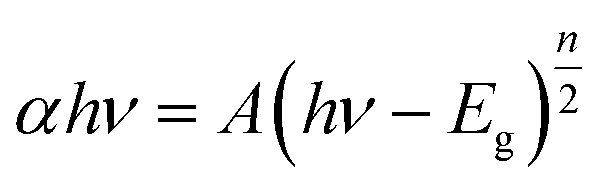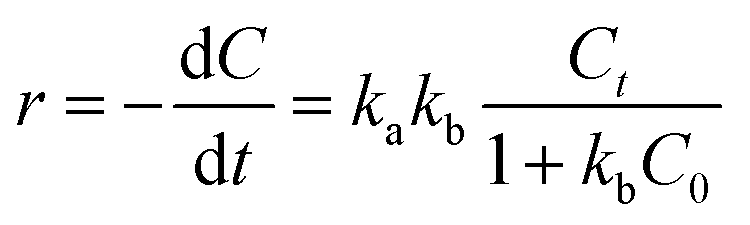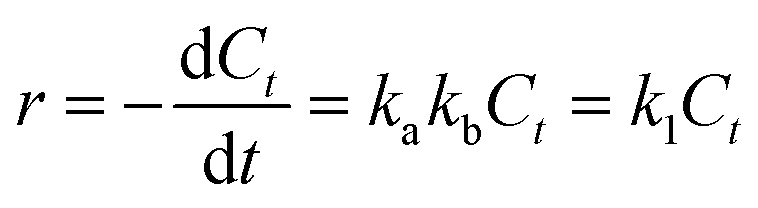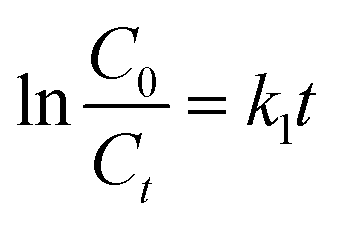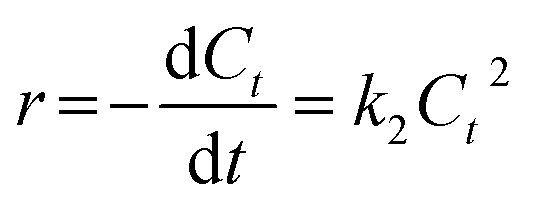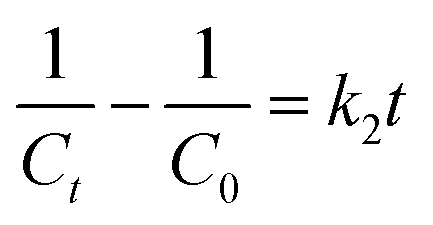DOI:
10.1039/C9RA04445K
(Paper)
RSC Adv., 2019,
9, 27768-27779
Synthesis and application of Bi2WO6 for the photocatalytic degradation of two typical fluoroquinolones under visible light irradiation†
Received
13th June 2019
, Accepted 9th August 2019
First published on 3rd September 2019
Abstract
Bismuth tungstate (Bi2WO6) was successfully synthesized by a method combining ultrasonic solvothermal treatment and high-temperature calcination. The products were affirmed by X-ray diffraction, scanning electron microscopy, UV-vis diffuse reflection spectroscopy, X-ray photoelectron spectroscopy, and Fourier transform infrared spectroscopy. The characterization results indicated that calcination could improve the crystallinity and visible light utilization capacity of Bi2WO6. The photodegradation experiments showed that Bi2WO6 calcined at 450 °C for 3 h exhibited better photocatalytic activity for the degradation of norfloxacin and enrofloxacin under visible light irradiation than the catalyst prepared without calcination or calcined at other temperatures. Meanwhile, the effects of the amount of 450-Bi2WO6, the initial concentration of targets, and the pH of the solutions on the degradation were studied. Under the optimal conditions, the removal ratios reached to 92.95% (for norfloxacin) and 94.58% (for enrofloxacin) within 75 min. Furthermore, h+ and ·O2− were identified to affect the photodegradation process significantly, and the possible photocatalytic mechanism was proposed. The as-prepared sample was verified to possess good stability and reusability, suggesting its potential application prospect in the treatment of fluoroquinolone antibiotics.
1 Introduction
Fluoroquinolone antibiotics (FQs), as common clinical antibiotics, have been widely used in the medical industry, animal husbandry and aquaculture.1 They have been detected in the environment all over the world, such as Asia,2,3 European regions,4 the United States,5 and Australia6 in recent years. The ecological risk of FQs is of great concern mainly due to the potential long-term impacts on the environment even at low concentrations.7–9 They could increase the tolerance of bacteria,10,11 cause the generation of resistant genes,12,13 inhibit the photosynthesis of plants,14 and gradually threaten both environmental and human health.13,15–17 However, the stability of FQs makes their effective removal by conventional treatment processes challenging, such as biodegradation,18,19 biosorption,20 sludge digestion,21 adsorption removal,22,23 and so on.
To date, photocatalytic degradation technology has been deemed as one of the most promising treatments for the refractory pollutants.24–26 Modern chemistry aims to identify high-efficiency and environmental-friendly photocatalytic materials which could utilize solar energy efficiently to mineralize organic pollutants thoroughly.27–30 In the earlier study, bismuth (Bi) was used to modify TiO2.31 Gradually, excellent photocatalytic properties have been found in various Bi-based compounds, such as Bi2O3,32 BiOX (X = Cl, Br, I),33–36 Bi2WO6,37–39 BiVO4,40 Bi2MoO6,41 and so on.
Bismuth tungstate (Bi2WO6) is one of the simplest Aurivillius oxides with a layered structure and visible light response (the forbidden bandwidth is about 2.8 eV). As early as 1999, Kudo and Hijii obtained Bi2WO6 by a solid-state method and proved that the product had the photocatalytic activity for the first time.42 Tang et al.43 confirmed its photocatalytic activity for mineralization of chloroform and acetaldehyde. Then the application market of Bi2WO6 in photocatalysis was opened. But there were still some limits for Bi2WO6 to perform photocatalysis due to the high recombination rate of the photogenerated electron–hole pairs and the boundedness of visible light response region.30,38,39 In order to enhance the photocatalytic activity and widen the visible light response region of Bi2WO6, some preparation methods have been developed and optimized to control the crystal form, crystallinity, particle size, specific surface area, and so on.30,44 Therein, hydrothermal and solvothermal synthesis have got into the researcher's good graces in recent years, because the corresponding reaction conditions were comparatively mild, and the obtained products had good dispersibility with a controlled particle size distribution. Factors that would affect the crystal structure of Bi2WO6 were usually adjusted to obtain the excellent photocatalytic activity, such as the pH and temperature of the reaction system. Meanwhile, a variety of auxiliary methods were also introduced into the preparation processes to improve the photocatalytic activity. Ultrasonic was introduced for its strong penetrability and excellent rapid mixing capacity,45 and calcination was reported to increase the crystallinity and reduce the particle size.46,47 However, long reaction time (up to 20–48 h) was usually needed for the hydrothermal/solvothermal stage.37–39,48,49
In this study, a combined method of ultrasonic solvothermal treatment and high-temperature calcination was attempted to synthesize Bi2WO6 with a shorter duration. The calcination temperatures were also investigated. The structure, morphology, chemical status and optical properties of the prepared samples were characterized by powder X-ray diffraction (XRD), scanning electron microscopy (SEM), field emission transmission electron microscope (FETEM), X-ray photoelectron spectroscopy (XPS), Fourier transformation infrared spectra (FT-IR) and UV-vis diffuse reflectance spectroscopy (DRS). The photocatalytic capacities were verified by degrading norfloxacin (NOR) and enrofloxacin (ENR) under visible light irradiation. The degradation kinetics were investigated, and the impacts of different reaction factors on the degradation were examined, such as the amount of photocatalyst, the initial concentration of drugs, and initial pH of the degradation system. Additionally, the dominant active species and probable photocatalytic degradation mechanism of NOR were also discussed.
2 Experimental
2.1 Materials
Bismuth(III) nitrate pentahydrate (Bi(NO3)3·5H2O, 99.0%), sodium tungstate dihydrate (Na2WO4·2H2O, 99.5%), ethylene glycol ((CH2OH)2, EG, 96.0%), sodium hydroxide (NaOH, 96.0%), ethanol (C2H5OH, 99.7%), isopropanol ((CH3)2CHOH, IPA, 99.7%), and triethanolamine ((CH2CH2OH)N, TEOA, 99.0%) were purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). Norfloxacin (C16H18FN3O3, NOR, 98.0%), enrofloxacin (C19H22FN3O3, ENR, 98.0%), and p-benzoquinone (C6H4O2, p-BQ, 97.0%) were supplied by Aladdin Company (Shanghai, China). Degussa P25-TiO2 nanoparticle was purchased from Degussa Corporation (Germany). Indium-tin oxide glass (ITO) was obtained from South China Science & Technology Co., Ltd (Guangzhou, China) All chemicals used in this research were analytical reagent grade without further purification. Milli-Q water (a minimum resistivity of 18.25 MΩ cm) was used as the solvent for all the solutions or dispersions.
2.2 Synthesis of Bi2WO6 samples
The solvothermal method was carried out to synthesize Bi2WO6 according to a previous study with modification.37 Initially, 0.9701 g Bi(NO3)3·5H2O was dissolved in 30 mL distilled water to form a homogeneous solution (solution A, 2 mM) and stirred vigorously for 10 min and sonicated for 20 min; 0.3298 g Na2WO4·2H2O was dissolved in 20 mL EG and stirred for 30 min to form solution B (1 mM). The solution B was added dropwise into solution A under ultrasonication for 60 min, and a white suspension was formed. After adjusting the pH of the mixture solution to 6, the solution was further stirred for 60 min. Then, the mixture solution was transferred into a 100 mL Teflon-lined stainless steel autoclave to undergo a solvothermal treatment at 180 °C for 12 h. After that, the autoclave was cooled down naturally to room temperature. The resulting precipitate was rinsed with ultrapure water and ethanol for three times. The obtained product was dried at 80 °C and denoted as bulk-Bi2WO6. Subsequently, the bulk-Bi2WO6 samples were calcined at different temperatures (T = 350 °C, 450 °C, and 550 °C) for 3 h. The corresponding products were named 350-Bi2WO6, 450-Bi2WO6, and 550-Bi2WO6, respectively.
2.3 Characterization
The crystal structures and phase identification analyses were characterized by XRD (Bruker D8 Advance, Germany) at the angle range of 2θ = 10–90° using Cu-Kα irradiation (λ = 0.15418 nm). The morphologies and structures were examined by SEM (Zeiss Sigma HD, Germany) and FETEM (Tecnai G2 F20 S-TWIN TMP, USA). FT-IR was performed on Nicolet iS50 (Thermo Fisher Scientific, USA) spectrophotometer in the range of 400–4000 cm−1. The chemical status and elemental compositions were analyzed by XPS (Thermo Fisher Scientific, USA) with monochromatic Al-Kα source (hν = 1486.6 eV, 6 mA × 12 kV), and the deconvolution of the spectrum was performed using the XPS PEAK 41 program with Gaussian functions after the subtraction of a Shirley background. The optical properties of as-prepared samples were investigated by UV-vis DRS (TU-1901, China) with the range of 200–800 nm, and BaSO4 was used as a reflectance standard. The specific surface areas were recorded by the Brunauer–Emmett–Teller (BET) technique (Quadrasorb SI-3MP, Quantachrome, USA) with nitrogen adsorption–desorption isotherm.
2.4 Photocatalytic degradation of NOR and ENR
The photocatalytic activities of the as-prepared Bi2WO6 samples were investigated by the degradation of NOR and ENR at room temperature under visible light irradiation. A Xenon arc lamp (300 W) was used as the visible light source with a UV cutoff filter (λ > 400 nm). In each experiment, 50 mg of Bi2WO6 sample was suspended into 100 mL of NOR or ENR aqueous solution (10 mg L−1). Prior to irradiation, the suspension was stirred in the dark for 60 min to ensure the adsorption–desorption equilibrium. After that, 2 mL of suspensions were withdrawn and centrifuged (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, 10 min) to produce the supernatant which was analyzed on a TU-1901 spectrophotometer, and the obtained result acted as the initial concentration, C0. Then, the irradiation was conducted with continuous magnetic stirring, and the sample solutions were withdrawn every 15 min and centrifuged. The concentrations of NOR or ENR (generally labeled as Ct) were monitored with the spectrophotometer. In this study, 0.1 mol L−1 NaOH was used as a reference solution, and the detected λmax of NOR and ENR in the solutions were 273 and 271 nm, respectively (Fig. S1 and S2†). The degradation efficiencies were calculated in the form of (C0 − Ct)/C0.
000 rpm, 10 min) to produce the supernatant which was analyzed on a TU-1901 spectrophotometer, and the obtained result acted as the initial concentration, C0. Then, the irradiation was conducted with continuous magnetic stirring, and the sample solutions were withdrawn every 15 min and centrifuged. The concentrations of NOR or ENR (generally labeled as Ct) were monitored with the spectrophotometer. In this study, 0.1 mol L−1 NaOH was used as a reference solution, and the detected λmax of NOR and ENR in the solutions were 273 and 271 nm, respectively (Fig. S1 and S2†). The degradation efficiencies were calculated in the form of (C0 − Ct)/C0.
2.5 Detection of active species
In order to understand the active species and possible photocatalytic mechanism, the quenching experiments were carried out. The experimentation was similar to the photocatalytic test except that scavengers were incorporated into the reaction solutions prior to the addition of photocatalyst. In this work, 20 mg L−1 p-benzoquinone (p-BQ), 0.01 mol L−1 triethanolamine (TEOA), and 0.01 mol L−1 isopropanol (IPA) were used as the scavengers in the photodegradation systems to quench superoxide radicals (·O2−), photo-holes (h+), and hydroxyl radicals (·OH), respectively.
2.6 Electrochemical characterization
The Mott–Schottky plots were measured in Na2SO4 electrolyte (0.1 mol L−1) at room temperature by an electrochemical workstation (CHI604E, China) at a constant frequency of 1000 Hz using a 5 mV amplitude signal. And the potential ranged from 0.7 V to − 0.8 V at a rate of 0.01 V s−1.
The ITO (10 mm × 10 mm × 1 mm) modified with photocatalyst was used as the working electrode. The Pt plate and saturated calomel electrode (SCE, 0.2415 V vs. NHE) were acted as the counter electrode and reference electrode, respectively.
2.7 Recycling experiments
Recycling experiments were programmed to investigate the reusability and stability of the prepared Bi2WO6 samples. The tests were conducted in four sequential cycles. Multiple sets of parallel experiments were performed at the same time, and finally the remaining solutions were merged to recovery the catalysts. After centrifugation and filtration, the recovered catalysts were washed and dried at 80 °C. The obtained powders were used in the next cycle experiment. The amount of catalyst used in each experiment was guaranteed to be the same (50 mg).
3 Results and discussions
3.1 Characterizations of the as-prepared samples
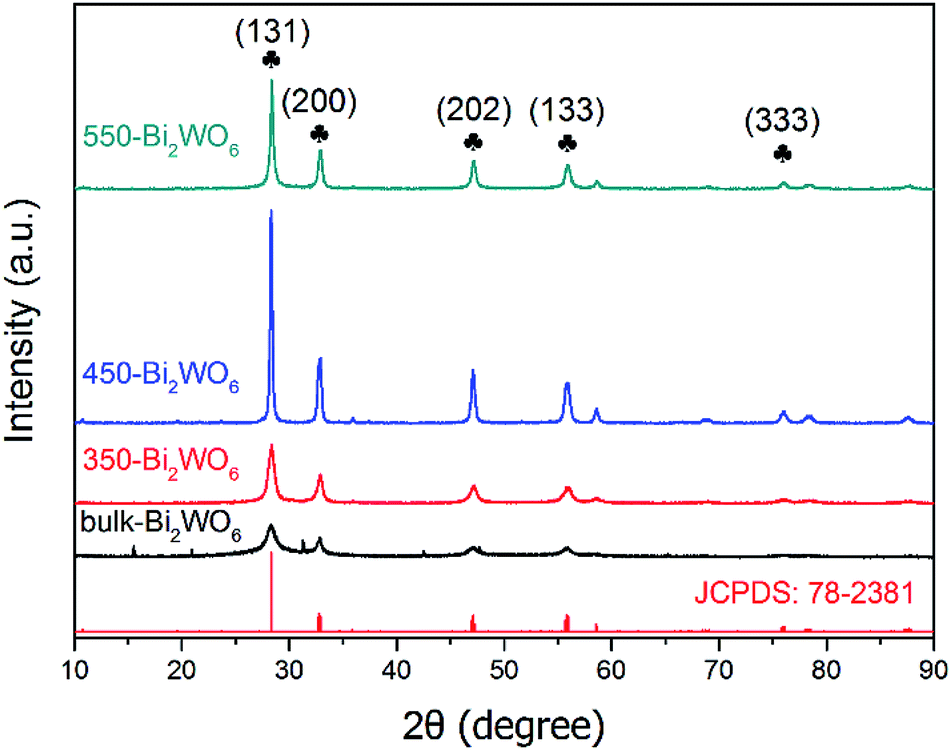
Powder XRD patterns of the prepared Bi2WO6 samples were shown in Fig. 1. There were similar diffraction peaks for the four samples, indicating that they might have the same crystal form. The main characteristic diffraction peaks situated at 2θ = 28.3°, 32.9°, 47.1°, 55.8°, 76.1° are related to the (131), (200), (202), (133) and (333) crystal planes of orthorhombic phase of Bi2WO6 (PDF #79-2381), respectively; which were consistent with those in other literatures,39,44,48 implying that the as-prepared samples were Bi2WO6 with high purity. Although some additional peaks were observed in bulk-Bi2WO6, all of these peaks were disappeared after calcination. As for the bulk-Bi2WO6, the shortened hydrothermal reaction time may affect the crystallinity of the products. However, the characteristic diffraction peaks became more intensive and acute with the increase of calcination temperature, indicative of the growth of Bi2WO6 nanocrystals. Fumiaki, et al.47 and Sheng, et al.50 had also affirmed that calcination would affect the crystallinity of Bi2WO6.
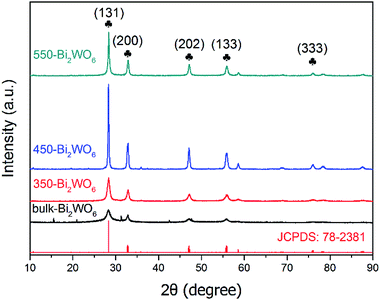 |
| | Fig. 1 XRD patterns of the as-prepared Bi2WO6 samples. | |
The morphology and structure were investigated by SEM (Fig. 2). Agglomerated small particles were observed for bulk-Bi2WO6 (Fig. 2(a)), while flower-like, lamellar-like and rod-like nanostructures were observed for 350-Bi2WO6, 450-Bi2WO6, and 550-Bi2WO6 (Fig. 2(b–d)), respectively. It suggested that calcination could affect the morphologies of Bi2WO6 samples efficiently. The surface areas of bulk-Bi2WO6, 350-Bi2WO6, 450-Bi2WO6, and 550-Bi2WO6 gauged by BET analysis were 3.73, 9.53, 15.83, and 15.27 m2 g−1, respectively. Among them, 450-Bi2WO6 had the largest surface area. FETEM was used to study the ultrastructure of 450-Bi2WO6 sample, and the typical image was presented in Fig. S3.† The lattice fringe spacing was about 0.315 nm, which was corresponding to the (131) crystal plane lattice fringes of orthorhombic Bi2WO6.39,51
 |
| | Fig. 2 SEM images of (a) bulk-Bi2WO6, (b) 350-Bi2WO6, (c) 450-Bi2WO6, (d) 550-Bi2WO6. | |
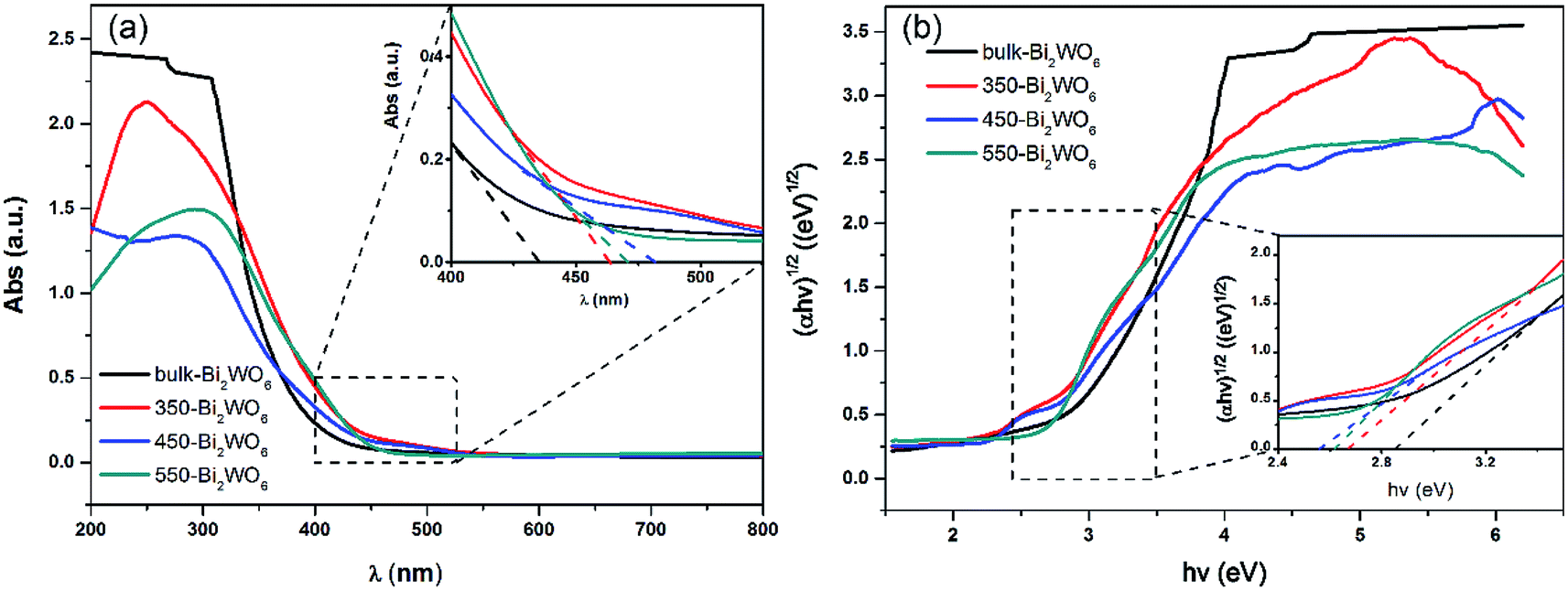
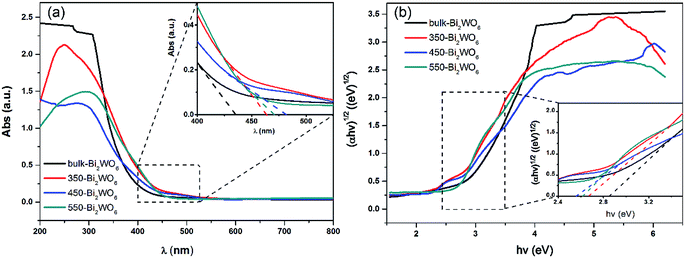
UV-vis DRS was used to analyze the optical property of catalysts and deduce their forbidden bandwidth (Eg). Fig. 3 showed the UV-vis absorption spectra and Tauc plots ((αhν)1/2 vs. hν) of the samples. There were strong broad absorption bands in the range from 200 to about 450 nm for all the as-prepared samples (Fig. 3(a)), indicating their potential photocatalytic activity under the visible light. The optical absorption edge of bulk-Bi2WO6 was about 435 nm and its Eg value was estimated to be 2.85 eV, which was in agreement with the results from other literatures.48,51 Nevertheless, for 350-Bi2WO6, 450-Bi2WO6, and 550-Bi2WO6, the optical absorption edges were 464, 482, and 471 nm, respectively. Undoubtedly, the absorption edges of the calcined samples were red-shifted compared with that of bulk-Bi2WO6. The forbidden bandwidths (Eg) of 350-Bi2WO6, 450-Bi2WO6, and 550-Bi2WO6 were also calculated with the Kubelka–Munk equation (eqn (1)) based on the optical absorption edge from the UV-vis DRS spectrum. The values were 2.67, 2.57, and 2.63 eV, respectively (Fig. 3(b)). It was validated that the visible light utilization capacity of the Bi2WO6 samples could be improved by calcination. Among the four as-prepared samples, 450-Bi2WO6 held the widest absorption region and the lowest band gap. Following is the Kubelka–Munk equation:
| |
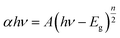 | (1) |
where
α is the absorption coefficient,
h is Planck's constant, 6.626 × 10
−34 J s,
ν is the optical frequency (s
−1),
A is a constant (generally 1),
Eg is the forbidden bandwidth (eV), and
n is decided by the optical transition type of typical semiconductors. Ordinarily,
n is 1 or 4, and 1 was selected in this research.
52
 |
| | Fig. 3 (a) UV-vis DRS spectra and (b) forbidden bandwidths of Bi2WO6 samples. | |
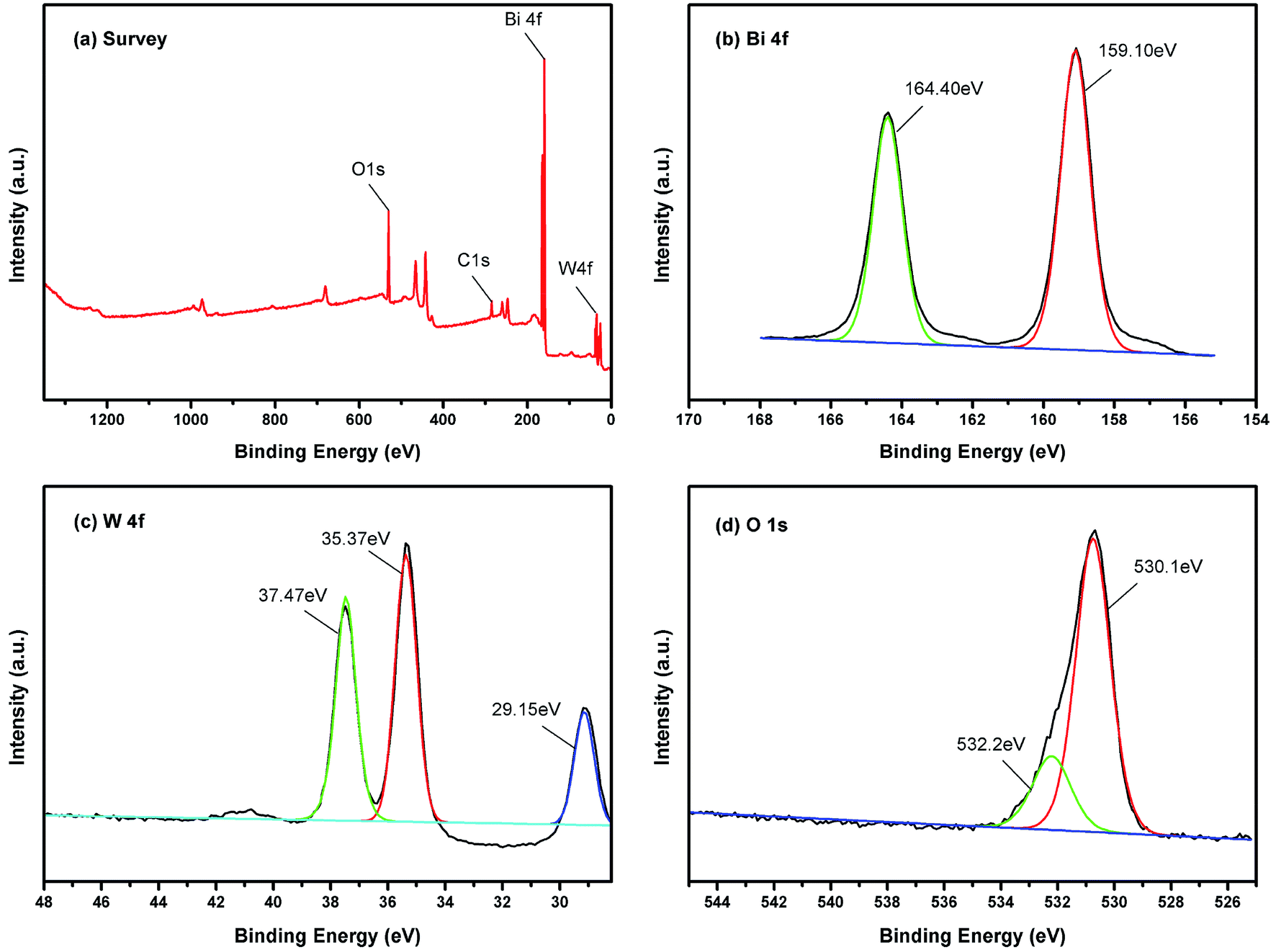
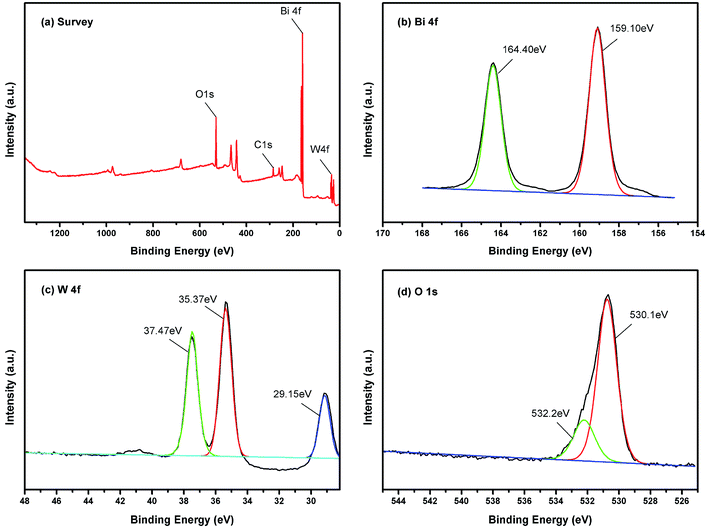
Surface status and elemental composition of 450-Bi2WO6 were examined by XPS spectra (Fig. 4). As shown in Fig. 4(a), the presence of Bi, W, O, and C elements could be identified in the sample, and their mole ratio was estimated to be 1.93:1:6.04 (Bi:W:O) based on the XPS results, which were consistent with the results of other literature.38 The C element might be derived from the pollutants in the XPS instrument or from the air. The binding energy of C 1s was used as the correction standard (284.8 eV). The two strong peaks located at 159.10 and 164.40 eV were attributed to Bi 4f7/2 and Bi 4f5/2 of Bi3+, respectively (Fig. 4(b)) (with the splitting energy Δ = 5.40 ± 0.10 eV).51 As shown in Fig. 4(c), the peaks at 35.37, 37.47, and 29.15 eV should be indexed to the W6+ in 450-Bi2WO6 sample.53 The XPS spectrum in the O 1 s region could be fitted into two peaks (Fig. 4(d)), located at 530.01 and 532.20 eV, which were corresponded to the [WO4]2− layers and crystal lattice oxygen [Bi2O2]2+ of 450-Bi2WO6.38,54
 |
| | Fig. 4 High-resolution XPS of 450-Bi2WO6 (a) total survey, (b) Bi 4f, (c) W 4f, (d) O 1 s. | |
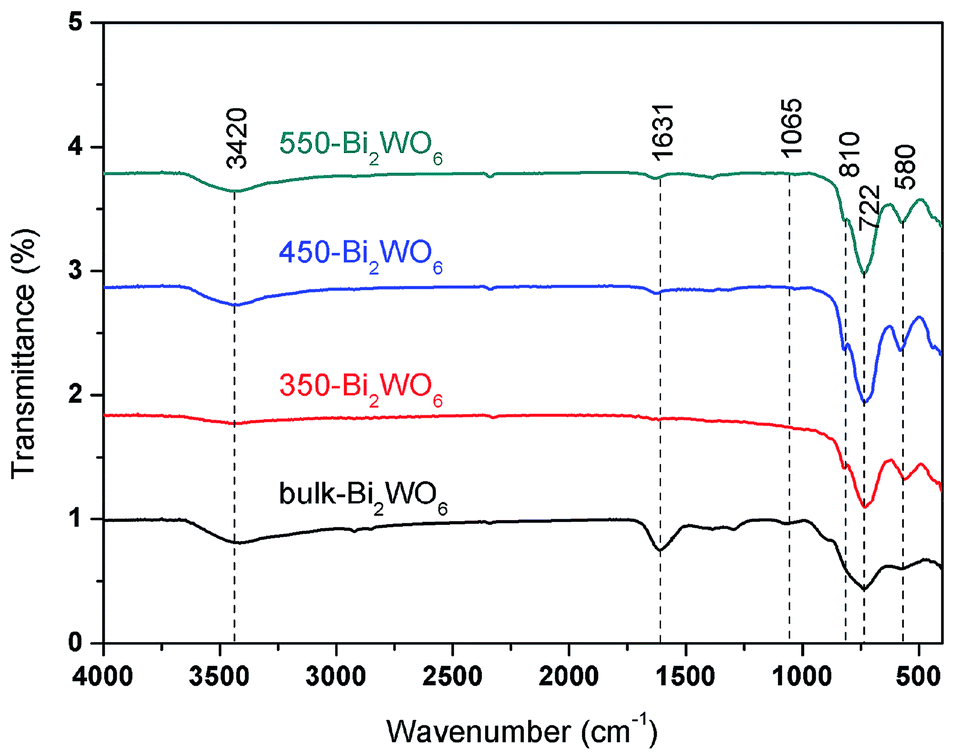
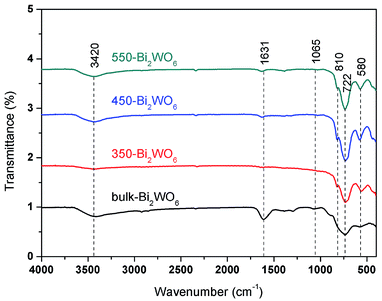
The FT-IR spectra of the Bi2WO6 samples were displayed in Fig. 5. The Bi–O–Bi asymmetric stretch located at 580 cm−1, while W–O was stretching at 722 and 1065 cm−1, and Bi–O was stretching at 810 cm−1, which were matched well with the literatures.55,56 The absorption bands at 1631 and 3420 cm−1 could be ascribed to the stretching and bending modes of O–H on the surface of the samples.48
 |
| | Fig. 5 FT-IR spectra of Bi2WO6 samples. | |
3.2 The kinetics of photocatalytic degradation
In order to investigate the kinetics, the as-prepared Bi2WO6 samples (50 mg) were put into 100 mL of 10 mg L−1 NOR or ENR solution to undergo photocatalytic degradation, and P25-TiO2 was used for comparison. Before irradiation, the adsorption–desorption experiment was firstly conducted in the dark, and the adsorption could reach equilibrium in 60 min (shown in Fig. S4†). The results also indicated that the adsorption ratio of the as-prepared catalyst was so small that the amount could be neglected. The Langmuir–Hinshelwood kinetic equation was usually used to calculate the rate constant of photocatalytic degradation, which was demonstrated in eqn (2):57,58| |
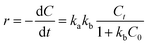 | (2) |
where r is the degradation rate of substrates (mol (L−1 min−1)), ka is the physical constant of the reaction system (i.e., the rate constant of solute molecules adsorbed on the surface of the catalyst (mol min−1)), kb is the photodegradation rate constant of reactive substrates (mol−1), Ct and C0 are the concentrations of the reactants at time t and “t = 0” (mg L−1).
When the initial concentration of substrates was sufficiently low, kbC0 ≪ 1, the eqn (2) could be reduced into eqn (3):
| |
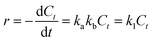 | (3) |
The eqn (4) was obtained from eqn (3) by integral processing:
| |
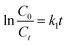 | (4) |
where
k1 =
kakb, which is the pseudo-first-order rate constant (min
−1). The slope of ln(
C0/
Ct)
vs. t is
k1.
When there were secondary reactants or two first-order reactants, the reaction could be pseudo-second-order kinetics. The eqn (2) was simplified and integral processed to eqn (5) and (6):
| |
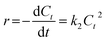 | (5) |
| |
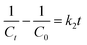 | (6) |
where
k2 is the pseudo-second-order rate constant (L (mol
−1 min
−1)). The fitted slope of
1/
Ct vs. t is
k2.
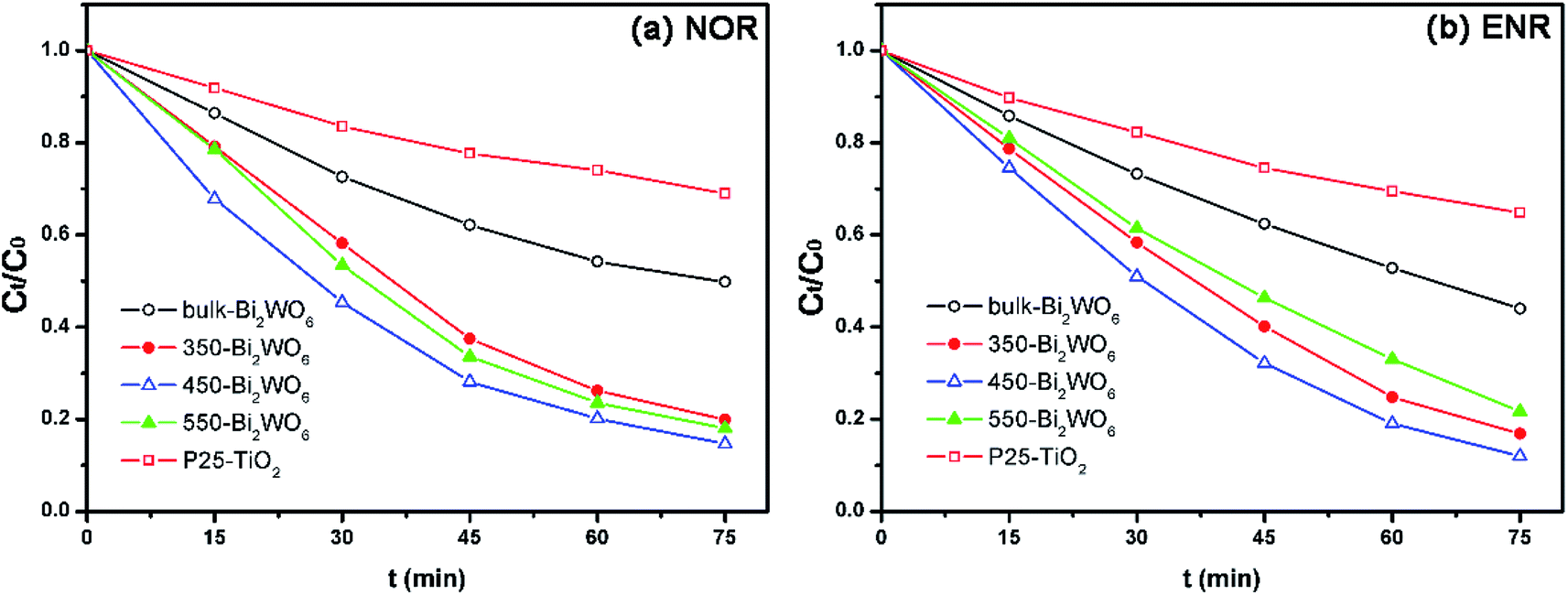
The degradation results were shown in Fig. 6 and Table 1. As shown in Fig. 6, all prepared Bi2WO6 samples exhibited higher activity than P25-TiO2. Moreover, the removal ratios of NOR and ENR with the presence of 450-Bi2WO6 could reach 85.36% and 88.01% within 75 min, respectively; suggesting an excellent photocatalytic activity for the degradation of NOR and ENR. The coefficients (R2) of pseudo-first-order kinetic model were ca. 0.99, which were much better than that of the pseudo-second-order kinetic model for the calcined Bi2WO6 samples. It implied that the pseudo-first-order kinetic model was more suitable for the prepared Bi2WO6 samples in this test. The sample of 450-Bi2WO6 had the highest rate constant (0.0261 min−1 of NOR and 0.0289 min−1 of ENR), and then it was chosen as the catalyst in the subsequent experiments.
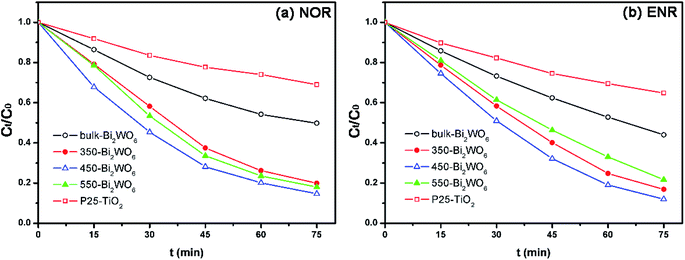 |
| | Fig. 6 Photocatalytic degradation of (a) NOR and (b) ENR with different photocatalysts (10 mg L−1 drug solution, 0.5 g L−1 photocatalyst, initial pH of 10.2 for NOR and of 10.7 for ENR). | |
Table 1 Kinetics parameters for degradation of NOR and ENR with photocatalystsa
| Drugs |
Materials |
Pseudo first-kinetic |
Pseudo second-kinetic |
| k1 (min−1) |
R2 |
k2 (L (mol−1 min−1)) |
R2 |
| The operating conditions were given in the caption of Fig. 6. |
| NOR |
Bulk-Bi2WO6 |
0.0096 |
0.9885 |
1.4287 × 10−5 |
0.9953 |
| 350-Bi2WO6 |
0.0225 |
0.9910 |
4.6973 × 10−5 |
0.9312 |
| 450-Bi2WO6 |
0.0261 |
0.9954 |
6.9244 × 10−5 |
0.9438 |
| 550-Bi2WO6 |
0.0241 |
0.9905 |
5.5157 × 10−5 |
0.9410 |
| P25-TiO2 |
0.0049 |
0.9861 |
0.5437 × 10−5 |
0.9946 |
| ENR |
Bulk-Bi2WO6 |
0.0109 |
0.9890 |
1.7459 × 10−5 |
0.9727 |
| 350-Bi2WO6 |
0.0243 |
0.9840 |
6.4410 × 10−5 |
0.8671 |
| 450-Bi2WO6 |
0.0289 |
0.9894 |
6.9452 × 10−5 |
0.8494 |
| 550-Bi2WO6 |
0.0202 |
0.9840 |
4.4378 × 10−5 |
0.8685 |
| P25-TiO2 |
0.0058 |
0.9913 |
0.6309 × 10−5 |
0.9986 |
3.3 Photocatalytic activity
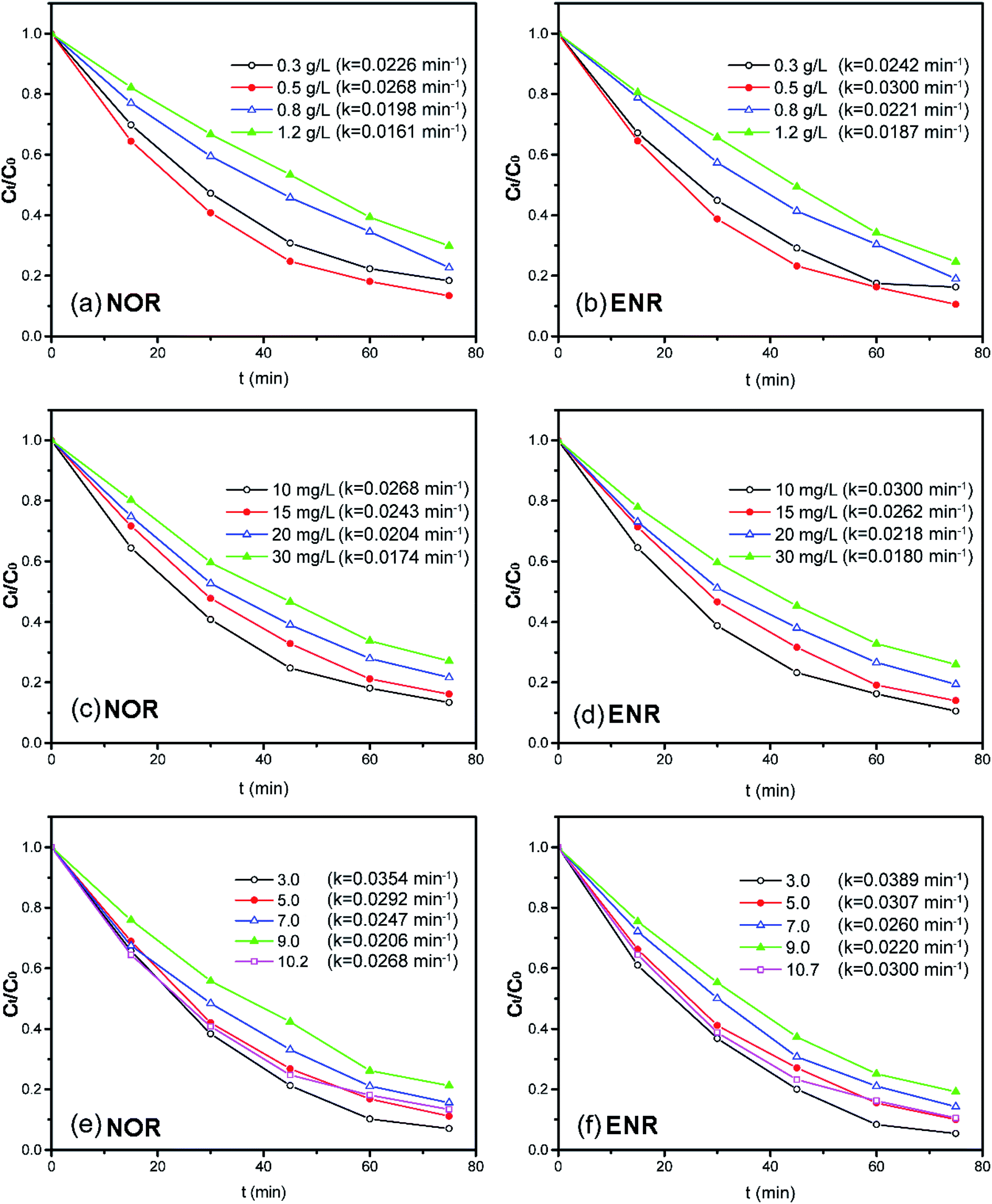
3.3.1 Effect of dosage. The effect of the photocatalyst dosage (ranging from 0.3 to 1.2 g L−1) on the NOR or ENR degradation was investigated with 10 mg L−1 NOR or ENR solution at the original pH. As shown in Fig. 7(a) and (b), both of the photocatalytic degradation efficiencies of NOR and ENR increased firstly and then decreased with the increase of photocatalyst dosage (the pseudo-first-order kinetic results were shown in Fig. S4(a) and (b)†). When the dosage was 0.5 g L−1, the degradation efficiencies of NOR and ENR were the maximum with the values of 86.62% and 89.44% within 75 min, respectively. When the amount of catalyst was not enough, the given light energy cannot be converted into chemical energy entirely so that the light energy failed to be fully utilized. With the increase of the catalyst, there were more active centers, which could effectively utilize the light energy to produce more charge carriers and active species. However, when the amount of catalyst exceeded 0.5 g L−1, the degradation efficiencies of NOR and ENR were slightly reduced. This might be attributed to the light shielding effect since more Bi2WO6 powders suspended in the solution and scattered the light. Correspondingly, the efficiency of the light and the degradation ratios were decreased.59,60 In this test, 0.5 g L−1 450-Bi2WO6 was selected as the optimum dosage.
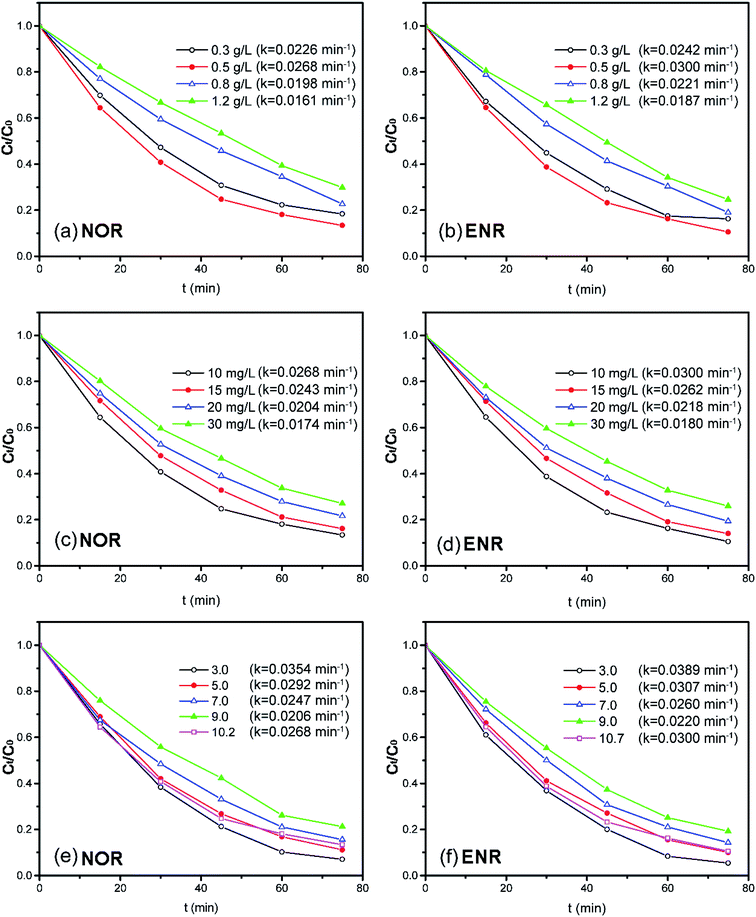 |
| | Fig. 7 Effects of the operation parameters on the degradation of NOR or ENR (different amounts of 450-Bi2WO6 for the solutions with the initial concentration of 10 mg L−1 at original pH; solutions with different initial concentrations at 450-Bi2WO6 dosage of 0.5 g L−1 and original pH; solutions with the initial concentration of 10 mg L−1 at 450-Bi2WO6 dosage of 0.5 g L−1 and different initial pH). | |
3.3.2 Effect of initial concentration. The effect of initial NOR or ENR concentration (ranging from 10 mg L−1 to 30 mg L−1) on the degradation efficiency was studied in the solutions with 0.5 g L−1 450-Bi2WO6 at original pH, and the results were shown in Fig. 7(c) and (d) (the pseudo-first-order kinetic results were shown in Fig. S4(c) and (d)†). With the initial concentration increasing from 10 mg L−1 to 30 mg L−1, the degradation efficiencies decreased from 86.62% to 72.92% for NOR, and from 89.44% to 74.07% for ENR within 75 min, respectively. This may be due to that the continuous increase of the substrates would cause the increase of light scattering. Moreover, with the increase of the initial concentration, more specific active centers would be occupied. Then, the adsorption of H2O, O2, OH−, and other molecules on active centers would reduce. Accordingly, the photocatalytic degradation efficiencies would also decrease.37 Generally, the photocatalytic oxidation technology was more suitable for the treatment of pollutants with lower concentrations.
3.3.3 Effect of pH. The initial pH of the substrate solutions could have a significant influence on the charge properties of the catalyst surface, the adsorption of the solute molecules on the catalyst surface, as well as the dispersion and dissociation of the solvent molecules. Thus, the pH would play a key role in affecting the photocatalytic degradation.61 As shown in Fig. 7(e) and (f), with the presence of 0.5 g L−1 450-Bi2WO6 and 10 mg L−1 NOR or ENR, the degradation efficiencies of NOR (ENR) were 92.95% (94.58%), 88.84% (89.90%), 84.32% (85.73%), 78.74% (80.83%) and 86.62% (89.44%) within 75 min at pH 3.0, 5.0, 7.0, 9.0, 10.2/10.7 (the original pH values of NOR/ENR solutions), respectively. The pseudo-first-order kinetic results were shown in Fig. S4(e) and (f).† The degradation efficiencies in acidic conditions were significantly higher than that in alkaline conditions for both targeted substances. The highest degradation efficiencies of NOR and ENR were obtained at pH 3.0, and the rate constants were 0.0354 and 0.0389 min−1, respectively. Firstly, H+ could be trapped by O2 on the surface (O2sur) of 450-Bi2WO6 in the acidic condition to form ·OH through a series of oxidation–reduction reactions. The ·OH had a strong oxidation activity to react with the organics in water. Secondly, the pHpzc of 450-Bi2WO6 was 3.5–4.5, which was obtained from the mass titration method and other literatures.62,63 The surface of 450-Bi2WO6 was positively charged at pH 3.0. It facilitated the formation of ·O2− (e− + O2sur → ·O2−) from the reactions of photo-electrons (e−) and O2sur of Bi2WO6, which had a strong oxidizing property for degrading the organics. The reactions were also benefited for the separation of e− and h+, which could improve photocatalytic activity. Therefore, pH 3.0 was selected as the optimal pH during the degradation experiments.
3.3.4 Comparison of the as-prepared Bi2WO6 to the already reported ones. As we known, Bi2WO6 is a kind of photocatalyst with good photocatalytic performance. Since its first synthesis, the preparation methods were adjusted by different researchers to improve its photocatalytic activity or utilizability. Table 2 summarized some applications of Bi2WO6 on the photodegradation of antibiotics. As can be seen in the table, though the solvothermal reaction time was shortened in this study, the photocatalytic capacity of the prepared catalysts was comparable with that of the reported Bi2WO6. It may be indicated that calcination could remedy the deficiency caused by the shortened hydrothermal reaction time. It had been ever confirmed that calcination at an appropriate temperature could promote the further growth of Bi2WO6 and improve the photodegradation activity.50
Table 2 Comparison of the as-prepared Bi2WO6 to that in the already reported literatures
| Material |
Preparation method |
Solvothermal/hydrothermal time |
Morphology |
Substrate solution |
Solid-to-liquid ratio |
Degradation efficiency |
Reference |
| Not mentioned. |
| Bi2WO6 |
Hydrothermal method withthe reaction solution at pH 4 |
20 h |
Ultrathin nanoflakes |
100 mL of 10 mg L−1 norfloxacin at pH 9 |
1 g L−1 |
90% (60 min) |
1 |
| Sponge-loaded Bi2WO6 |
Solvothermal method |
20 h |
—a |
70 mg L−1 tetracycline hydrochloride aqueous solution |
8 g L−1 |
94% (90 min) |
32 |
| Bi2WO6 |
Ultrasonic assisted hydrothermal method |
20 h |
Nanocuboids |
100 mL of 10 mg L−1 levofloxacin at pH 7.14 |
0.75 g L−1 |
80% (150 min) |
47 |
| Bi2WO6 |
Hydrothermal process |
20 h |
Nanoflowers |
100 mL 10 mg mL−1 ceftriaxone sodium aqueous solution |
1 g L−1 |
70.18% (240 min) |
62 |
| Bi2WO6 |
Ultrasonic assisted hydrothermal method combined with calcination |
12 h |
Lamellar-like nanostructures |
100 mL of 10 mg L−1 norfloxacin or enrofloxacin at pH 3 |
0.5 g L−1 |
92.95% for norfloxacin (75 min) and 94.58% for enrofloxacin (75 min) |
The present study |
3.4 Detection of active species
To evaluate the role of active species (·O2−, h+, and ·OH) during the photocatalytic degradation, the quench experiments were carried out for NOR by adding different scavengers. As can be seen in Fig. 8, the maximum removal ratio of NOR was achieved in the absence of any scavenger species. When p-BQ, TEOA, and IPA were added into the reaction systems, the removal ratios of NOR decreased from 90.62% (without scavenger) to 41.94%, 29.13%, and 73.78%, respectively. The pseudo-first-order rate constants were also decreased to 0.0072, 0.0045 and 0.0178 min−1 with the presence of p-BQ, TEOA, or IPA from 0.0354 min−1, respectively. Thus, it could be reasonably deduced that the h+, ·O2−, and ·OH were generated during the visible light irradiation with the presence of 450-Bi2WO6, and participated in the photocatalytic reaction. Compared with ·OH, h+ and ·O2− played a significant role in the photodegradation process.
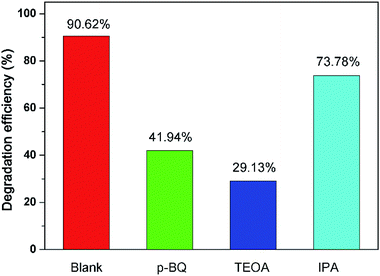 |
| | Fig. 8 Effect of different scavengers on the degradation efficiency of NOR with 450-Bi2WO6. | |
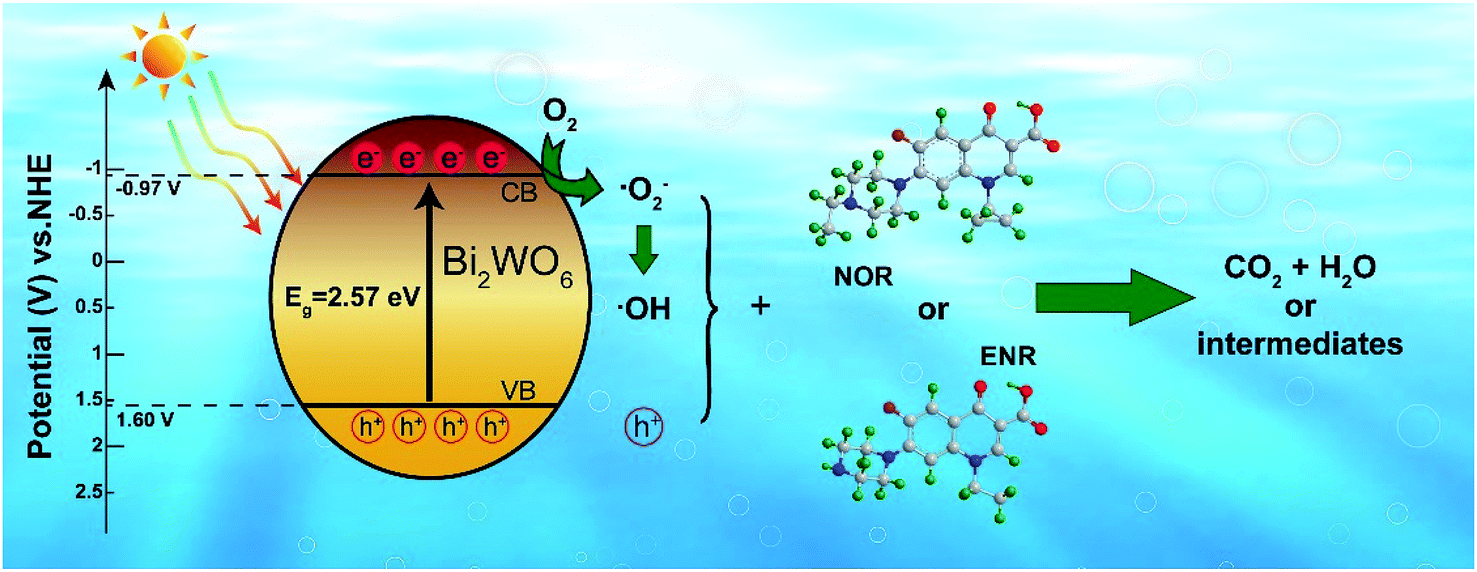
3.5 Possible mechanism of photodegradation
The band edge positions of the valence band (VB) and conduction band (CB) of the semiconductor could be estimated by the following empirical equations:where χ is the absolute electronegativity of compounds, Ee is the relative potential energy of free electron vs. NHE. EVB and ECB are the energy of VB and CB of semiconductor, respectively.
According to the principle of electronegativity equalization, the χ of 450-Bi2WO6 was calculated to be 6.39 eV, and the Ee was about 4.50 eV according to the previous research.37 Fig. 3(b) showed the Eg of 450-Bi2WO6 sample was about 2.57 eV. Thus, the EVB and ECB of 450-Bi2WO6 sample were calculated to be 3.17 eV and 0.60 eV according to eqn (7) and (8), respectively, which were in accordance with the results from the literatures.48,64
In general, when the visible light irradiated on the 450-Bi2WO6 particles suspended in aqueous solution, the photocatalyst absorbed the light energy (hν) and the electrons in the VB were excited to the CB, resulting in e− in the CB and h+ in the VB. The e− and h+ migrated to the surface of the catalyst and contributed to a series of reactions to produce active species.
From the view of thermodynamics, the amount of ·O2− generated in the system should be greatly lower than that of ·OH, since ECB of 450-Bi2WO6 was about 0.60 eV, which was positive than −0.33 eV (vs. NHE).55 And the EVB of 450-Bi2WO6 was calculated to be 3.17 eV, more positive than the potential of OH−/·OH (2.38 V vs. NHE) and H2O/·OH (2.72 V vs. NHE). It seemed that the ·OH could be formed from h+ and OH− or H2O.36,65,66 This reasoning contradicted with the results of the quench experiments, which showed that ·O2− exhibited a strong degradation effect than ·OH, and h+ was the most important active species in this study. The possible reason might be the lack of rigor in calculation of the ECB and EVB of the catalyst, since the formula was based on the condition of pH = 1,67 while pH of the investigated system was 10.2. In view of the effects of pH on surface charge and even the position of energy band, the ECB and EVB of 450-Bi2WO6 were likely to undergo a negative shift compared with the calculated values.67 For proving the conjecture, the Mott–Schottky test (Fig. 9) was performed, and it was found that the Fermi energy level of the 450-Bi2WO6 was −1.21 V vs. SCE (i.e. −0.97 V vs. NHE).68,69 Correspondingly, the ECB value of 450-Bi2WO6 should be −0.97 eV, suggesting that the reaction of producing ·O2− from e− and O2sur could occur and cause a significant reduction in the production of ·OH. This was agree with the actual experiments. Based on the experimental results and the theory analyses above, the mechanism of photocatalytic degradation of NOR/ENR with 450-Bi2WO6 photocatalyst was presented in Scheme 1.
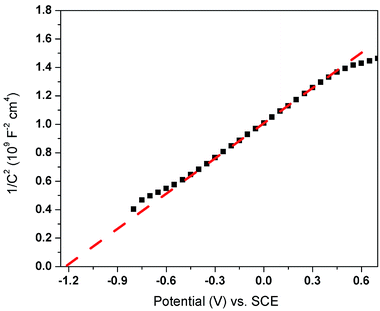 |
| | Fig. 9 Mott–Schottky plots of 450-Bi2WO6. | |
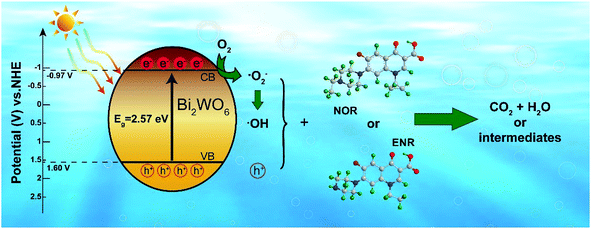 |
| | Scheme 1 Possible photocatalytic mechanism of the 450-Bi2WO6 sample. | |
Therefore, the photocatalytic degradation of NOR or ENR with the as-prepared 450-Bi2WO6 catalyst was likely to occur through the reactions as followed:
| | |
Bi2WO6 + hν → h+(VB) + e−(CB)
| (9) |
| | |
h+(VB) + NOR/ENR → small molecules
| (10) |
| | |
e−(CB) + O2sur → ·O2−
| (11) |
| | |
·O2− + NOR/ENR → small molecules
| (12) |
| | |
h+(VB) + OH−/H2O → ·OH
| (13) |
| | |
2e−(CB) + O2 + 2H+ → H2O2
| (14) |
| | |
H2O2 + ·O2− → ·OH + OH− + O2
| (15) |
| | |
·OH + NOR/ENR → small molecules
| (16) |
3.6 Reusability and mineralization ability of 450-Bi2WO6
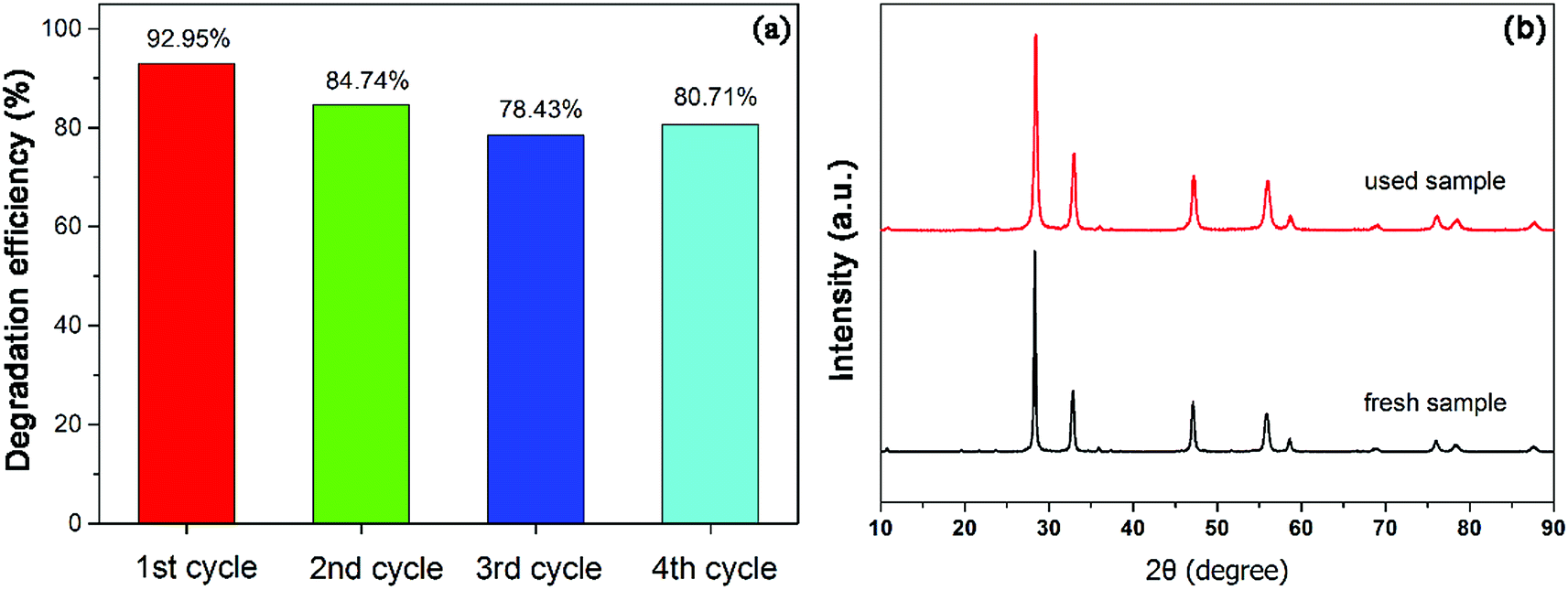
In order to study the reusability and stability of 450-Bi2WO6 photocatalyst, the stability test was examined. Four repetitive cycles of photodegradation experiments were carried out under the optimum conditions. The degradation efficiencies of NOR for the four successive cycles were 92.95%, 84.74%, 78.43% and 80.71%, respectively (Fig. 10(a)). The XRD pattern of the sample after been used for four cycles (Fig. 10(b)) was similar to that of the primary. It meant that the prepared 450-Bi2WO6 photocatalyst was stable under several reactions and was suitable for practical application.
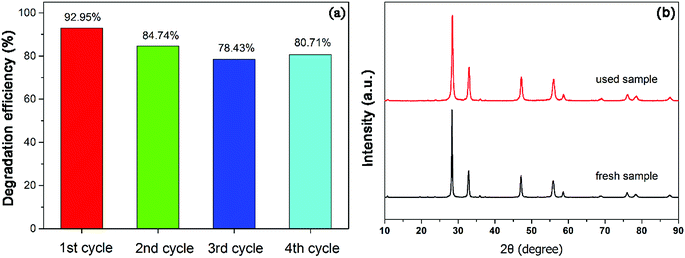 |
| | Fig. 10 (a) Results of recycling tests and (b) XRD of 450-Bi2WO6 after photocatalytic degradation of NOR for four cycles. | |
The mineralization ability of the 450-Bi2WO6 was also investigated by measuring the decrease of TOC during the photocatalytic processes. As shown in Fig. S6,† under the optimal degradation conditions, the TOC removal efficiencies of ENR and NOR solutions could reach 42.17% and 53.24% after 75 min respectively, indicating that the 450-Bi2WO6 photocatalyst could mineralize the ENR and NOR molecules.
4 Conclusion
The Bi2WO6 photocatalysts were successfully synthesized by the combined method of ultrasonic solvothermal treatment and high-temperature calcination, and the calcination played a crucial role in enhancing the crystallinity and photocatalytic activity of Bi2WO6. The 450-Bi2WO6 photocatalyst had a favorable photocatalytic performance for the degradation of NOR and ENR. The rate constant for the degradation of NOR with 450-Bi2WO6 catalyst was 2.72 and 5.33 times higher than those of bulk-Bi2WO6 and P25-TiO2, respectively (for ENR, the rate constant was 2.65 and 4.98 times higher than those of bulk-Bi2WO6 and P25-TiO2, respectively). The degradation efficiencies of NOR and ENR could reach to 92.95% and 94.58%, respectively (with the initial concentration of 10 mg L−1 in presence of 0.5 g L−1 450-Bi2WO6 at initial pH 3.0). The quench experiments demonstrated that h+ and ·O2− were the dominant active species for the photocatalytic degradation of NOR and a possible mechanism was proposed. In addition, 450-Bi2WO6 showed enough stability and maintained the activity for the photocatalytic degradation of NOR after four cycles. This work could provide practical methods for synthesis of excellent photocatalyst and removal of fluoroquinolones antibiotics in water.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
This work was supported by the National Special Fund for Agro-scientific Research in the Public Interest of China (No. 201503108) and Science & Technology Project of Hunan Province (No. 2017WK2091).
References
- J. J. Wang, L. Tang, G. M. Zeng, Y. Y. Zhou, Y. C. Deng, C. Z. Fan, J. L. Gong and Y. N. Liu, Trans. Nonferrous Met. Soc. China, 2017, 27, 1794–1803 Search PubMed.
- B. J. Dang, D. Q. Mao, Y. Xu and Y. Luo, Water Res., 2017, 111, 81–91 CrossRef CAS PubMed.
- V. N. Binh, N. Dang, N. T. K. Anh, L. X. Ky and P. K. Thai, Chemosphere, 2018, 197, 438–450 CrossRef CAS PubMed.
- I. T. Carvalho and L. Santos, Environ. Int., 2016, 94, 736–757 CrossRef PubMed.
- H. Y. Magee, M. M. Maurer, A. Cobos, B. F. G. Pycke, A. K. Venkatesan, D. Magee, M. Scotch and R. U. Halden, Sci. Total Environ., 2018, 643, 460–467 CrossRef CAS PubMed.
- K. Y. L. Chua, A. Bustamante, P. Jelfs, S. C. A. Chen and V. Sintchenko, Pathology, 2015, 47, 678–682 CAS.
- K. H. Wammer, A. R. Korte, R. A. Lundeen, J. E. Sundberg, K. McNeill and W. A. Arnold, Water Res., 2013, 47, 439–448 CAS.
- K. Kümmerer, Chemosphere, 2009, 75, 417–434 Search PubMed.
- K. Kümmerer, Chemosphere, 2009, 75, 435–441 CrossRef PubMed.
- R. T. Pei, J. M. Cha, K. H. Carlson and A. Pruden, Environ. Sci. Technol., 2007, 41, 5108–5113 CAS.
- T. Zhang, M. Zhang, X. X. Zhang and H. H. Fang, Environ. Sci. Technol., 2009, 43, 3455–3460 CrossRef CAS PubMed.
- J. J. Macauley, Z. M. Qiang, C. D. Adams, R. Surampalli and M. R. Mormile, Water Res., 2006, 40, 2017–2026 CrossRef CAS PubMed.
- X. H. Liu, G. D. Zhang, Y. Liu, S. Y. Lu, P. Qin, X. C. Guo, B. Bi, L. Wang, B. D. Xi, F. C. Wu, W. L. Wang and T. T. Zhang, Environ. Pollut., 2019, 246, 163–173 CrossRef CAS PubMed.
- L. Aristilde, A. Melis and G. Sposito, Environ. Sci. Technol., 2010, 44, 1444–1450 CAS.
- S. J. Jiao, S. R. Zheng, D. Q. Yin, L. H. Wang and L. Y. Chen, Chemosphere, 2008, 73, 377–382 CrossRef CAS PubMed.
- K. Kümmerer, Chemosphere, 2001, 45, 957–969 CrossRef.
- S. Zhang, H. L. Song, X. L. Yang, H. Li and Y. W. Wang, Bioresour. Technol., 2018, 256, 224–231 CrossRef CAS PubMed.
- F. Santos, A. P. Mucha, D. A. M. Alexandrino, C. M. R. Almeida and M. F. Carvalho, J. Environ. Manage., 2019, 231, 1145–1153 CAS.
- L. Wang, Z. M. Qiang, Y. G. Li and W. W. Ben, J. Environ. Sci., 2017, 56, 263–271 CrossRef PubMed.
- E. M. Golet, I. Xifra, H. Siegrist, A. C. Alder and W. Giger, Environ. Sci. Technol., 2003, 37, 3243–3249 CrossRef CAS PubMed.
- R. H. Lindberg, U. Olofsson, P. Rendahl, M. I. Johansson, M. Tysklind and B. A. V. Andersson, Environ. Sci. Technol., 2006, 40, 1042–1048 CrossRef CAS PubMed.
- Y. F. Wang, J. X. Zhu, H. O. Huang and H. H. Cho, J. Membr. Sci., 2015, 479, 165–174 CrossRef CAS.
- F. Tan, D. M. Sun, J. S. Gao, Q. Zhao, X. C. Wang, F. Teng, X. Quan and J. W. Chen, J. Hazard. Mater., 2013, 244–245, 750–757 CrossRef CAS PubMed.
- Y. Li, J. F. Niu and W. L. Wang, Chemosphere, 2011, 85, 892–897 CrossRef CAS PubMed.
- M. Sturini, A. Speltini, F. Maraschi, A. Profumo, L. Pretali, E. Fasani and A. Albini, Environ. Sci. Technol., 2010, 44, 4564–4569 CrossRef CAS PubMed.
- S. Y. Dong, J. L. Feng, M. H. Fan, Y. Q. Pi, L. M. Hu, X. Han, M. L. Liu, J. Y. Sun and J. H. Sun, RSC Adv., 2015, 5, 14610–14630 RSC.
- M. J. Chen and W. Chu, Appl. Catal., B, 2015, 168–169, 175–182 CrossRef CAS.
- D. H. Lan, H. T. Wang, L. Chen, C. T. Au and S. F. Yin, Carbon, 2016, 100, 81–89 CrossRef CAS.
- D. Liu, J. Wang, X. J. Bai, R. L. Zong and Y. F. Zhu, Adv. Mater., 2016, 28, 7284–7290 CrossRef CAS PubMed.
- R. a. He, S. W. Cao, P. Zhou and J. G. Yu, Chin. J. Catal., 2014, 35, 989–1007 CrossRef CAS.
- S. Rengaraj, X. Z. Li, P. A. Tanner, Z. F. Pan and G. K. H. Pang, J. Mol. Catal. A: Chem., 2006, 247, 36–43 CrossRef CAS.
- S. Gong, Q. F. Han, J. W. Zhu, X. Wang and L. D. Lu, Mater. Res. Bull., 2016, 76, 222–228 CrossRef CAS.
- J. L. Hu, W. J. Fan, W. Q. Ye, C. J. Huang and X. Qiu, Appl. Catal., B, 2014, 158–159, 182–189 CrossRef CAS.
- M. X. Du, Y. Du, Y. B. Feng, K. Yang, X. J. Lv, N. Jiang and Y. Liu, Carbohydr. Polym., 2018, 195, 393–400 CrossRef CAS PubMed.
- H. B. Li, Z. J. Yang, J. N. Zhang, Y. C. Huang, H. B. Ji and Y. X. Tong, Appl. Surf. Sci., 2017, 423, 1188–1197 CrossRef CAS.
- C. Y. Wang, X. Zhang, X. N. Song, W. K. Wang and H. Q. Yu, ACS Appl. Mater. Interfaces, 2016, 8, 5320–5326 CrossRef CAS PubMed.
- A. Kaur and S. K. Kansal, Chem. Eng. J., 2016, 302, 194–203 CrossRef CAS.
- Y. K. Huang, S. F. Kang, Y. Yang, H. F. Qin, Z. J. Ni, S. J. Yang and X. Li, Appl. Catal., B, 2016, 196, 89–99 CrossRef CAS.
- J. J. Wang, L. Tang, G. M. Zeng, Y. C. Deng, Y. N. Liu, L. L. Wang, Y. Y. Zhou, Z. Guo, J. J. Wang and C. Zhang, Appl. Catal., B, 2017, 209, 285–294 CrossRef CAS.
- D. Cao, Y. B. Wang, M. Qiao and X. Zhao, J. Catal., 2018, 360, 240–249 CrossRef CAS.
- S. Jonjana, A. Phuruangrat, T. Thongtem and S. Thongtem, Mater. Lett., 2016, 172, 11–14 CrossRef CAS.
- A. Kudo and S. Hijii, Chem. Lett., 1999, 1999, 1103–1104 CrossRef.
- J. W. Tang, Z. G. Zou and J. H. Ye, Catal. Lett., 2004, 92, 53–56 CrossRef CAS.
- L. Y. Liang, Y. Tursun, A. Nulahong, T. Dilinuer, A. Tunishaguli, G. Gao, A. Abulikemu and K. Okitsu, Ultrason. Sonochem., 2017, 39, 93–100 CrossRef CAS PubMed.
- S. W. Zhu, C. Y. Yang, F. Li, T. H. Li, M. Zhang and W. Cao, Mol. Catal., 2017, 435, 33–48 CrossRef CAS.
- G. Zhao, S. W. Liu, Q. F. Lu and L. J. Song, Ind. Eng. Chem. Res., 2012, 51, 10307–10312 CrossRef CAS.
- F. Amano, K. Nogami and B. Ohtani, J. Phys. Chem. C, 2009, 113, 1536–1542 CrossRef CAS.
- Y. Y. Zhao, Y. B. Wang, E. Z. Liu, J. Fan and X. Y. Hu, Appl. Surf. Sci., 2018, 436, 854–864 CrossRef CAS.
- M. Shang, W. Z. Wang, S. M. Sun, L. Zhou and L. Zhang, J. Phys. Chem. C, 2008, 112, 10407–10411 CrossRef CAS.
- J. Y. Sheng, X. J. Li and Y. M. Xu, Acta Phys.-Chim. Sin., 2014, 30, 508–512 CAS.
- X. L. Song, H. D. Wang, Y. Y. Li, S. Y. Ye and D. D. Dionysiou, Appl. Surf. Sci., 2018, 439, 815–822 CrossRef CAS.
- Z. J. Zhang, W. Z. Wang, L. Wang and S. M. Sun, ACS Appl. Mater. Interfaces, 2012, 4, 593–597 CrossRef CAS PubMed.
- Y. Tian, L. D. Zhang and J. X. Zhang, J. Alloys Compd., 2012, 537, 24–28 CrossRef CAS.
- Y. Y. Zhao, X. H. Liang, Y. B. Wang, H. X. Shi, E. Z. Liu, J. Fan and X. Y. Hu, J. Colloid Interface Sci., 2018, 523, 7–17 CrossRef CAS PubMed.
- F. Chen, D. Li, B. F. Luo, M. Chen and W. D. Shi, J. Alloys Compd., 2017, 694, 193–200 CrossRef CAS.
- J. G. Yu, J. F. Xiong, B. Cheng, Y. Yu and J. B. Wang, J. Solid State Chem., 2005, 178, 1968–1972 CrossRef CAS.
- R. W. Matthews, M. Abdullah and K. C. Low, Anal. Chim. Acta, 1990, 233, 171–179 CrossRef CAS.
- J. Gamage McEvoy, W. Cui and Z. Zhang, Appl. Catal., B, 2014, 144, 702–712 CAS.
- W. F. Yao, H. Wang, X. H. Xu, J. T. Zhou, X. N. Yang, Y. Zhang and S. X. Shang, Appl. Catal., A, 2004, 259, 29–33 CrossRef CAS.
- J. Grzechulska and A. W. Morawski, Appl. Catal., B, 2002, 36, 45–51 CrossRef CAS.
- H. J. Zhang, P. Zhang, Y. Ji, J. Tian and Z. X. Du, Chem. Eng. J., 2015, 262, 1108–1115 CrossRef CAS.
- O. Moumeni, O. Hamdaoui and C. Pétrier, Chem. Eng. Process., 2012, 62, 47–53 CrossRef CAS.
- K. Bourikas, C. Kordulis and A. Lycourghiotis, Environ. Sci. Technol., 2005, 39, 4100–4108 CrossRef CAS PubMed.
- H. H. Wang, J. Lu, F. Q. Wang, W. H. Wei, Y. Chang and S. J. Dong, Ceram. Int., 2014, 40, 9077–9086 CrossRef CAS.
- G. J. Leigh, J. Organomet. Chem., 1983, 251, c37 CrossRef.
- A. Akhundi and A. Habibi-Yangjeh, Mater. Chem. Phys., 2016, 174, 59–69 CrossRef CAS.
- B. Wang, H. Yang, T. Xian, L. J. Di, R. S. Li and X. X. Wang, J. Nanomater., 2015, 16, 194–200 Search PubMed.
- R. M. Fernández-Domene, R. Sánchez-Tovar, B. Lucas-granados, M. J. Muñoz-Portero and J. García-Antón, Chem. Eng. J., 2018, 350, 1114–1124 CrossRef.
- D. Ma, J. Wu, M. C. Gao, Y. J. Xin, Y. Y. Sun and T. J. Ma, Chem. Eng. J., 2017, 313, 1567–1576 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra04445k |
| ‡ Cong Huang and Leilei Chen contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Yanguang Muab and
Zhaoguang Yang*ab
*ab,
Yanguang Muab and
Zhaoguang Yang*ab