 40 | 40 |
| Retrorsine | 8.15 | 352.17 | 120.08, 138.09 | 30 | 40 |
| Heliotrine | 8.78 | 314.19 | 120.08, 138.09 | 20 | 25 |
| Heliotrine N-oxide | 9.1 | 330.2 | 120.08, 138.09 | 25 | 30 |
| Lindelofine | 9.15 | 286.19 | 124.08 | 30 | 35 |
| Lindelofine N-oxide | 9.7 | 302.31 | 124.08 | 35 | 35 |
| Senecionine | 11.00 | 336.18 | 120.08, 138.09 | 20 | 30 |
| Echimidine | 11.40 | 398.21 | 120.08, 138.09 | 30 | 30 |
| Senecionine N-oxide | 11.51 | 352.17 | 120.08, 138.09 | 40 | 30 |
| Senkirkine | 11.67 | 366.19 | 168.10 | 35 | 25 |
| Echimidine N-oxide | 11.71 | 414.20 | 120.08, 138.09 | 30 | 35 |
2.5 Method validation
Method validation was conducted for five PAs (heliotine, monocrotaline, jacobine, jacobine N-oxide and senkirkine) in both soil and water matrix samples. Sandy soil from Vejle, clay loam soil from Taastrup (near Borup and Holte, Denmark), inert glass material and deionized water were used. It was tested whether interfering compounds from the soil were extracted with PAs in an experiment comparing yield of PAs from spiked soil and spiked glass fibre. For quantification, the calibration curves (9-point) were obtained using an external standard calibration. The curves were constructed by plotting the peak area versus the concentration of each analyte, as y = ax + b with weighting factor of 1/x, in duplicate, (Table 2), to minimize the distortion of concentration. Precision and accuracy were evaluated for intra- and inter-day variations. For intra-day variation, 3 concentration levels 25, 50 and 100 μg L−1 of five PAs in triplicate were spiked to matrix samples. Then, the spiked samples were extracted as described in Section 2.3 on the same day. For the inter-day variation test, new solutions were prepared in parallel and analysed for three consecutive days. Precision was calculated by relative standard deviation. The method recovery was used to calculate accuracy. Matrix effect (ME) was determined by dividing peak area of matrix samples spiked on peak area of standards. Two concentration levels (25 and 100 μg L−1) of five PAs were used, in triplicate. Linearity was tested by expanding calibration curve, in triplicate. To calculate the limit of detection (LOD) and limit of quantification (LOQ), 7 injection of 25 μg L−1 standards solution is used. They are calculated as 3 and 10 times respectively the standard deviation of peak areas divided by the slope of the calibration curve for each PAs.27 Overall the method was validated within limits specified in SANCO document 12495/2011.28| PAs | Correlation coefficient (R) | Sensitivitya (slope) | LOD (μg L−1) | LOQ (μg L−1) | Recovery% ± SDb | ME%d | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Soil | Waterc | Soil | Water | |||||||
| Sandy soil | Clay loam soil | Glass fiber | ||||||||
| a Average of linear regression of three injections of all PAs. b Average recovery ± standard deviation. c De-mineralized water. d ME = matric effect. | ||||||||||
| Monocrotaline | 0.990 | y = 44x | 5.5 | 8.7 | 85 ± 6 | 76 ± 9 | 94 ± 3 | 89 ± 10 | 88 | 94 |
| Monocrotaline N-oxide | 0.990 | y = 40x | 4.6 | 7.5 | 88 ± 10 | 72 ± 8 | 96 ± 4 | 94 ± 3 | 82 | 91 |
| Erucifoline | 0.990 | y = 82x | 3.0 | 6.3 | 82 ± 12 | 75 ± 9 | 92 ± 4 | 91 ± 8 | 81 | 95 |
| Jacobine | 0.995 | y = 21x | 2.6 | 7.6 | 89 ± 6 | 81 ± 9 | 96 ± 5 | 96 ± 6 | 81 | 91 |
| Jacobine N-oxide | 0.993 | y = 40x | 4.7 | 6.2 | 91 ± 7 | 80 ± 7 | 97 ± 5 | 93 ± 7 | 90 | 91 |
| Retrorsine | 0.991 | y = 52x | 6.5 | 9.3 | 80 ± 13 | 80 ± 7 | 90 ± 4 | 96 ± 6 | 91 | 93 |
| Heliotrine | 0.989 | y = 6x | 6.9 | 9.6 | 81 ± 11 | 78 ± 12 | 96 ± 4 | 91 ± 8 | 85 | 89 |
| Heliotrine N-oxide | 0.992 | y = 11x | 4.2 | 7.6 | 83 ± 9 | 76 ± 10 | 98 ± 3 | 90 ± 6 | 86 | 93 |
| Lindelofine | 0.988 | y = 14x | 6.2 | 9.6 | 86 ± 8 | 80 ± 9 | 96 ± 2 | 91 ± 8 | 89 | 90 |
| Lindelofine N-oxide | 0.990 | y = 22x | 5.6 | 8.5 | 88 ± 12 | 78 ± 8 | 97 ± 4 | 92 ± 5 | 89 | 91 |
| Senecionine | 0.990 | y = 22x | 5.4 | 9.7 | 90 ± 7 | 68 ± 9 | 97 ± 3 | 91 ± 7 | 89 | 98 |
| Senecionine N-oxide | 0.991 | y = 227x | 5.6 | 9.5 | 81 ± 12 | 73 ± 6 | 90 ± 5 | 95 ± 6 | 80 | 96 |
| Echimidine | 0.980 | y = 78x | 4.5 | 8.1 | 89 ± 8 | 80 ± 9 | 93 ± 5 | 90 ± 5 | 87 | 91 |
| Senkirkine | 0.994 | y = 99x | 5.0 | 5.8 | 82 ± 9 | 65 ± 9 | 89 ± 7 | 95 ± 4 | 82 | 95 |
| Echimidine N-oxide | 0.986 | y = 96x | 5.1 | 9.6 | 88 ± 7 | 78 ± 8 | 94 ± 6 | 91 ± 5 | 86 | 92 |
3. Results and discussion
We tested different mobile phase and pH to choose optimal one for LC separation, MeCN and MeOH with two pH (2.7 and 8) were tested (Fig. 2). During separation, the acidic MeCN yielded by far best separation. Acidic condition improve full width at half maximum and increase the peak capacity (from 22 to 93 peaks per gradient) during the separation gradient. We decided to add 1% FA for both mobile phases. Elution time of PAs did not change with pH, but the retention times were longer for MeOH eluents (both for acidic or basic) compared to MeCN. The retention factor (k) for both solvents was in the range 1–5, however MeOH always had higher k value indicating that PAs are retained more and has spent more time interacting with the stationary phase. The k data helped to obtain the optimum resolution, by having resolution value of 1.5 or greater between two peaks, to ensure that the sample components are well separated to a degree at which the area or height of each peak may be accurately measured. Separation with MeCN eluents produce sharper peaks, less tailing, more stable baseline, higher S/N ratio, and 2.5 fold higher peak capacity. In addition, MeCN and MeOH with different buffer systems and modifiers (FA and NH4OH with NH4HCO3) were tested. For ammonium formate buffers, ammonium adducts for all PAs were checked, by assuming the concentration of ammonium in the eluent correlated with ammonium adducts. However, they are not correlated. The response was higher with low concentration (1 mM) of ammonium formate, it seems the fragmentation in collision cell is suppressed by increasing the concentration of ammonium formate in the mobile phase. Then to generate protonated adduct of PAs, 0.1% FA showed higher S/N ratio and precision was two times higher than the weakest buffer system.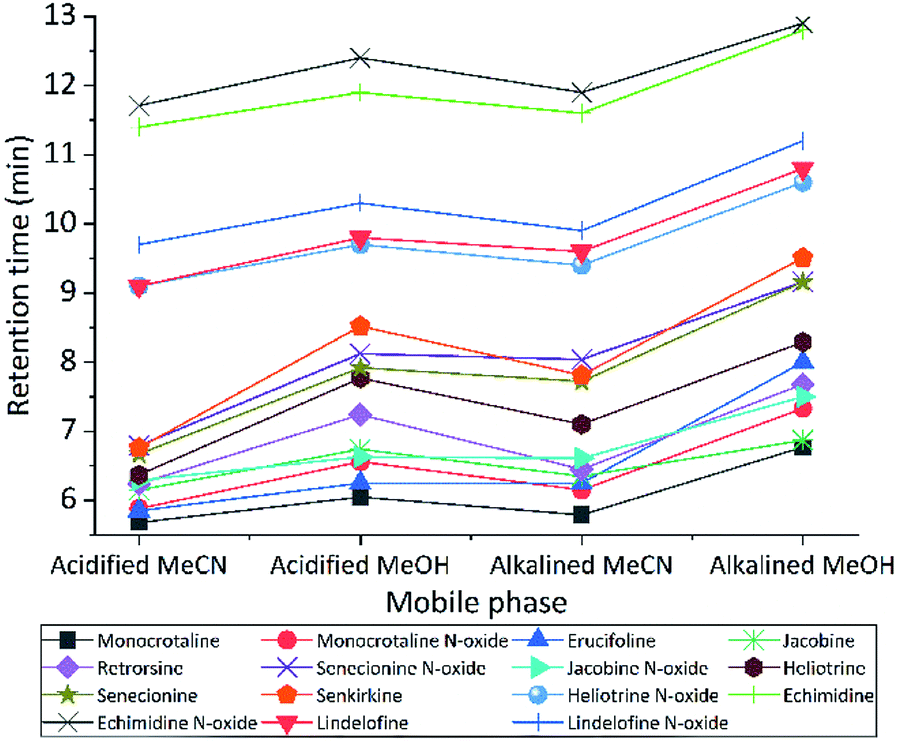 | ||
| Fig. 2 The impact of pH and additives on tR of PAs. The sequence was running at flow rate 0.45 mL min−1, gradient 3. Acidified MeCN and MeOH are acidified to pH = 2.7 with FA, also alkalined MeCN and MeOH are basic to pH = 8 with NH4OH with NH4HCO2. Unless specified the LC parameters used as final version of optimized method. | ||
Subsequently, different gradients were used to choose the optimal gradient time and determine the effect of gradient on S/N ratio. Acidified (pH = 2.7) MeCN is used with gradient 1, 2 and 3. The longer gradient the higher S/N ratio for all PAs (Fig. 3). Lastly with some modification gradient 3 were chosen for chromatographic separation. Using longer gradient didn't show further improvements. For chromatographic separation, no co-eluting compounds were detected for all PA standards and samples.
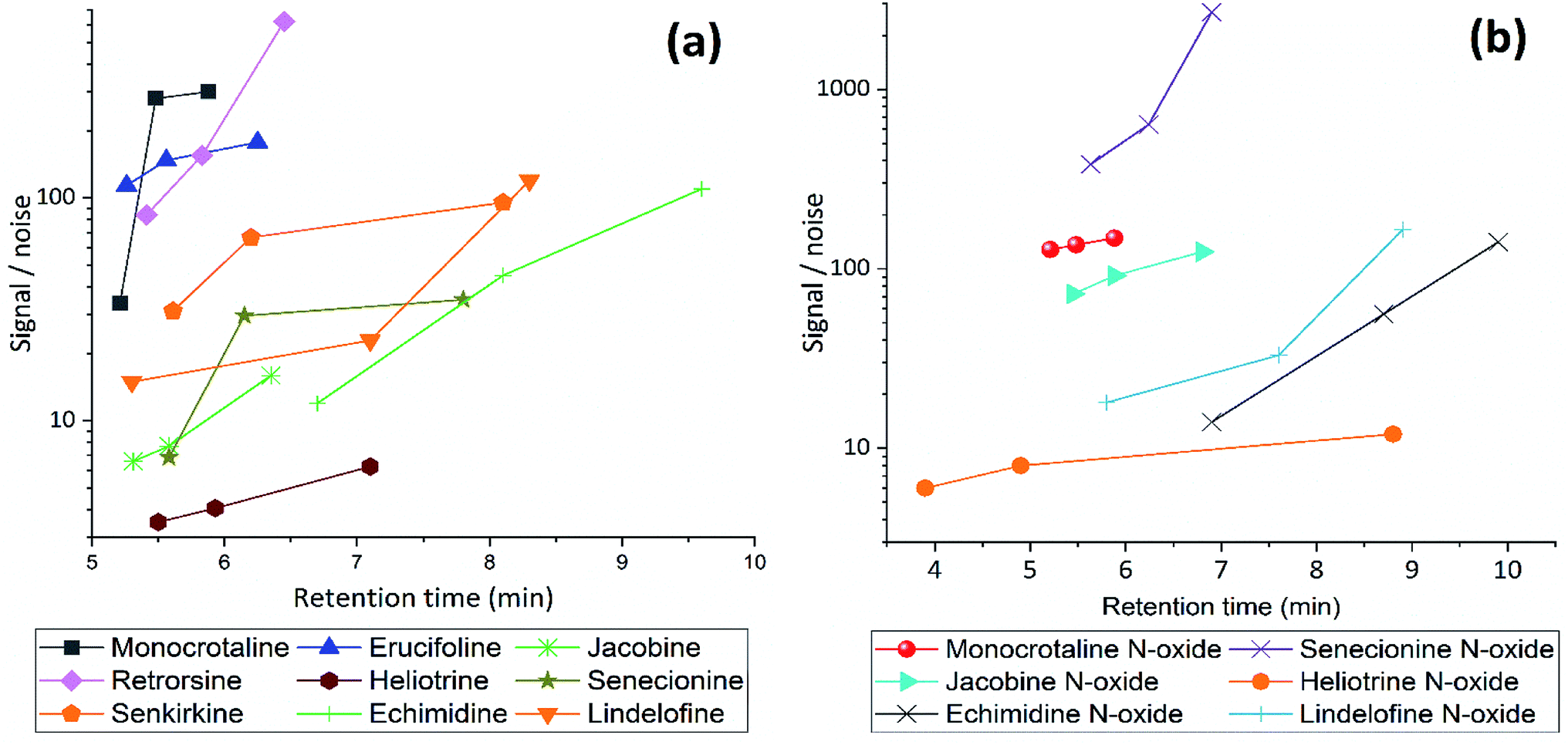 | ||
| Fig. 3 The effect of gradient condition on Signal/Noise ratio of (a) free PAs and (b) PANOs, using gradient 1, 2 and 3 with acidified MeCN (pH = 2.7), flow rate 0.45 mL min−1. | ||
Correlation coefficient (R), regression equation, LOD and LOQ for all PAs are listed in Table 2. Overall method detection limits are estimated to be about 2–7 μg L−1 for corresponded PAs, including sample preconcentration and recoveries, even this is comparable to other reported LC-MS/MS method.1,15,29–31 LOD can be improved by using larger amount of sorbent and dissolving samples in less than 1 mL of solvent before injection. All PAs showed a good linearity; they were linear from LOD up to 1000 μg L−1. MS/MS parameters optimized to increase the response and S/N ratio. CV allows to ionize the compound but does not induce fragmentation in electrospray ionization and can be used to monitor parent ion. It varied with PA type and structure (see ESI Fig. S1 and S2†), ranging from 20–40 V. With the optimal CV, the CE is changed to produce product ions in MRM run.32 CEs were used to investigate fragmentation patterns and intensities (see ESI Fig. S3 and S4†) in collision cell. Optimal CV and CE are listed in Table 1. Desolvation temperatures and cone flows optimized to 600 °C and 50 L, respectively. Their effect were linear on instrument's response and peak area.
Two types of sorbents has been employed to clean-up PAs, which are non-polar phases such as octylsilane or octadecylsilane [C8]32–34 or [C18],35 and cation-exchanges phases.14–17,30 Cation-exchange sorbent is widely used based on the suitability to retain base and PA like compounds. Therefore Oasis MCX was selected as a SPE cartridge. Demineralised water (100 mL) was spiked with 100 μg L−1 of PAs, and they were percolated at 10 mL min−1 through the cartridges. Under these conditions, the recoveries ranged between 60% and 85%.
The pH effect of water was evaluated by adjusting the samples (pH: 3, 5.5, 7, 8.5 and 10), with aliquots of FA and NH4OH. For all PAs, the efficiency of the extraction decreases dramatically when the pH increases except for monocrotaline, illustrated in Table 3. All PANOs exhibited high recovery at low pH. However, at high pH all PANOs showed low recovery, meanwhile corresponding free base PAs were still satisfactory, this may be due to the hydrolysis of PANOs to corresponding PAs at high pH. PAs with closed ester of retronecine, otonecine and platynecine showed better recovery at low pH.
| PAs | pH of distilled water used as loading solution | ||||
|---|---|---|---|---|---|
| pH 3 | pH 5.5 | pH 7 | pH 8.5 | pH 10 | |
| Monocrotaline | 89 | 88 | 75 | 83 | 93 |
| Monocrotaline N-oxide | 85 | 88 | 95 | 5 | 3 |
| Erucifoline | 91 | 90 | 78 | 79 | 86 |
| Jacobine | 87 | 85 | 64 | 67 | 57 |
| Jacobine N-oxide | 86 | 79 | 69 | 13 | 11 |
| Retrorsine | 84 | 89 | 82 | 74 | 75 |
| Heliotrine | 91 | 81 | 81 | 57 | 62 |
| Heliotrine N-oxide | 89 | 89 | 55 | 30 | 25 |
| Lindelofine | 90 | 84 | 48 | 28 | 14 |
| Lindelofine N-oxide | 93 | 86 | 61 | 32 | 21 |
| Senecionine | 85 | 64 | 75 | 70 | 83 |
| Senecionine N-oxide | 94 | 89 | 82 | 74 | 60 |
| Echimidine | 94 | 81 | 67 | 58 | 50 |
| Senkirkine | 91 | 81 | 83 | 74 | 71 |
| Echimidine N-oxide | 92 | 87 | 68 | 53 | 44 |
For acid wash step, hydrochloric acid, sulfuric acid and FA were used with different concentration (0.01, 0.05, 0.065, 0.1 and 0.2 M). FA (0.065 M) showed better response, and the baseline noise very much reduced. In organic wash step, MeOH, MeCN were used different concertation and volume. MeOH (50%, 5 mL) showed better response. For PA elution, different concentrations of NH4OH (5, 7.5, 10 and 12.5%) in MeOH (1:3, 1:2 and 1:1, (v/v)) and different volumes (5, 7.5, 10, 15 mL) were used. The solution of (10% NH4OH 1:3 MeOH (v/v)) with 10 mL can simultaneously elute all PAs with recoveries ranging from 84% to 95%. The SPE method improved S/N ratio and decreased baseline noise. The recovery of the methods is increased compared with reported methods, for retronecine, otonecine and heliotridine types by 14%, 30% and 59%, respectively.10,17,24
The sensitivity of the methods was improved by using different loading volume (0.25, 0.5, 1, 1.25, 1.5 and 2 L), to maximize the sample volume without including any breakthrough. Full recovery of PAs was obtained until 1.0 L sample volume, hereafter that recovery decreased. A breakthrough was observed when the loading volume exceeded 1000 mL. Recovery of three PA types in different sample volumes shown in Fig. 4.
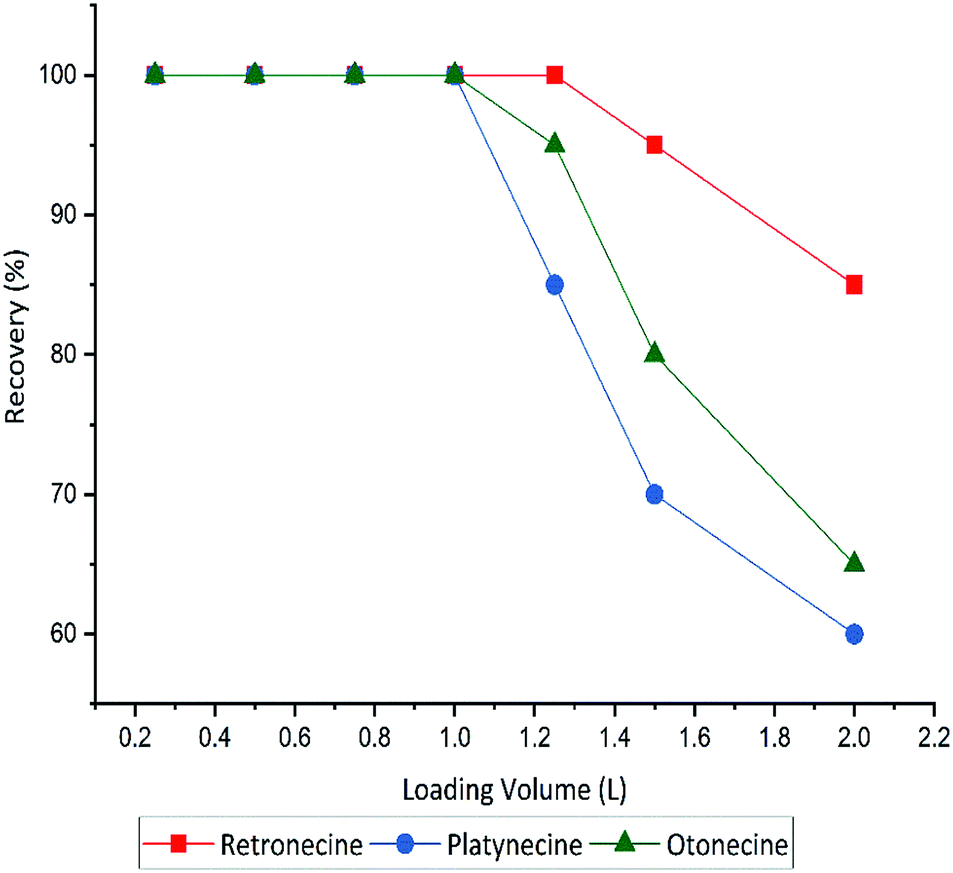 | ||
| Fig. 4 Influence of loading volume on recovery yields of three PA types retronecine, otonecine and platynecine for the optimised SPE method with MCX cartridge (150 mg), and total spiked amount 100 μg L−1 of each compound. | ||
3.1 Application on environmental samples
PAs were determined in environmental samples of soil and surface water, their concentrations listed in Table 4. To our knowledge, the concentration of PAs in water and soil are reported for the first time here, therefore no comparison can be drawn. The concentration of PAs in water and soil possibly correlated with the PAs in ragwort as there is abundant vegetation at the sample locations. In the soil samples, more than 86% of the 15 PAs were quantified, however in water 60% of the PAs were quantified. Thus, may explain that the PAs released from the plants into the soils, and then into water possibly with degradation. Also, 17 out of 90 PA concentrations (18%) exceeded 100 μg L−1. In topsoil, Holte had the highest concentration of PAs; senecionine N-oxide (1349 μg kg−1) and seneciphylline N-oxide (600 μg kg−1), followed by senecionine N-oxide (514 μg kg−1) in Vejle and integerrimine N-oxide (438 μg kg−1) in Holte. In Vejle sub-soil, the PAs concentration ranged 3–370 μg kg−1, jacobine N-oxide is the predominant PA. However, PA concentration in surface water were lower compare to the soils, they were ranged 6–270 μg L−1, jacobine was the highest PA (270 μg L−1) followed by seneciphylline (213 μg L−1) and jacoline (82 μg L−1). Also, some of the PAs were not detected.| Location | Vejle | Borup | Holte | Vejle | Standard used for quantification | ||
|---|---|---|---|---|---|---|---|
| Type of sample | Soil mean ± SD (μg kg−1) | Water mean ± SD (μg L−1) | |||||
| Topsoil | Sub-soil (below 2 cm) | Topsoil | Topsoil | Pond – surface water | |||
| Date | 4. 4. 2018 | 4. 4. 2018 | 28. 5. 2018 | 20. 5. 2018 | 4. 4. 2018 | 11. 5. 2018 | |
| a ND: not detected or under LOD. | |||||||
| Jacoline | 4 ± 1 | 3 ± 1 | 14 ± 1 | 26 ± 2 | 34 ± 2 | 82 ± 2 | Jacobine |
| Jacoline N-oxide | 47 ± 8 | 11 ± 2 | 70 ± 2 | 82 ± 4 | 5 ± 1 | 29 ± 1 | Jacobine N-oxide |
| Riddelline | 3 ± 1 | ND | ND | 5 ± 1 | 11 ± 1 | ND | Erucifoline |
| Erucifoline | ND | 3 ± 1 | 4 ± 1 | 3 ± 1 | 23 ± 1 | ND | Erucifoline |
| Seneciphylline N-oxide | 134 ± 1 | 48 ± 2 | 106 ± 5 | 600 ± 22 | ND | 6 ± 1 | Senecionine N-oxide |
| Erucifoline-N-oxide | 19 ± 1 | 13 ± 1 | 74 ± 2 | 369 ± 12 | 4 ± 1 | 18 ± 1 | Erucifoline-N-oxide |
| Riddelline N-oxide | 3 ± 1 | 4 ± 1 | 3 ± 1 | 10 ± 2 | ND | ND | Erucifoline-N-oxide |
| Jacobine | 6 ± 1 | 6 ± 1 | 226 ± 4 | 288 ± 9 | 270 ± 4 | 122 ± 9 | Jacobine |
| Integerrimine N-oxide | 41 ± 2 | 17 ± 1 | 138 ± 3 | 439 ± 21 | ND | 6 ± 1 | Senecionine N-oxide |
| Senecionine N-oxide | 514 ± 12 | 103 ± 1.3 | 45 ± 3 | 1349 ± 19 | 6 ± 1 | 6 ± 1 | Senecionine N-oxide |
| Jacobine N-oxide | 141 ± 2 | 370 ± 5.1 | 91 ± 3 | 232 ± 8 | 7 ± 1 | 47 ± 2 | Jacobine N-oxide |
| Seneciphylline | 6 ± 1 | 21 ± 1 | 36 ± 2 | 92 ± 2 | 6 ± 1 | 213 ± 10 | Senecionine |
| Integerrimine | ND | 8 ± 2 | 6 ± 1 | 42 ± 2 | ND | ND | Senecionine |
| Senecionine | 22 ± 2 | 17 ± 1 | 44 ± 3 | 296 ± 6 | ND | ND | Senecionine |
| Acetylerucifoline | 191 ± 1 | 110 ± 4 | 22 ± 1 | 82 ± 10 | ND | ND | Erucifoline |
4. Conclusion
The new UPLC-MS/MS method was developed to quantify PAs in soil and water from the environment with limit of detection for the UPLC-MS/MS 2–7 μg L−1 for ten selected PAs. In the method, parameters of both LC and MS part optimized separately. The MS part operated with full scan mode and MRM mode combined in one measurement. As a result, all three types of PAs (free base and N-oxide) could be quantified concurrently, in a considerable shorter runtime compared with previous methods. In addition, full validated SPE for clean-up and pre-concentration up to 1000 times provide 90–100% recovery for different PAs. To the best of our knowledge this is the first study on measuring PAs in soil and water, as the result 15 PAs were quantified, in a range of 3 to 1349 μg kg−1 in soil and 4 to 270 μg L−1 in surface water. The method will be an efficient platform to further study PAs in natural water and aquifers, and to follow their fate after being released or washed into the environment.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and Innovation Programme under the Marie Sklodowska-Curie grant agreement no. 722493 (NaToxAq). We wish to thank Bo Levesen from Vejle municipality for providing samples from Vejle, also Birgitte B. Rasmussen, Linus M. V. Malmquist and Ibrahim Nazem, for their technical assistance.References
- B. Avula, S. Sagi, Y. H. Wang, J. Zweigenbaum, M. Wang and I. A. Khan, Food Chem., 2015, 178, 136–148 CrossRef CAS
.
- J. Robertson and K. Stevens, Nat. Prod. Rep., 2017, 34, 62–89 RSC
- R. Moreira, D. M. Pereira, P. Valentão and P. B. Andrade, Toxicology and Food Safety, Int. J. Mol. Sci., 2018, 19, 1668–1690 CrossRef
- M. R. Gertruida, J. B. Christo and N. E. Jacobus, Phytochem. Lett., 2014, 8, 126–131 CrossRef
- P. P. Fu, Q. Xia, G. Lin and M. W. Chou, Drug Metab. Rev., 2004, 36, 1–55 CrossRef CAS
- S. L. Li, G. Lin, P. P. Fu, C. L. Chan, M. Li, Z. H. Jiang and Z. Z. Zhao, Mass Spectrom., 2008, 22, 591–602 CAS
- L. Kristanc and S. Kreft, Food Chem. Toxicol., 2016, 92, 150–164 CrossRef CAS
- M. Boppre, Food Addit. Contam., Part A, 2011, 28, 260–281 CrossRef CAS
- EFSA, EFSA J., 2011, 9, 1–134 Search PubMed
- M. Bolechová, J. Cáslavský, M. Pospíchalová and P. Kosubová, Food Chem., 2015, 170, 265–270 CrossRef
- H. P. van Egmond, Anal. Bioanal. Chem., 2004, 378, 1152–1160 CrossRef CAS PubMed
- BfR, Opinion No 018/2013 of 15.06.2013, 2013, https://www.bfr.bund.de/cm/349/pyrrolizidine-alkaloids-in-herbal-teas-and-teas.pdf, accessed 25th June 2019.
- JOINT FAO/WHO, 2012, http://www.fao.org/tempref/codex/Meetings/CCCF/CCCF5/cf05_INF.pdf, accessed 25th June 2019.
- C. Gottschalk, S. Ronczka, A. Preiß-Weigert, J. Ostertag, H. Klaffke, H. Schafft and M. Lahrssen-Wiederholt, Anim. Feed Sci. Technol., 2015, 207, 253–261 CrossRef CAS
- E. Kowalczyk and K. Kwiatek, J. Vet. Res., 2018, 62, 183–191 CAS
- C. T. Griffin, M. Danaher, C. T. Elliott, D. G. Kennedy and A. Furey, Food Chem., 2013, 136, 1577–1583 CrossRef CAS
- L. Zhu, J. Ruan, N. Li, P. P. Fu, Y. Ye and G. Lin, Food Chem., 2016, 194, 1320–1328 CrossRef CAS
- M. Rodriguez-Aller, R. Gurny, J. L. Veuthey and D. Guillarme, J. Chromatogr. A, 2013, 1292, 2–18 CrossRef CAS
- H. V. Botitsi, S. D. Garbis, A. Economou and D. F. Tsipi, Mass Spectrom. Rev., 2011, 30, 907–939 CAS
- F. Clauson-Kaas, H. C. B. Hansen and W. B. Strobel, Anal. Bioanal. Chem., 2016, 408, 7981–7990 CrossRef CAS PubMed
- C. Crews, F. Berthiller and R. Krska, Anal. Bioanal. Chem., 2010, 396, 327–338 CrossRef CAS
- A. These, D. Bodi, S. Ronczka, M. Lahrssen-Wiederholt and A. Preiss-Weigert, Anal. Bioanal. Chem., 2013, 405, 9375–9383 CrossRef CAS
- M. Kempf, M. Wittig, A. Reinhard, K. von der Ohe, T. Blacquière, K.-P. Raezke, R. Michel, P. Schreier and T. Beuerle, Food Addit. Contam., Part A, 2011, 28, 332–347 CrossRef CAS
- Y. Zhou, N. Li, F. F. Choi, C. F. Qiao, J. Z. Song, S. L. Li, X. Liu, Z. W. Cai, P. P. Fu, G. Lin and H. X. Xu, Anal. Chim. Acta, 2010, 681, 33–40 CrossRef CAS
- P. Petersson, A. Frank, J. Heaton and M. R. Euerby, J. Sep. Sci., 2008, 31, 2346–2357 CrossRef CAS
-
A. D. McNaught and A. Wilkinson, IUPAC. Compendium of Chemical Terminology, (the “Gold Book”), Blackwell Scientific Publications, Oxford, 2nd edn, 2014, vol. 1, ch. 1, pp. 1074–1075 Search PubMed
- G. L. Long and J. D. Winefordner, Anal. Chem., 1983, 55, 712A–724A CrossRef CAS
- EURL European Commission, Contract No: 26-09, 2013, https://ec.europa.eu/food/sites/food/files/plant/docs/pesticides_mrl_guidelines_wrkdoc_2017-11813.pdf, accessed on 25th June 2019.
- J. Tang, M. Cheng and M. Hattori, Anal. Methods, 2012, 4, 2797–2808 RSC
- W. Rozhon, L. Kammermeier, S. Schramm, N. Towfique, N. A. Adedeji, S. A. Ajayib and B. Poppenbergera, Phytochem. Anal., 2018, 29, 48–58 CrossRef CAS
- F. Kaltner, M. Rychlik, M. Gareis and C. Gottschalk, J. Agric. Food Chem., 2018, 66, 5221–5228 CrossRef CAS
- G. Hosch, H. Wiedenfeld, T. Dingermann and E. Roder, Phytochem. Anal., 1996, 7, 284–288 CrossRef
- BfR, BfR-PA-Honey-1.0/2013, 2013, https://www.bfr.bund.de/cm/349/determination-of-pyrrolizidine-alkaloids-pa-in-honey.pdf, accessed 20th June 2019.
- BfR, BfR-PA-Tea-2.0/2014, 2014, https://www.bfr.bund.de/cm/349/determination-of-pyrrolizidine-alkaloids-pa-in-plant-material.pdf, accessed 20th June 2019.
- T. Mroczek, K. Glowniak and A. Wlaszczyk, J. Chromatogr. A, 2002, 949, 249–262 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra05301h |
| This journal is © The Royal Society of Chemistry 2019 |