DOI:
10.1039/C9RA05664E
(Paper)
RSC Adv., 2019,
9, 27060-27068
Gallic acid-functionalized graphene hydrogel as adsorbent for removal of chromium (III) and organic dye pollutants from tannery wastewater†
Received
22nd July 2019
, Accepted 14th August 2019
First published on 29th August 2019
Abstract
The pollution caused by tannery wastewater containing high concentrations of trivalent chromium ions [Cr(III)] and organic dyes has severely restricted the sustainable development of the leather industry. To address this problem, a three-dimensional (3D) porous graphene-based hydrogel with good mechanical strength and large surface area was fabricated by self-assembly of graphene oxide (GO) sheets reduced and modified by gallic acid (GA) through π–π interactions. As an adsorbent, this GA-functionalized graphene hydrogel (GA-GH) can effectively capture Cr(III) by coordination complexation between Cr(III) and deprotonated carboxylic groups of GA at pH ∼ 4.0. Moreover, GA-GH could be easily regenerated by desorption of adsorbed Cr(III) at pH 2.0 and maintained its high adsorption capacity after multiple adsorption–desorption cycles, which was also helpful for reusing desorbed Cr(III) as tanning agent. In addition, compared with a graphene hydrogel (GH) without modification by GA, adsorption capacity of GA-GH for organic dye was significantly improved due to the enhanced π–π interactions between the GA-GH and aromatic dyes.
1. Introduction
Leather or leather goods, such as footwear, clothing and bags with different colors are necessities in most people's daily lives. At present, over 90% of leather products are tanned using Cr(III) salts, due to their excellent hydrothermal stability, and physical and mechanical properties.1–3 The chrome tanning mechanism is mainly based on coordination complexation between Cr(III) and deprotonated carboxyl groups of skin collagen under acidic (pH ∼ 4.0) conditions, which results in chemical cross-linking of collagen fibers and conversion of rawhide to stable leather.4–6 However, in the conventional chrome tanning process, only approximately 70% of the total chrome agent is absorbed by the leather, leading to accumulation of Cr(III) in tannery wastewater to concentrations higher than 3000 ppm.7,8 In China, according to relevant laws, this chromium-containing tannery wastewater is prohibited from being directly discharged to waterways until the concentration of Cr(III) is reduced to lower than 10 ppm.9 Unfortunately, current technologies for treating chromium wastewater, which normally utilize alkali chemicals to precipitate Cr(III) from wastewater to cause further pollution of sludge with Cr(OH)3, still lack efficiency and are less than satisfactory. Cr(III) in tannery sludge, on the one hand, is very difficult to collect and recycle, while on the other hand, it can be more easily oxidized into highly toxic hexavalent chromium Cr(VI) when the sludge is exposed to air, which will bring potential risk to human health.10–12 Besides the Cr(III) pollutant in the chrome tanning process, various cationic and anionic organic dyes that can combine with skin collagen through electrostatic interactions are also widely used in the subsequent dyeing process during leather making.13,14 In addition, most of these organic dyes are aromatic, resulting in tannery wastewater contaminated with aromatic pollutants, which are not only harmful to humans but also toxic to microorganisms.15–17 Indeed, tannery wastewater containing Cr(III) and organic dyes has become a serious environmental pollution, which has severely affected the sustainable development of the leather industry.18–20
For the removal of both Cr(III) ions and organic dyes from tannery wastewater, adsorption has proved to be an attractive disposal technique based on its low cost, simplicity of operation and recycling of the adsorbent.21–24 However, most traditional adsorbents, including polymeric resins,25 activated carbon,26 silica,27 and metal oxides,28,29 usually suffer from either low adsorption capacity or low efficiency. Over the past decade, free-standing graphene hydrogel (GH), fabricated by self-assembly of reduced GO sheets through π–π interactions, has attracted much attention as a super adsorbent for wastewater treatment, due to its unique properties, such as three-dimensional (3D) porous network structure, large surface area, low density, chemical stability and aromaticity.30–34 Up to now, the one-step hydrothermal method has been the most popular method to reduce GO sheets and subsequently obtain 3D GH.35–37 However, such hydrothermal process is usually time-consuming (∼12 h) and requires very high temperature (∼200 °C), which limit the large-scale production of GH.38–40 Recently, tremendous efforts have been devoted to fabricate 3D porous GH with the aid of chemical reduction under mild conditions, which is a process called in situ reducing-assembly.41 For instance, in the presence of a variety of reducing agent, GO sheets was reduced within 10 hours by heating at low-temperature below 100 °C and spontaneously self-assembled into a free-standing hydrogel through π–π interactions.42 To meet the requirements of GH as effective adsorbent to remove heavy metal ions and organic pollutants from wastewater, reducing agents have also been carefully selected to play multi-functional roles as reductant, surface functionalization agent and provider of active sites for anchoring heavy metal ions and organic pollutants.43 For example, Duan's group developed a polydopamine (PDA) modified GH (PDA-GH) by using dopamine as both a reductant and surface functionalization agent, which had high adsorption capacity for heavy metals, synthetic dyes and aromatic pollutants, benefiting from the abundant and aromatic catechol groups of the PDA on the graphene sheets.44
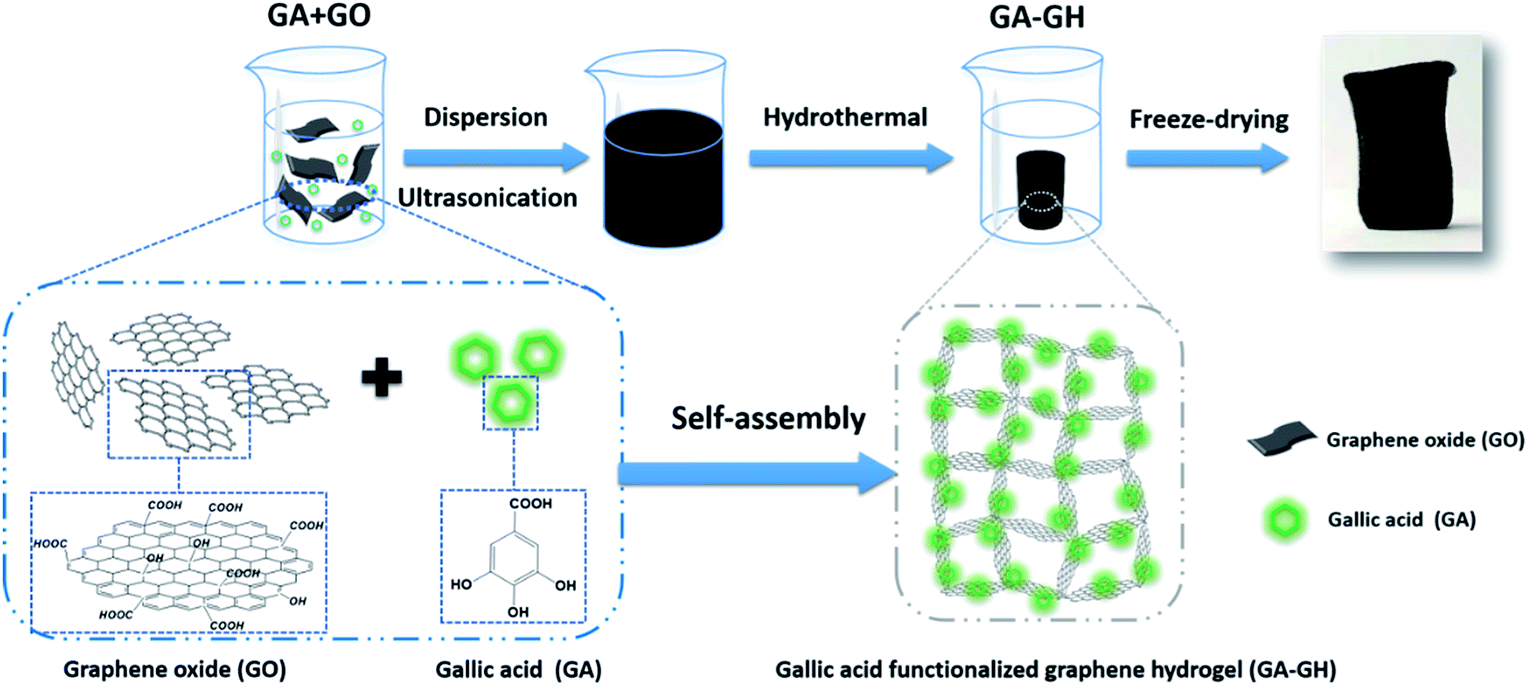
Here, to effectively remove Cr(III) ions and aromatic dyes in tannery wastewater, natural gallic acid (GA) extracted from plants, which contains carboxyl groups and aromatic catechol groups, was selected to reduce and modify GO into self-assembled 3D porous GA-functionalized graphene hydrogel (GA-GH) as adsorbent (as shown in Scheme 1). Compared with pristine GH, the GA-GH is supposed to improve the adsorption capacity for Cr(III) and aromatic dyes through complexation with the carboxyl group of GA, as well as enhanced π–π interactions with the phenolic group of GA, respectively. The unique features of GA-GH including multi-functional groups, 3D porous structure and large surface area and free-standing make it a suitable and promising adsorbent candidate for removal of Cr(III) and aromatic dyes from tannery wastewater.
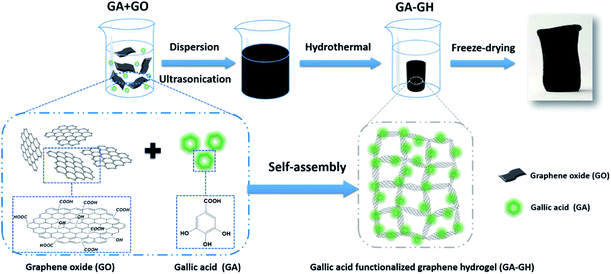 |
| | Scheme 1 Schematic illustration of the formation process of gallic acid functionalized graphene hydrogel (GA-GH). | |
2. Materials and method
2.1 Materials
GO were prepared from natural graphite powder according to a modified Hummer's method.45 Gallic acid (GA, 99%) was purchased from Aladdin Chemical Corporation (Shanghai, China). Chromium chloride hexahydrate (CrCl3·6H2O, 99%), methylene blue (MB) and methyl red (MR) dyes were purchased from Shudu Chemical Co. Ltd. (Chengdu, China). Other chemical materials were purchased from Jinshan Chemical Reagents Corporation (Chengdu, China) and all were analytical reagents.
2.2 Preparation of GA-GH
The aqueous dispersions of GO at a concentration of 2 mg mL−1 and a pH value of 9.0 were first prepared, and then GA was added at a concentration equal to that of GO. GA-GH was obtained by heating the above mixture at 100 °C for 8 hours without any disturbance. For comparison purposes, hydrothermally-reduced GH was also prepared by hydrothermal treatment of GO aqueous solution (2 mg mL−1) at 180 °C for 12 h. Then, the above as-prepared samples were dialyzed with deionized water for one week, followed by freeze-drying to obtain the dry GA-GH and GH.
2.3 Adsorption and desorption properties of GA-GH for Cr(III)
Batch adsorption experiments were performed at room temperature to investigate the adsorption behaviors of Cr(III) on GA-GH or GH samples at pH 3.0 or pH 3.8. Typically, 10 mg GA-GH or GH adsorbent was added into 100 mL aqueous solutions with different initial concentrations of Cr(III) at pH 3.0 or pH 3.8, shaking in a rotary shaker for a period of time or 10 h to reach adsorption equilibrium. After adsorption, the GA-GH or GH was removed, and the solution was further centrifuged at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 20 min. Inductively coupled plasma optical emission spectroscopy (ICP-OES) was used to determine the residual Cr(III) concentration in the solution. The adsorption capacities of the adsorbents were calculated according to the equation: qe = (C0 − Ce) V/m, where C0 and Ce represent the initial and equilibrium concentrations (mg g−1), respectively; V is the volume of the solutions (mL); m is the amount of adsorbent (mg). For desorption experiment, HCl was used as desorption agent to adjust the pH of the solution to pH 2.0 for regenerating the adsorbents of GA-GH or GH after adsorption.
000 rpm for 20 min. Inductively coupled plasma optical emission spectroscopy (ICP-OES) was used to determine the residual Cr(III) concentration in the solution. The adsorption capacities of the adsorbents were calculated according to the equation: qe = (C0 − Ce) V/m, where C0 and Ce represent the initial and equilibrium concentrations (mg g−1), respectively; V is the volume of the solutions (mL); m is the amount of adsorbent (mg). For desorption experiment, HCl was used as desorption agent to adjust the pH of the solution to pH 2.0 for regenerating the adsorbents of GA-GH or GH after adsorption.
2.4 Adsorption property of GA-GH for organic dye
Two typical dyes used in traditional leather processing, namely MB and MR were chosen for the adsorption test. For dye adsorption, a 10 mg amount of GA-GH or GH was added into a 20 mL MB solution with pH of 7.0 at a concentration of 10 ppm or a 20 mL MR solution with pH of 7.0 at a concentration of 20 ppm. The mixture was stirred continuously, and the sediment was collected and removed by centrifugation at 13000 rpm for 20 min. The concentration of residual dye solution was analyzed by ultraviolet-visible (UV-vis) spectroscopy at the maximum absorption wavelength to measure the absorbance. The concentration of each dye before and after adsorption was determined from the standard calibration curve.
2.5 Characterization
The morphology of the GA-GH and GH was examined by field-emission scanning electron microscopy (FESEM) on a JSM-7500F scanning electron microscope (JEOL Ltd., Tokyo, Japan). The chemical states of the element on the graphene aerogel surface were investigated by X-ray photoelectron spectroscopy, (XPS) using an XSAM 800 photoelectron spectroscope (Kratos Analytical, Ltd, Manchester, U.K.). X-ray diffraction (XRD) spectra were recorded on a D/max-2200/PC X-ray diffractometer (Rigaku Corporation, Tokyo, Japan) with Cu Kα radiation. Thermogravimetric analysis (TGA) was performed using a TG209F1 Libra Thermogravimetric Analyzer (Netzsch NETZSCH Group, GmbH, Selb, Germany), from room temperature to 750 °C at a heating rate of 20 °C min−1 in air. The concentration of Cr(III) was determined by ICP-OES, using an Optima 2100DV spectrometer (PerkinElmer, Waltham, MA, USA). UV-vis diffuse reflectance spectra (UV-vis DRS) were recorded using a UV-3600 UV-vis-NIR spectrophotometer (Shimadzu, Kyoto, Japan) equipped with an integrating sphere and using BaSO4 as reference. The specific surface areas of GA-GH and GH were calculated by the Brunauer–Emmett–Teller (BET) method.
3. Results and discussion
3.1 Preparation and characterization of GA-GH hydrogel
GA, extracted from plant, is a kind of phenolic acid, showing reducibility under alkaline condition due to the transformation of its phenol group to a benzoquinone group (Fig. S1†).46,47 As shown in Scheme 1, GA-GH was prepared by the one-step reduction-induced assembly of GO with the aid of GA, conducted at 100 °C for 8 hours at pH 9.0. However, as shown in Fig. 1A, the formation of the GH without GA using a similar hydrothermal reduction method required higher temperature (180 °C) and longer time (12 hours). This is due to the partial reduction of GO sheets by GA during the formation of GH, which weakened the hydrothermally synthesis process. At the same time, GA molecules were modified onto graphene sheets due to the catechol groups and strong affinity between aromatic rings.48,49 After freeze-drying, the GA-GH exhibited a well-defined aerogel structure compared to that of the GH, with obvious mechanical strength that can be capable of supporting a weight of 200 g (Fig. 1A and D). This might be due to the chemical reduction and modification of GO sheets by GA, and the increased π–π interactions between graphene sheets, which lead to the formation of a more compact 3D porous architecture of the GA-GH. Therefore, the porous structure and morphology of GA-GH and GH we investigated by SEM. The SEM images of GA-GH shown in Fig. 1E and F clearly reveal a honeycomb-like structure with pores connecting the surface to the interior, which exhibited a more compact and ordered 3D porous network than that of the GH shown in Fig. 1B and C. These results indicated that the modification by GA enhanced the π–π interactions between graphene sheets during the self-assembly process, leading to a well-defined and interconnected 3D porous structure (as shown in Scheme S1†).
 |
| | Fig. 1 Hydrothermally reduction of GO solutions with or without GA to form free-standing GH (A) and GA-GH (D). SEM images of GH (B and C) and GA-GH (E and F) with low and high magnifications. | |
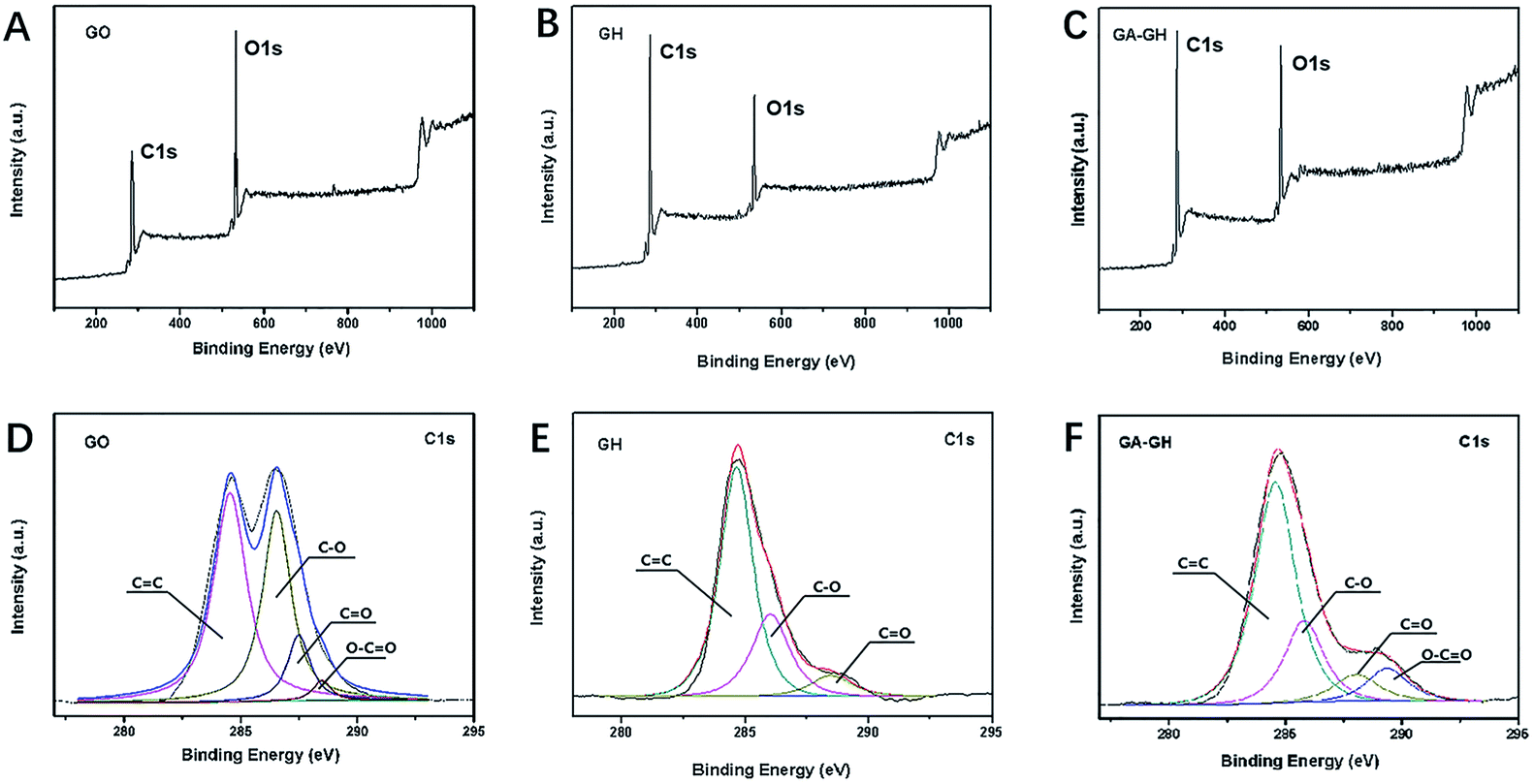
To further investigate the modification of GA on graphene sheets, the chemical composition of GO, GH and GA-GH was determined by XPS analysis and compared. As can be seen, the XPS survey spectra of GO, GH and GA-GH samples shown in Fig. 2A, B and C, respectively, all displayed two sharp and strong peaks located at 285.2 and 532.1 eV, which were assigned to the C 1s and O 1s, respectively. The element ratio of C and O for the GH sample that caculated from Fig. 2B is 4.48, which is much higher than C/O ratio of 1.67 for GO sample caculated from Fig. 2A, suggesting the efficient reduction of the GO sheets during the hydrogel formation process. However, the C/O ratio is 3.53 caculated from Fig. 2C for GA-GH sample, which is lower than that for GH sample, indicating the modification of GA on graphene sheets with carboxyl groups. To accurately confirm the compositional change of GO before and after reduction by the hydrothermal methods or GA, deconvolution of the C 1s signal of three samples was performed. The C 1s signal of the GO in Fig. 2D indicated the presence of heavily oxygenated carbon species, such as C–O (hydroxyl and epoxy, ∼286.5 eV), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (carbonyl, 287.5 eV) and O–CO (carboxyl, ∼288.6 eV). In contrast, after hydrothermal reduction, the intensity of the C–O and CO peaks for the GH (Fig. 2E) dramatically decreased with the disappearance of the O–CO peak, leading the peak associated with CC (∼284.7 eV) to become dominant. Instead, for the GA-GH sample that self-assembled by graphene oxide sheets reduced by GA, the O–CO peak that reappeared at 289.4 eV (Fig. 2F) corresponded to the carboxyl groups of GA, confirming the functionalization of GA on the GH.
O (carbonyl, 287.5 eV) and O–CO (carboxyl, ∼288.6 eV). In contrast, after hydrothermal reduction, the intensity of the C–O and CO peaks for the GH (Fig. 2E) dramatically decreased with the disappearance of the O–CO peak, leading the peak associated with CC (∼284.7 eV) to become dominant. Instead, for the GA-GH sample that self-assembled by graphene oxide sheets reduced by GA, the O–CO peak that reappeared at 289.4 eV (Fig. 2F) corresponded to the carboxyl groups of GA, confirming the functionalization of GA on the GH.
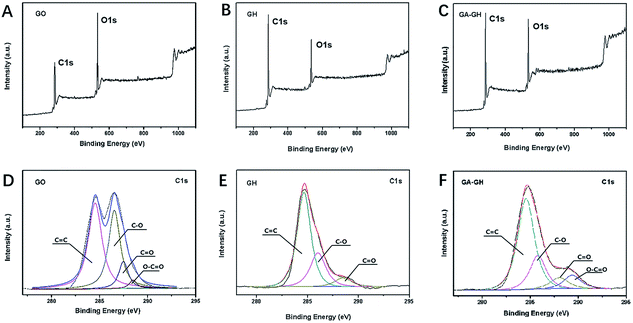 |
| | Fig. 2 XPS survey spectra of GO (A), GH (B) and GA-GH (C) samples. High-resolution XPS of C 1s peaks for GO (D), GH (E) and GA-GH (F) samples. | |
The successful reduction of GO by GA was also verified by XRD analysis, using GO and GH samples as control. As shown in Fig. 3A, the XRD spectrum of GO displayed an intense peak at 2θ = 9.80°, implying an interlayer distance (d-spacing) of 0.903 nm that can be attributed to the reflection of stacked GO sheets. After hydrothermal reduction of the GH sample, a new broad diffraction peak appeared at 26.30° with a d-spacing of 0.338 nm, which is almost the same as that of natural graphite (0.336 nm), indicating the sufficient removal of the oxygen-containing groups of GO and successful reduction of GO to graphene. For the GA-GH sample, a similar broad diffraction peak was found at 25.80° and the interlayer spacing of the GA-GH was calculated to be 0.345 nm, which is slightly higher than that of GH. The above results demonstrated the presence of π–π stacking interactions between reduced graphene oxide sheets in both hydrogel samples, which drive the overlapping and coalescing of flexible graphene sheets to form the 3D porous network structure that is observed by SEM (Fig. 1).37,44 In addition, GH and GA-GH also had similar specific surface areas, which were determined to be 282.1 and 298.6 m2 g−1, respectively, by the nitrogen adsorption–desorption test (Fig. S2†). However, in contrast to GH, the slightly larger d-spacing of GA-GH suggested the presence of GA with oxygenated functional groups (carboxyl) on graphene sheets, which is consistent with the results of the XPS analysis.
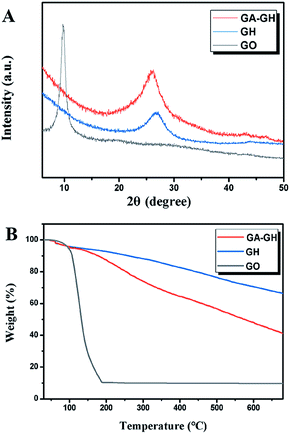 |
| | Fig. 3 XRD patterns (A) and TGA curves (B) of GO, GH and GA-GH samples. | |
Thermal stability is an important indicator of the adsorption capacity of GH as an adsorbent. Therefore, the thermal stability of GA-GH was evaluated by TGA (Fig. 3B). The GO sample exhibited a drastic mass loss of 87% at 200 °C, which was due to the decomposition of labile oxygen containing groups on GO sheets and evaporation of absorbed water. Clearly, the mass-loss curves of the GH and GA-GH samples both showed less than 4 wt% weight loss at 200 °C, indicating the improved thermal stability of the prepared GH. However, the weight loss of the GA-GH sample was higher than that of the GH sample when the temperature was above 250 °C, which might be attributed to the decomposition of the functionalized GA on graphene sheets.
3.2 The adsorption and desorption properties of GA-GH for Cr(III)
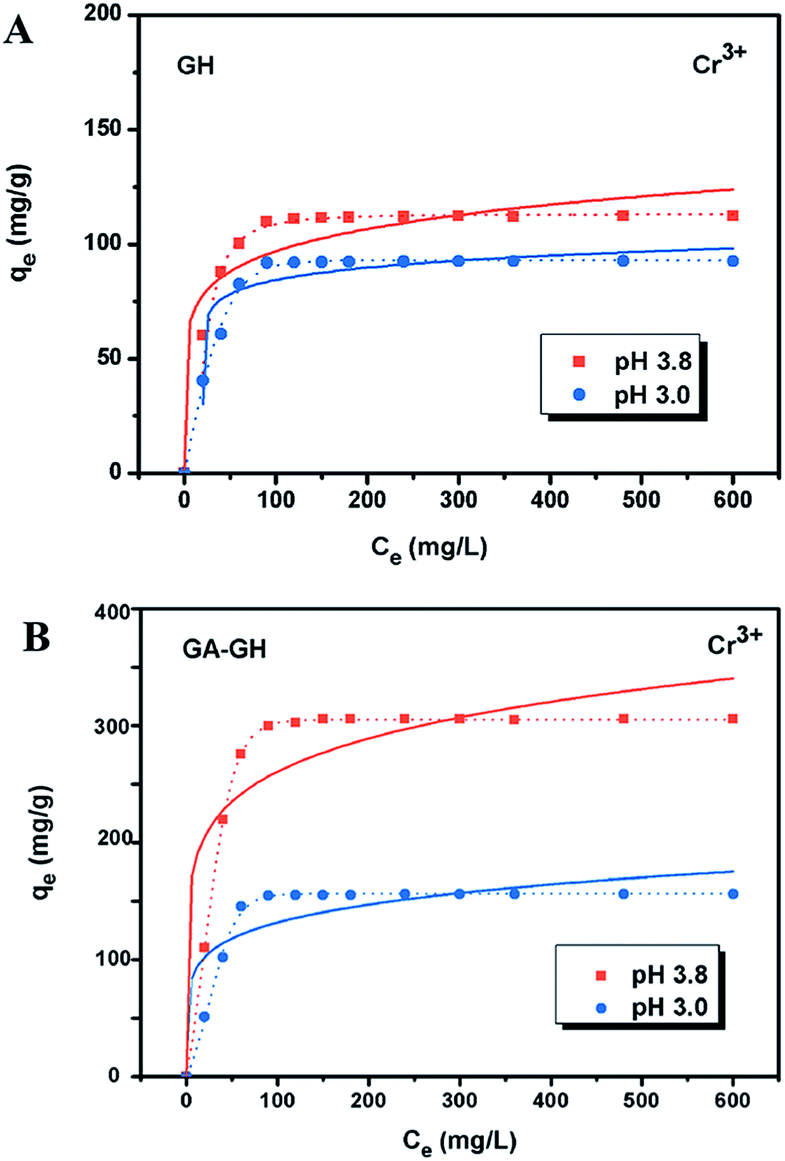
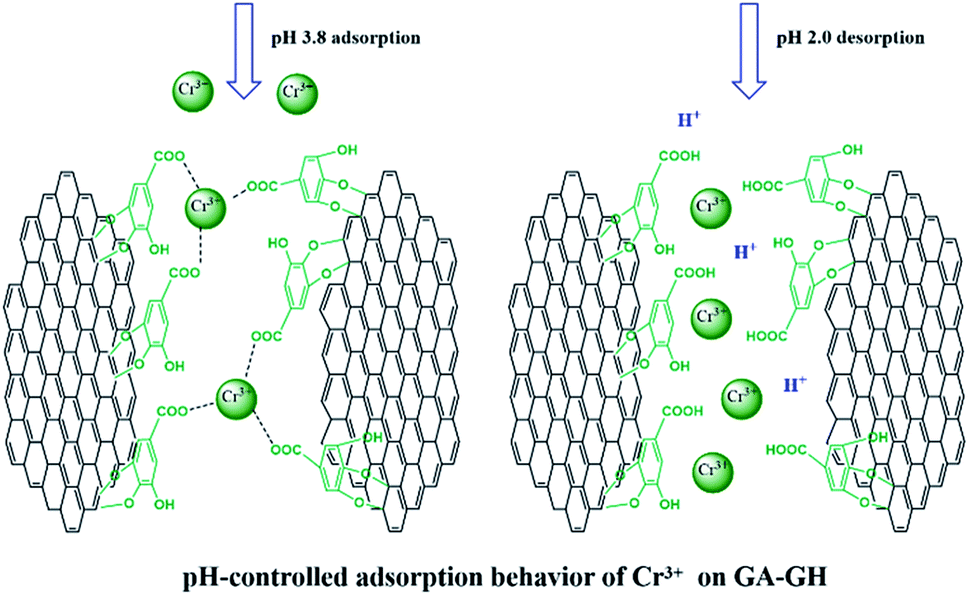
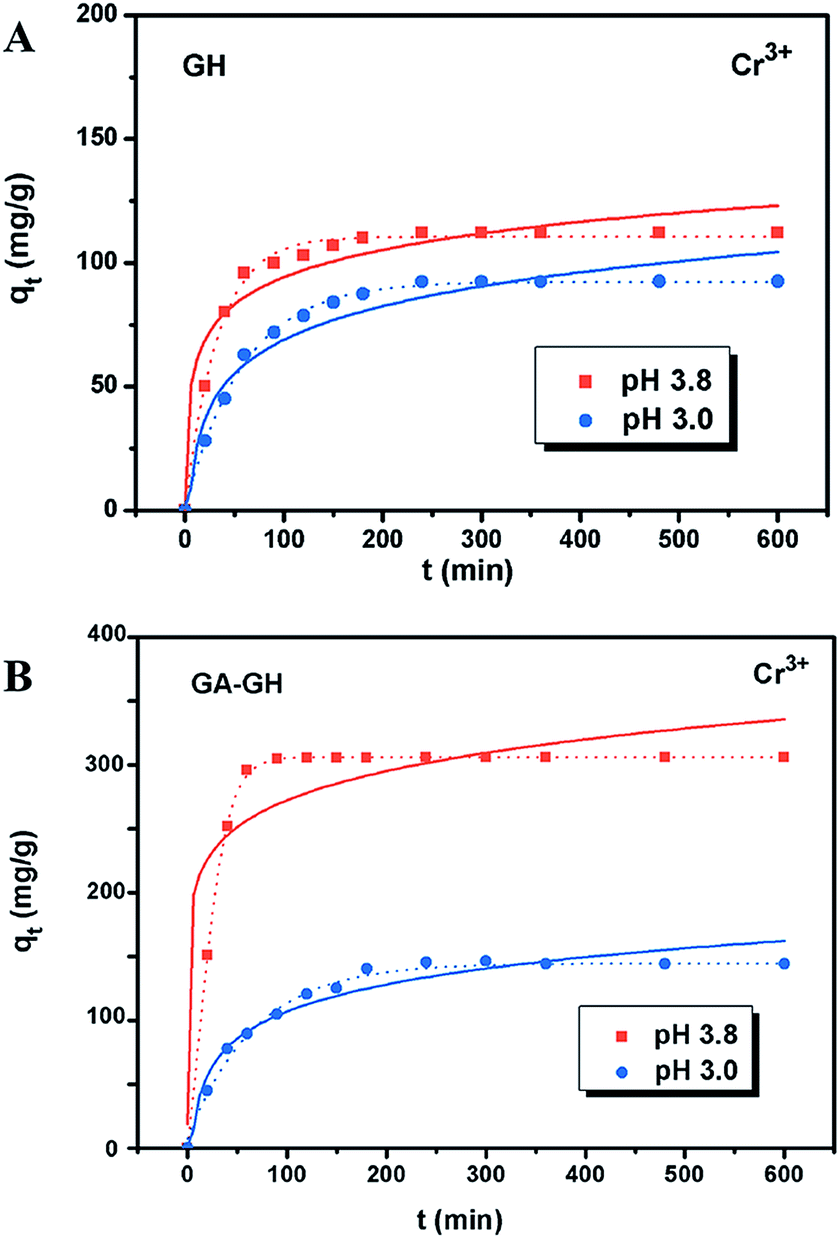
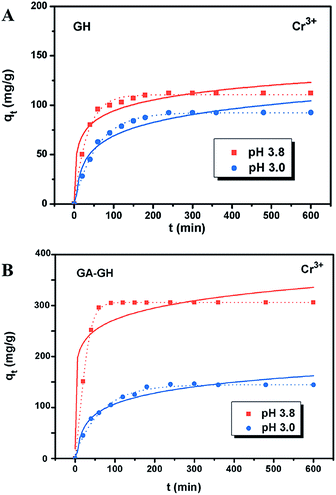
It is well-known that, in the conventional chrome tanning process, Cr(III) usually form Cr(OH)3 precipitates in aqueous solution at pH above 4.0, but can form strong coordination complex bonds with deprotonated carboxyl groups (–COO−) at around pH 4.0.4 However, below pH ∼ 3.0, most of the carboxyl groups are protonated and have very weak interaction with Cr(III) ions. Thus, the pH value of the solution becomes a critical factor that may affect the adsorption capacity of the GA-GH. In this study, due to the modification of GA with carboxyl groups, it was anticipated that the GA-GH will have a more enhanced adsorption capacity than that of the GH, and the adsorption performance of GA-GH will be better at the pH value around 4.0 than that at pH 3.0. Therefore, pH values of 3.8 and 3.0 were selected to evaluate the effect of the pH on the adsorption of Cr(III) onto GA-GH and GH, respectively. As shown in Fig. 4, the adsorption capacity of GH and GA-GH at pH 3.8 and pH 3.0, respectively, evaluated by equilibrium adsorption isotherm, both gradually increased with increasing concentration of Cr(III), eventually reaching saturation states, with adsorption data all fitted into the Langmuir and Freundlich isotherm models, respectively. Moreover, the adsorption data were found to be better fitted with the Langmuir isotherm model than that by the Freundlich model, indicating that the adsorption of Cr(III) took place at the functional groups or binding sites on the surface of the adsorbent in a monolayer manner. From the fitting results, the maximum adsorption capacity of Cr(III) onto GH was 111.9 and 92.5 mg g−1 at pH 3.8 and 3.0, respectively (Fig. 4A). For the GA-GH sample with equal adsorbent weight to that of GH, the maximum adsorption capacity of Cr(III) was 305.4 and 152.2 mg g−1 at pH 3.8 and 3.0, respectively (Fig. 4B). Evidently, the adsorption capacity of GA-GH for Cr(III) ions was much higher than that of GH, especially at pH 3.8. These results demonstrated the effective coordination complexation between Cr(III) and deprotonated carboxylic groups of GA modified on graphene sheets at pH value close to 4.0 during the adsorption process (illustrated in Scheme 2). In contrast, the adsorption capacity of the GA-GH for Cr(III) significantly decreased at pH 3.0, which showed the weaker binding affinity of protonated carboxylic groups to Cr(III).
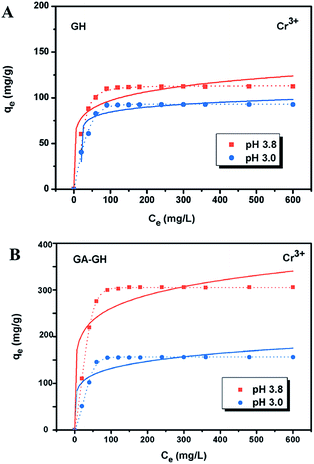 |
| | Fig. 4 Adsorption behaviors of Cr(III) on GH (A) and GA-GH (B) at pH 3.0 and 3.8, respectively. Adsorption isotherm plots and curves were fitted by Langmuir (dot line) and Freundlich (solid line) models, with the equations given as qe = qmaxKLCe/(1 + KLCe) and qe = KFCe1/n, respectively, where qe is the amount (mg g−1) of Cr3+ ions adsorbed at equilibrium, Ce is the equilibrium concentration (mg L−1) of Cr3+ ions, KL and qmax (maximum adsorption capacity) are the Langmuir constants of adsorption, and KF and n are the Freundlich constants of adsorption. | |
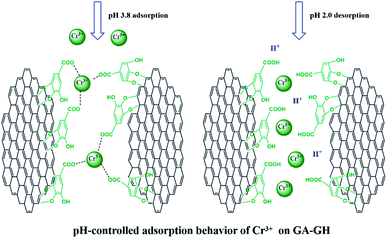 |
| | Scheme 2 pH-controlled adsorption and desorption behavior of Cr(III) ions on GA-GH. | |
The adsorption kinetics of Cr(III) by GH or GA-GH was also measured by using a metal ion concentration of 100 mg L−1 at pH 3.0 and pH 3.8, respectively. At different time intervals up to 10 h, the samples were collected to calculate the adsorption capacity of Cr(III). As shown in Fig. 5, the adsorption amount of Cr(III) for GH or GA-GH at pH 3.0 or 3.8 both increased dramatically within the initial 100 min, and gradually reached equilibrium in 240 min. At the same pH value, the adsorption rate of Cr(III) for GA-GH was much faster than that for GH. As anticipated, the adsorption rate of Cr(III) for GA-GH at pH 3.8 was faster and reached equilibrium within 60 min. These results further indicated that the adsorption mechanism of Cr(III) onto GA-GH was mainly based on the complexation between Cr(III) and functionalized GA at around pH 4.0. In addition, it is clear that the pseudo-second-order kinetic model shown in Fig. 5B, which usually means that chemisorption is the rate-determining step,4 provided a better correlation in contrast to the pseudo-first-order model for the adsorption of Cr(III) onto GA-GH. This result also confirmed the existence of chemical complexation between GA-GH and Cr(III) during the adsorption process.
 |
| | Fig. 5 Adsorption kinetic plots and curving fitting by pseudo-first-order (solid line) and pseudo-second-order (dot line) kinetic models of Cr(III) on GH (A) and GA-GH (B). The pseudo first order and pseudo second order kinetic models were according to the equations of ln(qe − qt) = ln(qe) − k1t and t/qt = 1/k2qe2 + t/qe, respectively, where qe and qt are the capacities (mg g−1) of metal ions adsorbed at equilibrium and time t (min), k1 is the rate constant of pseudo first order model (min−1), and k2 is the rate constant of the pseudo second order model of adsorption (g mg−1 min−1). | |
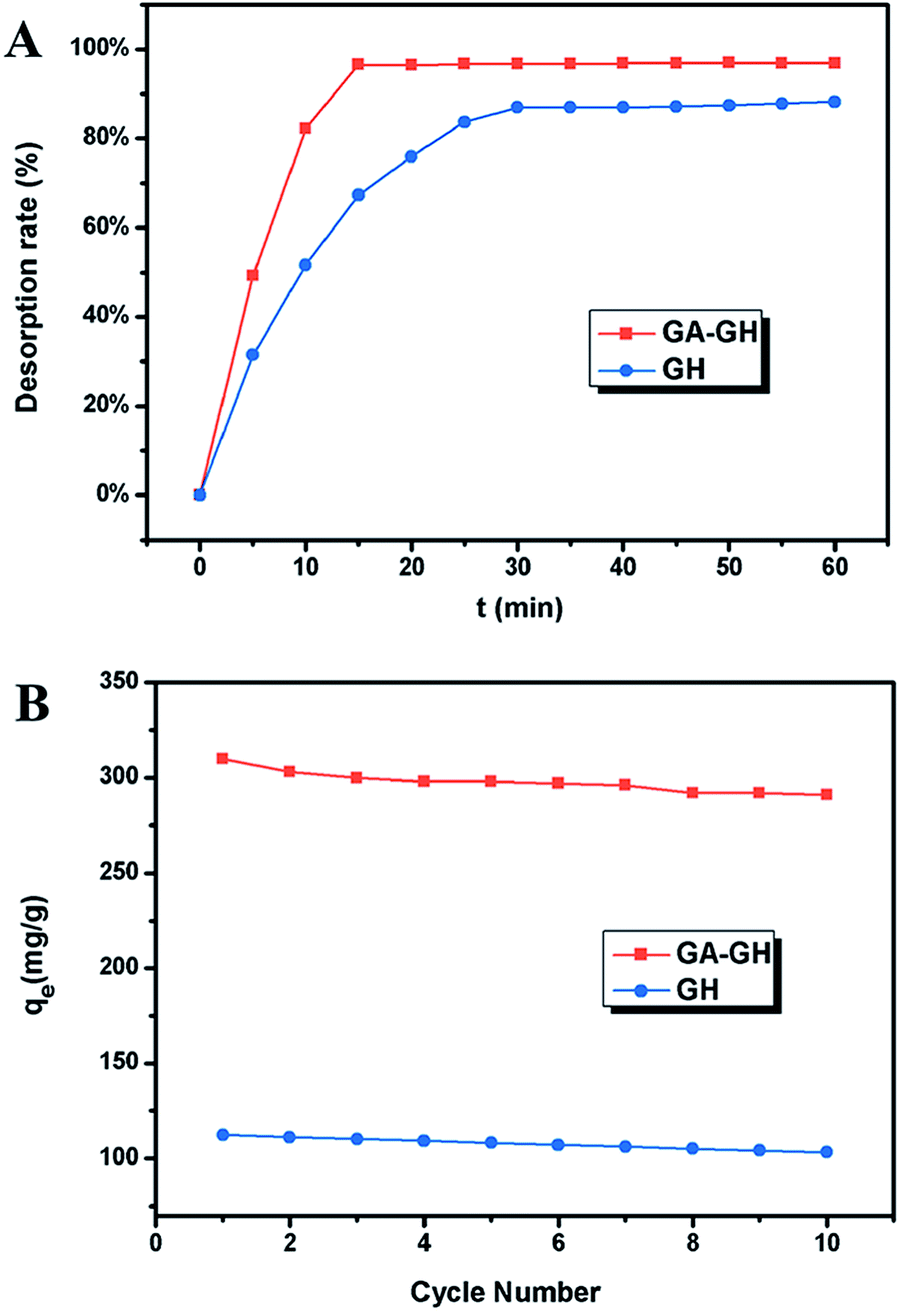
Since coordination complexation between Cr(III) and carboxyl groups is very weak at pH values below 3.0, the desorption of Cr(III) from GA-GH can be achieved under strong acidic condition. In our study, an HCl solution (0.1 M) was selected as the desorption agent to recover Cr(III) from GA-GH and GH at pH 2.0. As expected, the results shown in Fig. 6A reveal that the desorption rate of Cr(III) from GA-GH was higher than that from GH, due to the protonation of GA (Scheme 2). After 20 min, nearly 97% of the adsorbed Cr(III) was desorbed from the GA-GH. In comparison, at least 30 min was needed to desorb 89% of adsorbed Cr(III) from GH. These findings indicated the excellent desorption efficiency for the GA-GH. Furthermore, the regenerated GA-GH steadily maintained its adsorption capacity even after 10 repeated adsorption and desorption cycles, which was also much higher than that of GH adsorbents (Fig. 6B). The chemical state of adsorbed chromium was also investigated by XPS. As shown in Fig. S3,† the high resolution Cr 2p spectrum can be fitted into two peaks, which located at 578.8 eV and 588.3 eV, respectively, which confirmed the trivalent chemical state of the adsorbed chromium and demonstrated Cr(III) was not oxidized into Cr(VI) during the adsorption process. The result suggested that the desorbed Cr(III) from GA-GH is potentially to be reused for tanning leather, which will be beneficial to the leather industry. Therefore, based on these results, the GA-GH can be applied as an effective adsorbent for the removal of Cr(III) from tannery effluent.
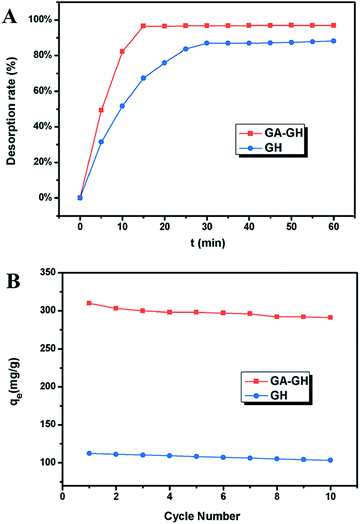 |
| | Fig. 6 (A) The desorption rate of Cr(III) from GA-GH and GH at pH 2.0; (B) adsorption capacities of GA-GH and GH as a function of repeated adsorption–desorption cycles for the removal of Cr(III). | |
3.3 The adsorption properties of GA-GH for organic dye
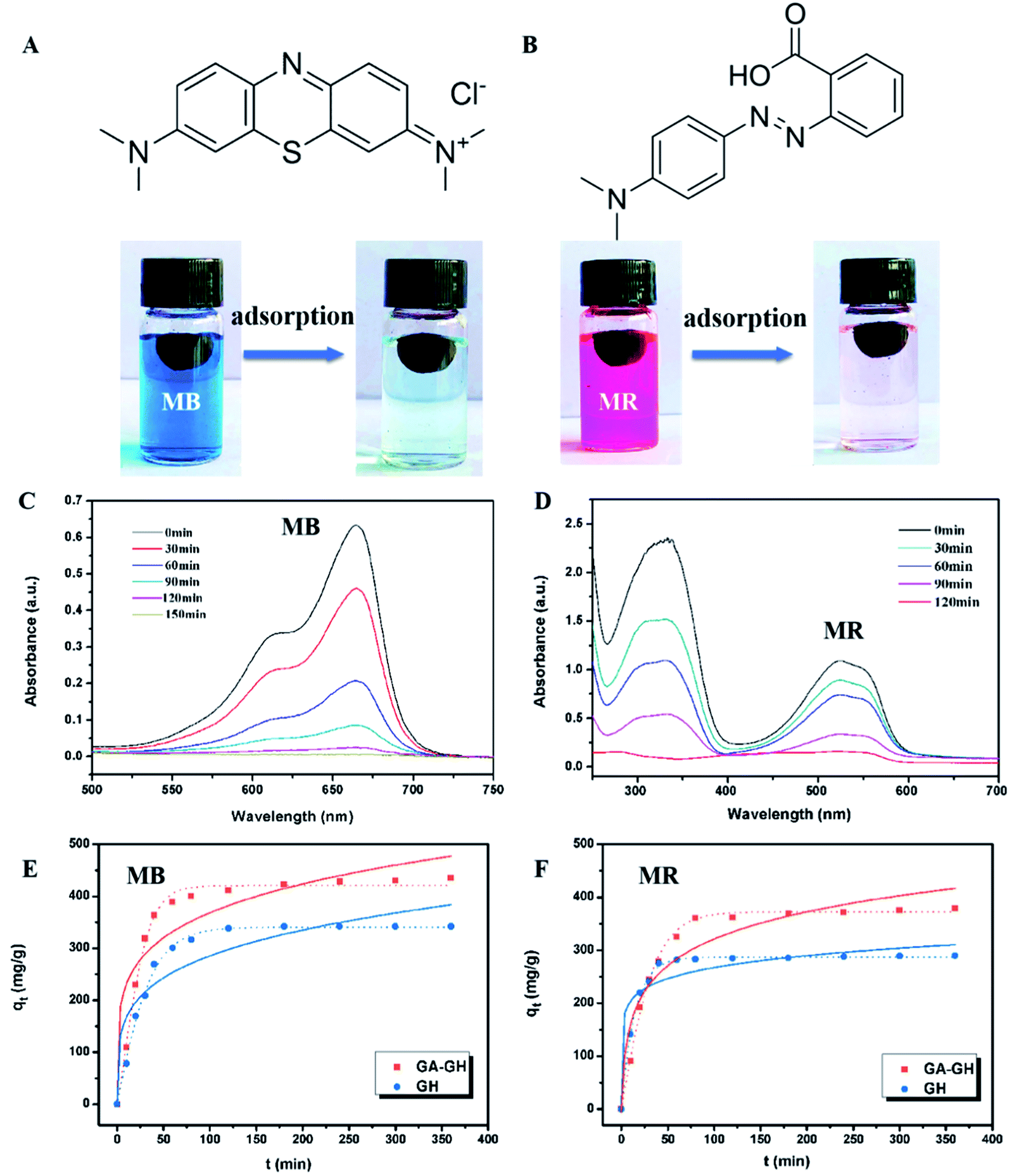
In this study, MB and MR were selected as models of cationic organic and anionic dyes, respectively, as they are commonly used in the dying process of leather making and are also the main source of organic pollutants in tannery effluent. As shown in Fig. 7A and B, the decolorization of the MB solution (20 mL, 10 ppm) and MR solution (20 mL, 20 ppm) in the presence of 10 mg GA-GH after 2 h was conspicuous, indicating the effective organic dye adsorption capacity of GA-GH. Fig. 7A and B also revealed that the characteristic UV-vis absorption peaks of MB at 664 nm and of MR at 520 nm decreased with the adsorption time. After 2 hours, almost 98% of MB and 100% of MR could be captured by the GA-GH adsorbent at room temperature. The excellent adsorption capacity of GA-GH for MB and MR can possibly be attributed to π–π interactions between the GA-GH and the aromatic organic dyes, as they drive the adsorbed organic dyes into the 3D porous hydrogel. To confirm this hypothesis, the adsorption kinetics of the aromatic MB or MR dye on GA-GH was further studied in comparison with that on GH. Based on the results presented in Fig. 7E and F, the equilibrated adsorption capacity of MB and MR on GA-GH were calculated to be 395 and 376 mg g−1 at 2 h, respectively, while for GH, the equilibrated adsorption capacity of MB and MR was 305 and 287 mg g−1, respectively. The results clearly demonstrated that the adsorption capacity of GA-GH for MB or MR was higher than that of GH, which confirm that the functionalized GA with a benzene ring enhanced the π–π conjugation between the GA-GH and the aromatic MB or MR dye. Therefore, the GA-GH can also be applied as a suitable absorbent for the removal of aromatic pollutants from tannery effluent.
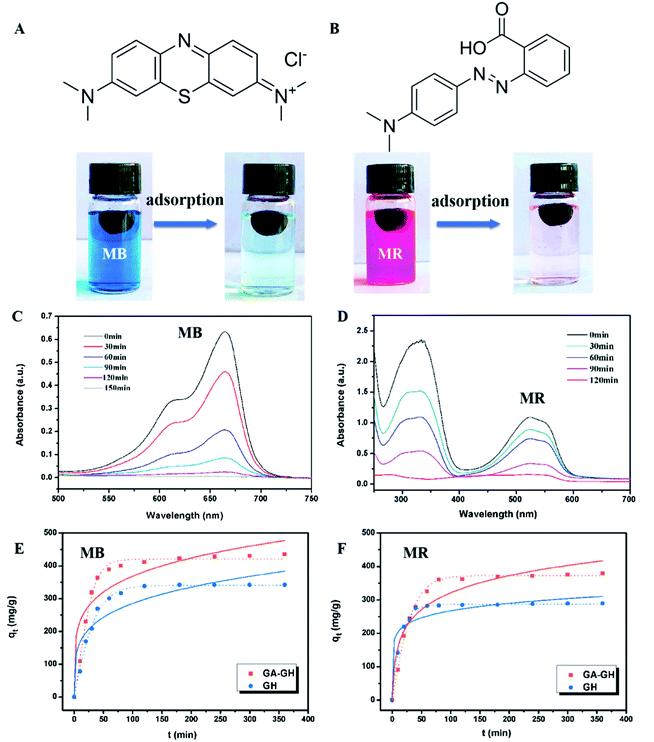 |
| | Fig. 7 (A and B) The chemical structure and color fading of MB and MR solution after adsorption by GA-GH; (C and D) the change of UV-vis absorption spectra of original MB (10 ppm) and MR (20 ppm) in the presence of GA-GH; (E and F) adsorption kinetic plots and curving fitting by pseudo-first-order (solid line) and pseudo-second-order (dot line) kinetic models of MB and MR on GH and GA-GH, respectively. | |
4. Conclusions
In order to effectively remove Cr(III) and organic dye from tannery wastewater, a GA-GH adsorbent with 3D porous architectures was successfully fabricated using a facile in situ reducing-assembly approach. Due to the carboxyl groups of GA, the GA-GH was able to effectively capture Cr(III) ions through coordination complexation at pH value close to 4.0, leading to a much higher adsorption capacity of Cr(III) than that of the GH. Importantly, GA-GH was easily regenerated at pH 2.0 with HCl and retained its high adsorption capacity after multiple adsorption–desorption cycles. Moreover, the modified GA on graphene sheets with benzene group was also able to improve the adsorption capacity of organic dyes, which was attributed to the enhanced π–π interactions between GA-GH and aromatic dyes. Taken together, the GA-GH is a promising adsorbent for treating tannery effluent.
Conflicts of interest
There are no conflicts declare.
Acknowledgements
This work was supported by Opening Project of Key Laboratory of Leather Chemistry and Engineering of Ministry of Education, Sichuan University (20826041C4159), Opening Project of State Key Laboratory of Hydraulics and Mountain River Engineering, Sichuan University and “the Fundamental Research Funds” for the Central Universities.
Notes and references
- A. J. Bailey, N. D. Light and E. D. T. Atkins, Nature, 1980, 288, 408–410 CrossRef CAS PubMed.
- A. D. Covington, Chem. Soc. Rev., 1997, 26, 111–126 RSC.
- K. Li, R. Yu, R. Zhu, R. Liang, G. Liu and B. Peng, ACS Sustainable Chem. Eng., 2019, 7, 8660–8669 CrossRef CAS.
- Y. Ding, D. Guo, J. Cheng, H. Zhang and M. Sui, Struct. Chem., 2016, 27, 1255–1263 CrossRef CAS.
- T. Lan, R. An, Z. Liu, K. Li, J. Xiang and G. Liu, J. Colloid Interface Sci., 2018, 532, 331–342 CrossRef CAS PubMed.
- Y. Zhang, B. W. Mansel, R. Naffa, S. Cheong, Y. Yao, G. Holmes, H. L. Chen and S. Prabakar, ACS Sustainable Chem. Eng., 2018, 6, 7096–7104 CrossRef CAS.
- J. Lu, J. Ma, D. Gao, B. Lyu and J. Zhang, J. Cleaner Prod., 2016, 139, 788–795 CrossRef.
- S. Cao, K. Wang, S. Zhou, Y. Wang, B. Liu, B. Chen and Y. Li, ACS Sustainable Chem. Eng., 2018, 6, 3957–3963 CrossRef CAS.
- J. Zhang, D. L. Mauzerall, T. Zhu, S. Liang, M. Ezzati and J. V. Remais, Lancet, 2010, 375, 1110–1119 CrossRef.
- H. Gu, S. B. Rapole, J. Sharma, Y. Huang, D. Cao, H. A. Colorado, Z. Luo, N. Haldolaarachchige, P. David, B. Walters, S. Wei and Z. Guo, RSC Adv., 2012, 2, 11007–11018 RSC.
- S. P. Goutam, G. Saxena, V. Singh, A. K. Yadav, R. N. Bharagava and K. B. Thapa, Chem. Eng. J., 2018, 336, 386–396 CrossRef CAS.
- L. Zhang, F. Fu and B. Tang, Chem. Eng. J., 2019, 356, 151–160 CrossRef CAS.
- S. Ramalingam and R. R. Jonnalagadda, ACS Sustainable Chem. Eng., 2017, 5, 5537–5549 CrossRef CAS.
- S. Ramalingam, K. J. Sreeram, J. R. Rao and B. U. Nair, ACS Sustainable Chem. Eng., 2016, 4, 2706–2714 CrossRef CAS.
- T. Abou Elmaaty, J. Ma, F. El-Taweel, E. Abd El-Aziz and S. Okubayashi, Ind. Eng. Chem. Res., 2014, 53, 15566–15570 CrossRef CAS.
- J. Kanagaraj and R. C. Panda, Ind. Eng. Chem. Res., 2011, 50, 12400–12408 CrossRef CAS.
- S. S. Sun, T. Xing and R. C. Tang, Ind. Eng. Chem. Res., 2013, 52, 8953–8961 CrossRef CAS.
- Y. Gao and J. Xia, Environ. Sci. Technol., 2011, 45, 8605–8606 CrossRef CAS PubMed.
- G. Liu, K. Li, Q. Luo, H. Wang and Z. Zhang, J. Colloid Interface Sci., 2017, 490, 642–651 CrossRef CAS PubMed.
- J. Xiang, L. Ma, H. Su, J. Xiong, K. Li, Q. Xia and G. Liu, Appl. Surf. Sci., 2018, 458, 978–987 CrossRef CAS.
- H. S. Shang, Y. J. Lu, F. Zhao, C. Chao, B. Zhang and H. S. Zhang, RSC Adv., 2015, 5, 75728–75734 RSC.
- P. L. Yap, S. Kabiri, D. N. H. Tran and D. Losic, ACS Appl. Mater. Interfaces, 2019, 11, 6350–6362 CrossRef CAS PubMed.
- A. J. Capezza, W. R. Newson, R. T. Olsson, M. S. Hedenqvist and E. Johansson, ACS Sustainable Chem. Eng., 2019, 7, 4532–4547 CrossRef CAS.
- L. Wen, K. Li, J. Liu, Y. Huang, F. Bu, B. Zhao and Y. Xu, RSC Adv., 2017, 7, 7688–7693 RSC.
- Y. Fang, D. Lin and S. Yao, J. Chem. Eng. Data, 2018, 63, 4418–4424 CAS.
- J. M. Ambrosy, C. Pasel, M. Luckas, M. Bittig and D. Bathen, Ind. Eng. Chem. Res., 2019, 58, 4208–4221 CrossRef CAS.
- Y. Zhao, Y. Zhu, T. Zhu, G. Lin, J. Huo, D. Lv, H. Wang and Y. Sun, Ind. Eng. Chem. Res., 2019, 58, 156–164 CrossRef CAS.
- X. Guan, J. Chang, Y. Chen and H. Fan, RSC Adv., 2015, 5, 50126 RSC.
- R. K. Upadhyay, N. Soin and S. S. Roy, RSC Adv., 2014, 4, 3823–3851 RSC.
- Y. Zhao, T. Sun, W. Liao, Y. Wang, J. Yu, M. Zhang, Z. Yu, B. Yang, D. Gui, C. Zhu and J. Xu, ACS Appl. Mater. Interfaces, 2019, 11, 22794–22800 CrossRef CAS PubMed.
- L. Zhu, R. Liu, Z. Fang, P. O. Agboola, N. F. Al-Khalli, I. Shakir and Y. Xu, ACS Appl. Mater. Interfaces, 2019, 11, 2218–2224 CrossRef CAS PubMed.
- Y. Wang, P. Zhang, C. F. Liu and C. Z. Huang, RSC Adv., 2013, 3, 9240–9246 RSC.
- X. Zhang, T. Zhang, Z. Wang, Z. Ren, S. Yan, Y. Duan and J. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 1303–1310 CrossRef CAS PubMed.
- S. Li, J. Zheng, J. Yan, Z. Wu, Q. Zhou and L. Tan, ACS Appl. Mater. Interfaces, 2018, 10, 42573–42582 CrossRef CAS PubMed.
- H. Sun, L. Cao and L. Lu, Nano Res., 2011, 4, 550–562 CrossRef CAS.
- C. Li and G. Shi, Nanoscale, 2012, 4, 5549–5563 RSC.
- J. Luo, J. Lai, N. Zhang, Y. Liu, R. Liu and X. Liu, ACS Sustainable Chem. Eng., 2016, 4, 1404–1413 CrossRef CAS.
- Y. Xu, K. Sheng, C. Li and G. Shi, ACS Nano, 2010, 4, 4324–4330 CrossRef CAS PubMed.
- B. Badhani, N. Sharma and R. Kakkar, RSC Adv., 2015, 5, 27540 RSC.
- L. Zhang, G. Chen, M. N. Hedhili, H. Zhang and P. Wang, Nanoscale, 2012, 4, 7038–7045 RSC.
- W. Chen and L. Yan, Nanoscale, 2011, 3, 3132–3137 RSC.
- Y. Lei, Z. Tang, R. Liao and B. Guo, Green Chem., 2011, 13, 1655–1658 RSC.
- J. Wang, Z. Shi, J. Fan, Y. Ge, J. Yin and G. Hu, J. Mater. Chem., 2012, 22, 22459–22466 RSC.
- H. Gao, Y. Sun, J. Zhou, R. Xu and H. Duan, ACS Appl. Mater. Interfaces, 2013, 5, 425–432 CrossRef CAS PubMed.
- S. W. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef.
- G. Liu, H. Gao, K. Li, J. Xiang, T. Lan and Z. Zhang, J. Colloid Interface Sci., 2018, 514, 338–348 CrossRef CAS PubMed.
- Q. Xia, L. Yang, K. Hu, K. Li, J. Xiang, G. Liu and Y. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 2352–2363 CrossRef CAS PubMed.
- H. Bai, C. Li, X. L. Wang and G. Q. Shi, J. Phys. Chem. C, 2011, 115, 5545–5551 CrossRef CAS.
- Y. Sun, M. Yang, F. Yu, J. Chen and J. Ma, Prog. Chem., 2015, 27, 1133–1146 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The schematic transformation of GA under alkali conditions, illustration of the formation process of GA-GH, N2 adsorption–desorption isotherms of GA-GH, high-resolution of XPS of Cr peaks of GA-GH after adsorption for Cr(III). See DOI: 10.1039/c9ra05664e |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Ruiquan Yuab,
Tianxiang Lanb,
Zheng Liuc,
Peng Zhangd and
Ruifeng Liang*d
ab,
Ruiquan Yuab,
Tianxiang Lanb,
Zheng Liuc,
Peng Zhangd and
Ruifeng Liang*d