DOI:
10.1039/C9RA05636J
(Paper)
RSC Adv., 2019,
9, 30976-30988
Encapsulation of echinomycin in cyclodextrin inclusion complexes into liposomes: in vitro anti-proliferative and anti-invasive activity in glioblastoma†
Received
21st July 2019
, Accepted 20th September 2019
First published on 30th September 2019
Abstract
Echinomycin, a DNA bis-intercalator peptide, belongs to the family of quinoxaline antibiotics. Echinomycin exhibits potent antitumor and antimicrobial activity. However, it is highly water insoluble and suffers from low bioavailability and unwanted side effects. Therefore, developing new formulations and delivery systems that can enhance echinomycin solubility and therapeutic potency is needed for further clinical application. In this study, echinomycin has been complexed into the hydrophobic cavity of γ-cyclodextrin (γCD) then encapsulated into PEGylated liposomes. The anti-proliferative and anti-invasive effect has been evaluated against U-87 MG glioblastoma cells. Echinomycin-in-γCD inclusion complexes have been characterized by phase solubility assay, TLC, and 1H-NMR. The echinomycin-in-γCD inclusion complexes have been loaded into liposomes using a thin film hydration method to end up with echinomycin-in-γCD-in-liposomes. Drug-loaded liposomes were able to inhibit cell proliferation with IC50 of 1.0 nM. Moreover, echinomycin-in-γCD-in-liposomes were found to inhibit the invasion of U-87 MG cells using the spheroid gel invasion assay. In conclusion, the current work describes for the first time γCD-echinomycin complexes and their encapsulation into PEGylated liposomes.
1. Introduction
Echinomycin (NSC526417) is a depsipeptide antibiotic that belongs to the quinoxaline family which was first isolated from fermentation cultures of Streptomyces echinatus in 1957 in Germany.1,2 Echinomycin has shown antibacterial, antiviral, and antitumor activity against different types of cancers including hepatocellular carcinoma, melanoma, leukemia, breast cancer, glioma, colon cancer, pancreatic cancer, gastric cancer, and cervical cancer.3–5 Interestingly, echinomycin can selectively kill the leukemia-initiating cells in relapsed acute myeloid leukemia (AML) without harming the normal stem cells. In 2015, echinomycin has received an orphan drug status for AML in the USA.6 The mechanism of action of echinomycin is attributed to the bis-intercalation and binding with high affinity to the 5′-CG bases of DNA, which prevent the unwinding of the double helical DNA; thus, inhibiting the replication of chromosomal DNA.4,7,8 Furthermore, it is a potent inhibitor of the hypoxia-inducible factor-1 (HIF-1) DNA-binding, a factor that controls genes necessary in tumor biology including cell growth, glycolysis, angiogenesis, metastasis, and invasion.7 Unfortunately, no significant response was observed in clinical trials conducted on patients suffering from metastatic soft tissue sarcoma, advanced breast cancer, squamous cell cervical carcinoma, advanced ovarian cancer, advanced renal carcinoma, central nervous system, and colorectal cancer due to its high dose-related toxicity.8–20 One of the most common side effects is related to echinomycin in gastrointestinal tract symptoms, including moderate to severe nausea, vomiting, and diarrhea, as well as the elevation of hepatic enzymes and a mild decrease in platelets and granulocyte counts which was life-threading in some cases.11,21 Accordingly, in all clinical trials, no dose escalation was possible to achieve a significant clinical response. Moreover, echinomycin is highly water insoluble and precipitates when added to water which complicates the formulation of proper dosage forms thereby suffering from low bioavailability. The available solvents for echinomycin are clinically unacceptable. Therefore, there is a need for new formulations and delivery systems that can enhance solubility and therapeutic potency of echinomycin.
Glioblastoma (GBM) is a common and aggressive type of malignant brain tumors.22 GBM accounts for 16% of all primal cases of brain tumors. It is a fatal and non-curable disease, with only less than 5% of patients that survive after 5 years of diagnosis.22,23 Different treatments are in use for GMB including surgery, radiation, chemotherapy, and immunotherapy.24 Nevertheless, these therapies don't bear a higher survival rate in GBM patients due to the aggressive nature of GBM that invades normal cells of the central nervous system and the protection advantage by the neurovascular niche.25
Liposomes have unique properties that expanded the scope of pharmacokinetic and pharmacodynamics of insoluble drugs.26,27 Liposomes are widely used in clinics as drug delivery systems.28 Liposomes have sophisticated properties including the biocompatibility, low toxicity, biodegradability, and the targeting capability.29 Due to its tiny size, liposomes have the ability to penetrate leaky blood vessels of tumor cell by passive targeting mechanism usually referred to enhanced permeability and retention effect.30 Additionally, Stealth® liposomes coated with hydrophilic polymers such as polyethylene glycol (PEG) showed prolong blood half-life by masking their detection and destruction by the mononuclear phagocyte system (MPS).31
Cyclodextrins (CDs) are biocompatible and water-soluble cyclic oligosaccharides. CDs consist of the lipophilic crater and hydrophilic outer exterior. These novel features of CDs can enhance the physiochemical properties of water-insoluble drugs. The natural α-, β- and γ-cyclodextrin consist of 6, 7, and 8 glucopyranose units, respectively.32 Although CDs are valuable solubilizers of hydrophobic drugs, the complex formation of gust drug and the host CD cavity is weak and the drug suffers dissociation after administration in vivo in which the blood components can displace the included drug. Moreover, drug-in-CD inclusion is challenged by rapid renal clearance, renal toxicity, and interaction with the cellular membrane components. Therefore, combining the dual models of drug-in-CD-in-liposomes can augment the fate of hydrophobic drugs in vivo.32 In the current work, the echinomycin-in-γCD-in-liposomes formulation has been developed. First, echinomycin (Fig. 1A) has been complexed into γCD (Fig. 1B) and the echinomycin-in-γCD complex inclusion has been characterized by phase solubility assay TLC, and 1H-NMR. Then, the echinomycin-in-γCD inclusion has been loaded into the aqueous core of PEGylated liposomes using thin-film hydration method. The resulting echinomycin-in-γCD-in-liposomes have been characterized for the encapsulation efficacy, size, charge, uptake, and the anti-proliferative and anti-invasive activity against U-87 MG glioblastoma cells.
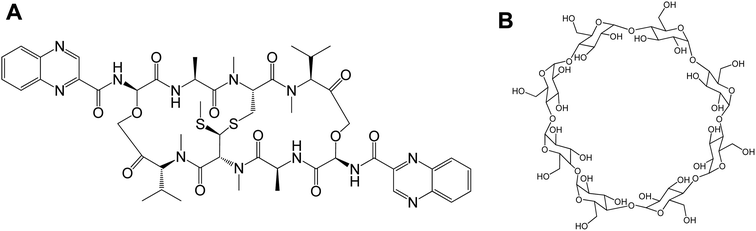 |
| | Fig. 1 The chemical structure of echinomycin (A) and γCD (B). | |
2. Materials and methods
2.1. Chemicals
Echinomycin (N-(2-quinoxalinylcarbonyl)-O-[N-(2-quinoxalinylcarbonyl)-D-seryl-L-alanyl-3-mercapto-N,S-dimethylcysteinyl-N-methyl-L-valyl]-D-seryl-L-alanyl-N-methylcysteinyl-N-methyl L-valine (81)-lactone cyclic (37)-thioester) obtained from TOCRIS® bioscience (Bristol, UK). The lipids; DPPC (1,2-dihexadecanoyl-sn-glycero-3-phosphocholine), DSPE-PEG2000 (1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethyleneglycol)-2000]). 1,2-Dioleoyl-sn-glycero-3-phosphoethanolamine -N-(lissamine rhodamine B sulfonyl) (Lissamine) (Rho-PE), and cholesterol were purchased from Avanti Polar Lipids (Alabaster, USA). γ-cyclodextrin was purchased from Sigma-Aldrich (USA). Other chemicals were obtained from different sources and were used as received.
2.2. Phase solubility and binding constant of echinomycin with γCD
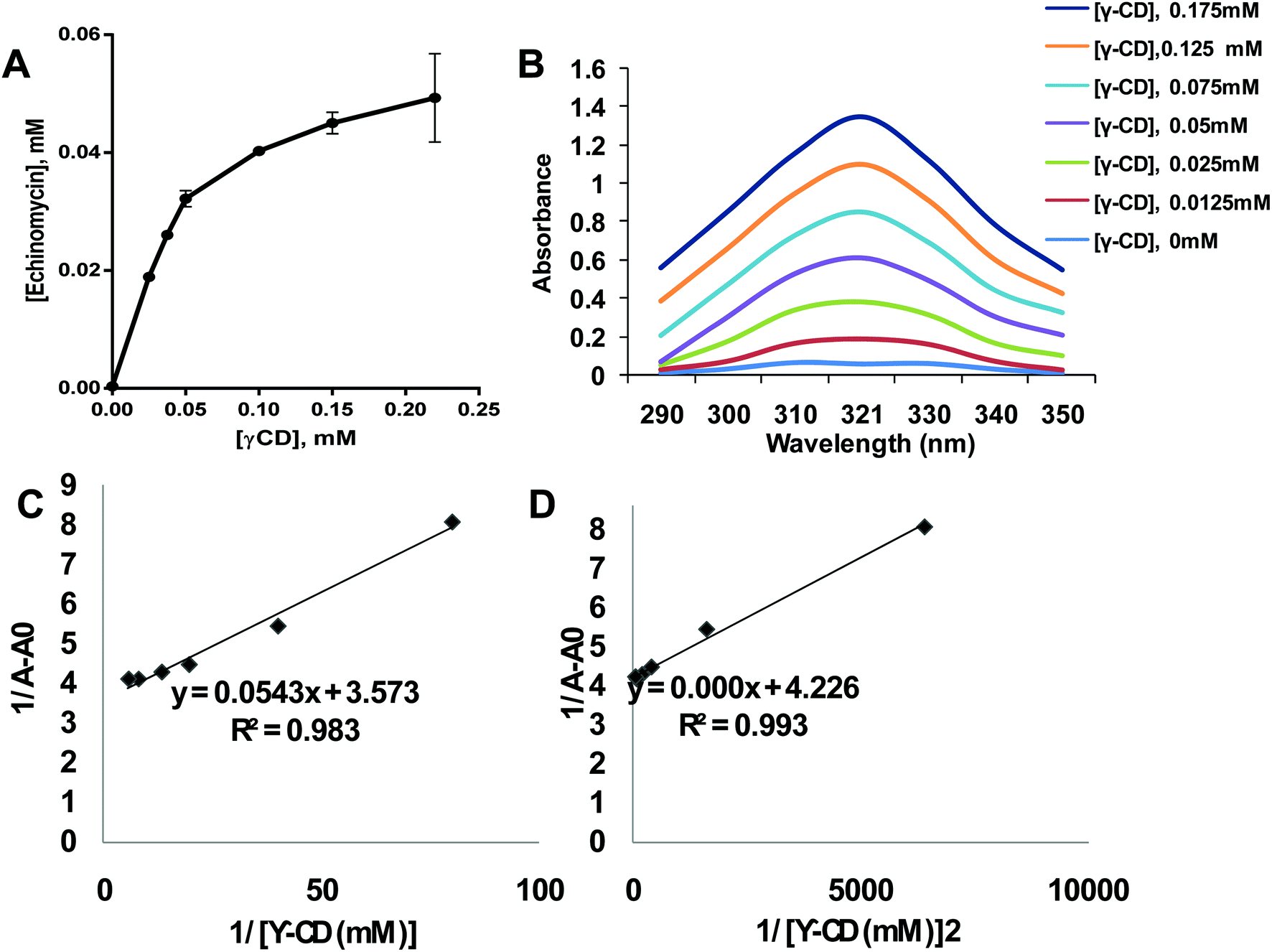
Phase solubility analysis was performed according to the method reported by Higuchi.33 A constant amount of echinomycin (55 μM) was added into 3 mL of γCD aqueous solutions with various concentrations (0.025–0.22 mM). Then all samples were mixed on a shaker at 25 °C for 24 h. After the equilibrium was reached, the mixtures were centrifuged for 5 min at 1000 rpm. The concentration of echinomycin in the supernatant was determined by a UV Nanodrop spectroscopy (Thermo Scientific) using a calibration curve. The binding constant (K1:1) and complexation efficiency (CE) were analyzed using equations were analyzed using the equations:
| K1:1 = slope/S0(1 − slope) = [Ech-γCD]/[Ech][γCD] |
| CE = [D − CD]/[CD] = S0K1:1 = slope/(1 − slope) |
where: S0 is drug solubility without cyclodextrin (i.e. drug intrinsic solubility in water) and the slope is the slope of the linear part of the phase solubility diagram.34
2.3. Determination of stoichiometry of echinomycin with γCD using Benesi–Hildebrand method
A constant amount of echinomycin (35 μM) was added into 3 mL of γCD polymer aqueous solutions with various concentrations (0.0125–0.175 mM). Then all samples were mixed on a shaker at 25 °C for 24 h. Then the mixtures were centrifuged for 5 min at 1000 rpm. The absorbance of echinomycin of each supernatant was scanned by a UV Nanodrop spectroscopy (Thermo Scientific TM Nanodrop 2000 and 200C) using the wavelengths 290–300 Benesi–Hildebrand correlation between 1/[γCD] and 1/[γCD]2vs. 1/(A − A0) were plotted.
2.4. Characterization of the echinomycin-in-γCD complex by 1H-NMR spectroscopy
The complex was prepared by solvent evaporation encapsulation technique. A 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 drug to the γCD ratio (0.22 μmol of echinomycin, 0.24 mg and 0.44 μmol of γCD, 0.57 mg) were dissolved separately in 1 mL ethanol and 2 mL deionized water, respectively. The solution was then mixed on a shaker at 280 rpm at room temperature for 24 h. The solution was dried completely at 50 °C under vacuum.35 The 1H-NMR analysis was performed using Bruker Avance III 500 mHz instrument (Bruker BioSpin, Switzerland).
:2 drug to the γCD ratio (0.22 μmol of echinomycin, 0.24 mg and 0.44 μmol of γCD, 0.57 mg) were dissolved separately in 1 mL ethanol and 2 mL deionized water, respectively. The solution was then mixed on a shaker at 280 rpm at room temperature for 24 h. The solution was dried completely at 50 °C under vacuum.35 The 1H-NMR analysis was performed using Bruker Avance III 500 mHz instrument (Bruker BioSpin, Switzerland).
2.5. Echinomycin-γCD inclusion complex preparation
The inclusion of echinomycin into γCD was performed using dispersion/co-evaporated dispersion method. A stock of echinomycin was prepared by dissolving 1 mg of echinomycin in 1 mL of chloroform to achieve 1 mg mL−1 final concentration. A stock solutions γCD was prepared by dissolving 1 mg of γCD powder in 1 mL PBS (pH 7.4) to achieve 1 mg mL−1 final concentration. The echinomycin-in-γCD inclusion was prepared by dropwise addition of 100 μL of echinomycin dissolved in chloroform into 235 μL of the γCD solution to get 1:2 molar ration of echinomycin-γCD. The complex solution was then stirred for 1 h at room temperature. Further, the chloroform was evaporated under vacuum for 1.5 h. The final volume of echinomycin-in-γCD inclusion then adjusted to 1 mL in PBS (pH 7.4) for further loading into liposomes.36
To investigate the echinomycin-in-γCD inclusion complexes, thin layer chromatography (TLC) has been used. TLC was performed on aluminum-backed, 10 cm × 5 cm silica gel plates. The plates were developed with n-hexane 95%, ethylacetate, and methanol (3:3:0.6 v/v) mobile phase. Echinomycin, γCD, echinomycin -in-γCD and “in situ” mixture were spotted on a baseline, 0.5 cm from the edge of the silica plate. The developed plates were air dried, to visualize echinomycin under UV light, and then sprayed with the 5% ethanolic sulfuric acid solution and dried on a hot plate to locate free and complexed γ-cyclodextrins.
2.6. Encapsulation of echinomycin-in-γCD inclusion into liposomes
Liposomes loaded with echinomycin-in-γCD inclusion were prepared by lipid thin-film-hydration method.37 A stock solution of lipids composed of DPPC, cholesterol, and DSPE-PEG2000 in a 63:35:3 molar ratio was dissolved in 5 mL chloroform. The thin-film was formed by evaporation of chloroform using rotary evaporator (IKA, Germany) for 1 h at 50 °C under decreased atmospheric pressure using BUCHI® vacuum controller. The thin film then was hydrated with echinomycin-in-γCD inclusion, followed by vortex mixing for 30 min until the thin lipid film was completely dissolved. Downsizing of the hydrated liposomes was performed by extruding the liposomes 13 times through 100 nm polycarbonate membrane (Nuclepore, Whatman plc, UK) using Mini-Extruder (Avanti Polar Lipids, Inc. USA). After that, the free echinomycin-in-γCD inclusion was removed by ultrafiltration using Amicon® filters (cut-off of 100 kDa) (Millipore, Germany).
2.7. Quantification of echinomycin by HPLC
Quantification of echinomycin was performed by HPLC system (DIONEX TM Ultimate TM 3000, Thermofisher) using reversed phase C18 column 150 × 4.6 mm, 5 μm (KNAUER). The mobile phase containing 60% acetonitrile (HPLC grade) and 40% water was prepared according to a method described by the National Center for Biotechnology Information (PubChem Database; Echinomycin). Chromatographic conditions were as per the following: the prepared mobile phase was provided in an isocratic mode at flow rate 1 mL min−1, detection by UV at 243 nm, 20 μL of each sample was used for injection, the retention time was 4.6 min. The calibration curve was constructed using known concentrations of standard echinomycin in the range 0.000587–0.3 mg mL−1 in absolute ethanol. Echinomycin has been quantified using the linearity regression equation was Y = 1015X − 1.0722 (n = 3, r = 0.9999) (Fig. S2A and B†). Echinomycin was released from liposomes using acetonitrile up to 1 mL to disintegrate the liposomes and then quantified by HPLC. The encapsulation efficiency (EE%) and drug loading (DL%) were calculated based on the following equation:
| EE (%) = (Drug in liposomes/Total drug added) × 100 |
| DL (%) = (Weight of loaded drug/Weight of lipids) × 100 |
2.8. Liposomes size and charge
The mean hydrodynamic diameter of the liposomes was determined by Dynamic Light Scattering (DLS) using a Nano ZS (Malvern Instruments, UK). The polydispersity index (PDI) has been used as an indicator of size distribution. The zeta potential was measured using the same apparatus. All liposomes were diluted in free deionized water to obtain a final sodium chloride concentration of 8 mM (pH 7.4).
2.9. Transmission electron microscopy (TEM)
TEM analysis for blank liposomes and echinomycin-in-γCD-in-liposomes were performed using the negative staining method.38 200-mesh formvar copper grids (SPI supplies, USA) were uniformly coated with carbon and glow discharged using low vacuum Leica EM ACE200 coating system (Leica, Austria) then coated using 1.5% vinyl K solution in chloroform. Further, 7 μL of liposomes were placed on the formvar copper grid and left to dry overnight. Next, the loaded grids were stained using 3% uranyl acetate dye solution. After 20 min, the grids were rinsed with distilled water and air-dried. The prepared grids were observed under Versa 3D (FEI, Netherlands) operated at 30 kV.
2.10.
In vitro stability of echinomycin-in-γCD-in-liposomes
The stability assay of echinomycin-in-γCD-in-liposomes was performed at 4 °C and 37 °C in PBS (pH 7.4), with storage times of 14 days. Liposomes samples (50 μL) were collected at 4 °C and 37 °C at different time intervals (0, 12, 24, 48, 168, and 336 h). The mean hydrodynamic diameter and polydispersity index (PDI) of echinomycin-in-γCD-in-liposomes were determined at each time point using a similar method described before. All liposomes were diluted in free deionized water to obtain a final sodium chloride concentration of 8 mM (pH 7.4).
The release assay of echinomycin from loaded liposomes was performed at 37 °C in PBS (pH 7.4). Samples of eechinomycin-in-γCD-in-liposomes (700 μL) were incubated at different times intervals (0, 12, 24, 48, and 72 h). The free echinomycin-in-γCD inclusions at each time point were removed by ultrafiltration using Amicon® filters (cut-off of 100 kDa) (Millipore, Germany). The concentration of echinomycin in the liposomal fraction was quantified by HPLC using the same method described before. The released echinomycin was calculated based on the initially loaded echinomycin and the remaining echinomycin in liposomes.
2.11. Echinomycin-cyclodextrin liposomes cellular uptake assessment
Labeled liposomes were prepared using 0.5 mol% of PE-Rhodamine by a thin film hydration method. 5 × 104 cells of glioblastoma cell-line U-87 MG were seeded in 12 wells plate (SPL, Korea) containing glass coverslips, and incubated at 37 °C for 24 hours. After cell attachment and having 50% confluency, the cells were treated with 20 μM of echinomycin-in-γCD -PE-Rhodamine complex liposomes and incubated at 37 °C for 4 h. After incubation, the cells were washed 3 times with 1 mL of PBS and then fixed by 1 mL of 4% formaldehyde for 10 min at room temperature in dark place. The cells then were washed 3 times using PBS and the coverslips were transferred onto a glass slide and dipped in mounting medium with 4′,6-diamidino-2-phenylindole, dihydrochloride (DAPI), and finally analyzed by confocal microscope system Zeiss LSM780.
2.12.
In vitro cytotoxicity assessment
U-87 MG cell line was obtained from American Type Culture Collection (ATCC), the cells were cultured in Eagle's Minimum Essential Medium (EMEM) (ATCC, Manassas, USA) supplemented with 10% fetal bovine serum (Gibco, USA), 1% 200 mM L-glutamine, and penicillin–streptomycin (Euroclone, Italy) (100 IU mL−1 to 100 μg mL−1). Cells were kept at 37 °C in a humidified atmosphere containing 5% CO2 and subculture was performed at 70–90% confluency.
The cytotoxicity of free echinomycin and liposomal preparations was determined using cell viability microculture tetrazolium (MTT) assay. Cells were seeded at an initial density of 8 × 104 cells per well in 96-well culture plate (Costar, USA) in 100 μL complete culture medium and incubated in a humidified atmosphere of 5% CO2 at 37 °C for 24 h. After that, for the U-87 MG cells, a stock solution of free echinomycin, echinomycin-in-γCD inclusion, echinomycin-in-γCD-in-liposomes were serially diluted in EMEM high glucose medium to reach desired concentrations started from 20 nM to 0.02 nM in 100 μL volume and incubated in a humidified atmosphere of 5% CO2 at 37 °C for 72 h. The solutions were then removed and replaced with 100 μL fresh EMEM and 10 μL of MTT solution was added to each well, followed by incubation at 37 °C for 3 hours. After that, MTT-media solution was removed and 50 μL of DMSO was added to solubilize the dark blue formazan crystals. Absorbance was read at 570 nm with a microplate reader (Synergy™ HTX by BioTek Instruments Inc, USA) and the IC50 values were calculated.
2.13. Invasiveness assay
Enough collagen with pH 7.4 was pipetted into each chamber of an 8-chamber cover glass (Nunc, Lab-Tek, Thermo scientific). After polymerization at 37 °C for 45 min, spheroids from hanging drops were carefully removed and 1 spheroid was placed in each chamber of the cover glass. Another 200 μL layer of collagen gel matrix was prepared as above and was carefully added to each chamber and then set in a 37 °C incubator (no CO2) for 45 min to polymerize. Finally, 200 μL of RPMI was added to each chamber and incubated at 37 °C, 5% CO2 for 4 days. The spheroids were observed using inverted light microscopy at 10× objective lens and images were captured daily using Nikon Eclipse camera (TE2000-4, Panasonic Lumix DMC-GF6).
3. Results and discussion
3.1. Echinomycin-in-γCD complex formation and characterization
The poor aqueous solubility is a challenging factor that affects the therapeutic applications of drugs. Most of the anticancer agents belong to class IV of biopharmaceutical classification system which is characterized by the low aqueous solubility and low membrane permeability.39 Therefore, different methods have been used to enhance the solubility, dissolution, and bioavailability of water-insoluble drugs such as the use of cosolvents, solubilization, and solid dispersion.40 Complexation of drugs with CDs provided an effective approach to enhance the aqueous solubility of poorly soluble drugs thereby improving their therapeutic applications.41–46 Echinomycin is known for the potent antitumor and antimicrobial activity. Interestingly, echinomycin can be intercalated into 5′-CG bases in an irreversible manner which interferes with chemoresistance mechanisms.47–51 However, echinomycin is water-insoluble and suffers from low bioavailability and severe side effects. In the current study, echinomycin has been complexed with γCD providing water soluble echinomycin-in-γCD inclusion complexes. The phase solubility diagrams for the complex formation between echinomycin and γCD showed in Fig. 2A. The plot shows the aqueous solubility of echinomycin increased linearly as the γCD concentration reaches 60 μM. However, the phase solubility diagram showed a negative deviation from the linearity when the γCD concentrations were above 60 μM. Therefore, the solubility diagram of echinomycin can be classified as AN type according to Higuchi classification.33 Several reports have described AN type of solubility diagram and can be explained by the changes to the solvent induced by the solubilizer at different concentrations and/or the self-association of drug molecules, drug/cyclodextrin inclusion, and formation of non-inclusion complexes.34,52 Furthermore, the echinomycin-in-γCD complexation constant (K1:1) was calculated based on the linear part of the phase solubility diagram and was 5.92 mM−1, which indicates stable echinomycin-in-γCD inclusion complexes. Moreover, the binding constant K1:1 was > 1 which means that the complexation is not a 1:1 ratio and can be 1:2 or more. This result was anticipated due to the very large size of echinomycin molecule (Fig. 1A). In fact, calculating S0 leads to an error due to difficulty in its measurement. In order to avoid S0 discrepancy, complexation efficiency(CE) is more convenient to be used.53 The solubilizing efficiency (CE) for 1:1 echinomycin-γCD was calculated and found to be 1.8. It's worth to mention that the less the amount of cyclodextrin being used is better for complexation purposes. According to the value of CE, it can be concluded that a small amount of γCD is needed to solubilize echinomycin.
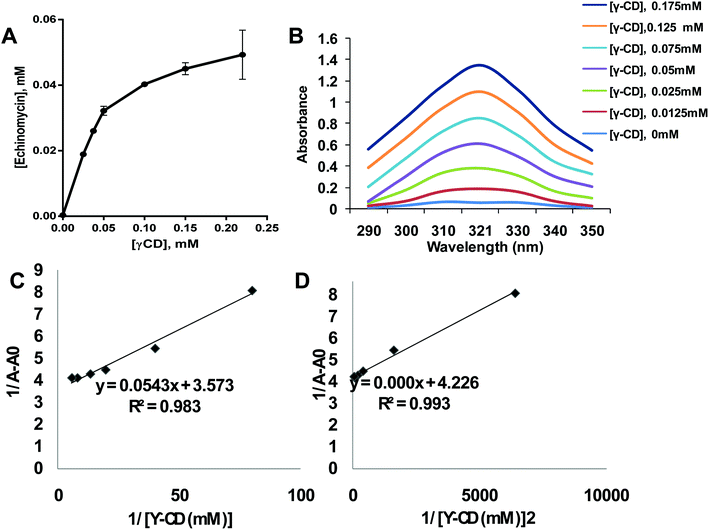 |
| | Fig. 2 Formation of echinomycin-in-γCD inclusion complexes. (A) Phase solubility diagram of echinomycin 0.055 mM concentration vs. different γCD concentration from 0.025–0.22 mM (n = 3). (B) Scanning for absorbance of Echi-γCD from 290–350 nm is showing an increase in UV spectra by increasing the concentration of γCD. (C) Echinomycin 0.035 mM concentration 1/(A − A0) vs. 1/[γCD] of different γCD concentration from 0.025–0.22 mM (n = 3). (D) Echinomycin 0.023 mM concentration 1/(A − A0) vs. 1/[γCD]2 of different γCD concentration from 0.012–0.1 mM (n = 3). | |
As concluded from the scanning of echinomycin-in-γCD complexes using UV visible spectroscopy, Fig. 2B shows a correlation between the enhanced solubility of echinomycin in water with increasing concentrations of γCD as indicated by the increased absorbance. Actually, due to the small changes in absorbance with increasing γCD concentration the Benesi–Hildebrand method was performed. The Benesi–Hildebrand correlation between 1/[γ-CD] and 1/[γ-CD]2vs. 1/(A − A0) was plotted (Fig. 1D). According to (Fig. 2C and D), the R2 of the second-order Benesi plot(1/[γ-CD]2vs. 1/(A − A0)) is higher than the R2 of the first order 0.991 and 0.987, respectively. This leads us to conclude that the stoichiometry of the inclusion complex is not 1:1 and can be 1:2 or more.54
For further evaluation of echinomycin-in-γCD inclusion, a thin layer chromatography assay was performed (Fig. S1, ESI†). Fig. S1A† showed different migration rates for non-polar echinomycin, polar γCD and an in-between spot for echinomycin-in-γCD complex migration. These observations and the different retention factor (Rf) values significantly proved the complex formation and the water solubility enhancement of echinomycin upon complexation with γCD. The “in situ” experiment, as shown in Fig. S1B,† exclude the possibility of direct complex formation on the chromatographic plate and indicating the existence of only a physical mixture between echinomycin and γCD.55
3.2. Characterization of the echinomycin-in-γCD complex by 1H-NMR spectroscopy
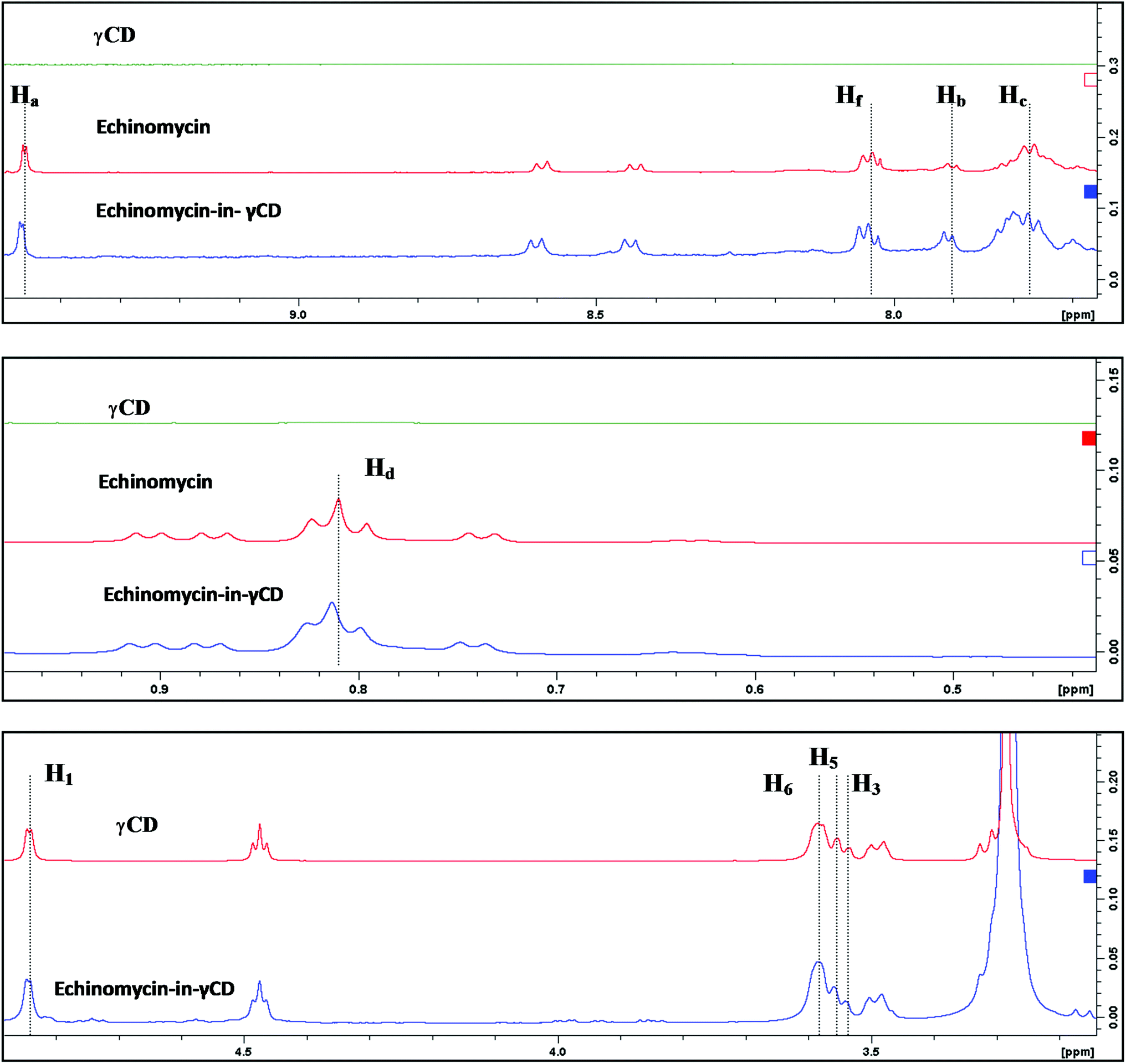
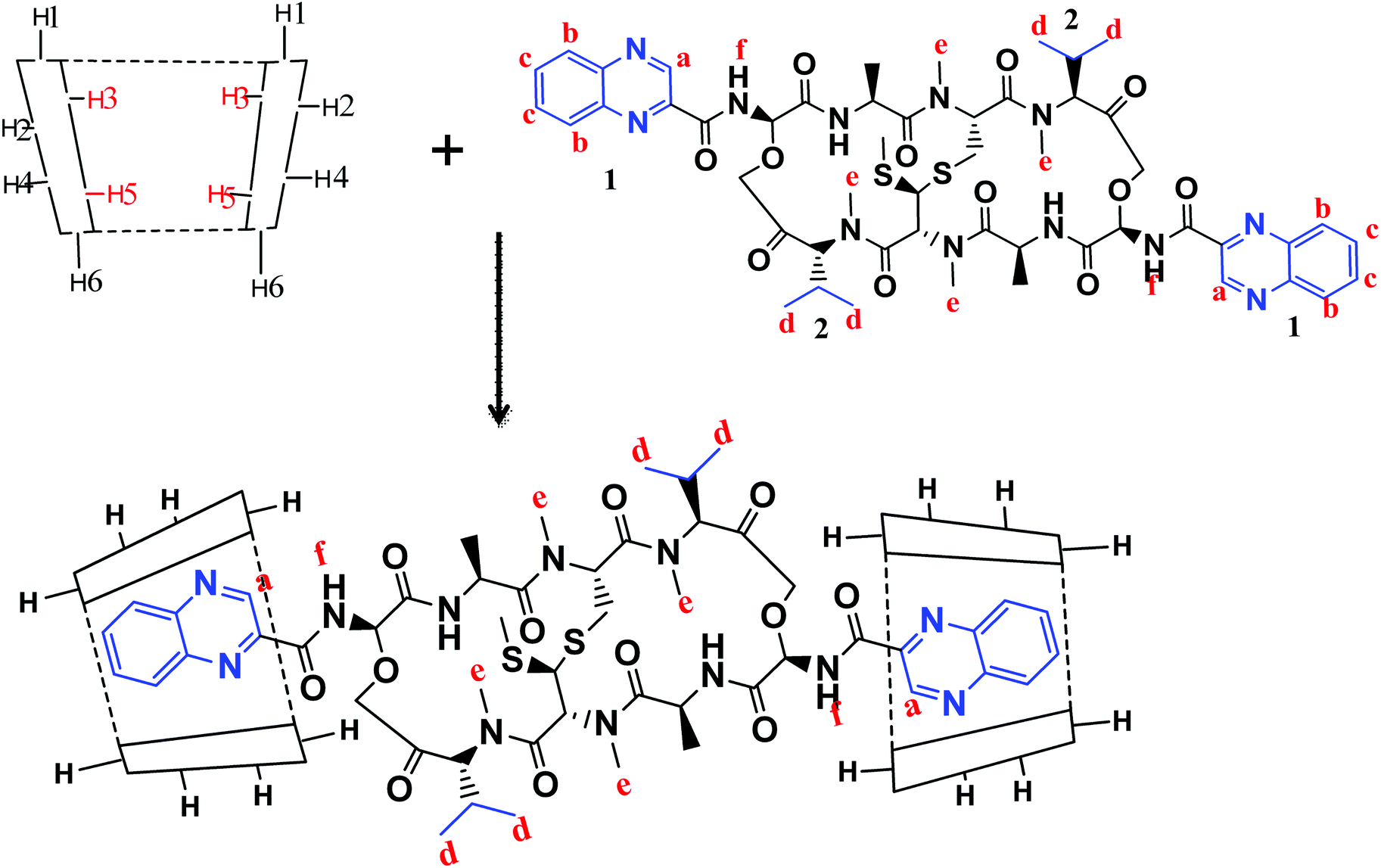
1H-NMR spectra is a useful technique to confirm the formation of an inclusion complex and can provide useful information on the inclusion mechanism of cyclodextrins (CDs) with the guest molecules. Chemical shift changes of specific nuclei in the host molecule can verify the formation of inclusion complex in solution since significant changes in the microenvironment are known to occur in CD of the inclusion complex.56 In general, if a guest molecules situated within the cyclodextrin (CD) cavity, then the hydrogen atoms located on the inner shell of the cavity (H3 and H5) will be considerably shielded by the guest, while the hydrogen atoms on the outer surface (H1, H2, H4) will be unaffected by the inclusion complex formation (Fig. 3). The noticeable upfield shift of the protons on the inner cavity of γCD, i.e., H3 and H5 were observed due to shielding by ring current from the aromatic rings of echinomycin (Table 1). The H1, H2 and H4 protons of γCD, on the outer part of the cavity also encounter a little upfield shift but do not show considerable changes upon addition of echinomycin. The same phenomenon is observed with the H6 proton with a downfield shift. A change of the echinomycin protons environment, when echinomycin enters the hydrophobic cavity of γCD, led to an upfield shift of the Ha, Hb, Hc and Hf protons of the quinoxaline ring as shown in Fig. 3. There is another inclusion probability from the isopropyl region due to the obvious upfield shift for Hd. The 1H signals for both γCD and echinomycin molecules could be observed and distinguished in 1H NMR spectrum from echinomycin-in-γCD inclusion complexes which strongly suggests that the new inclusion complex has been formed as appeared by a model proposed in Fig. 4.
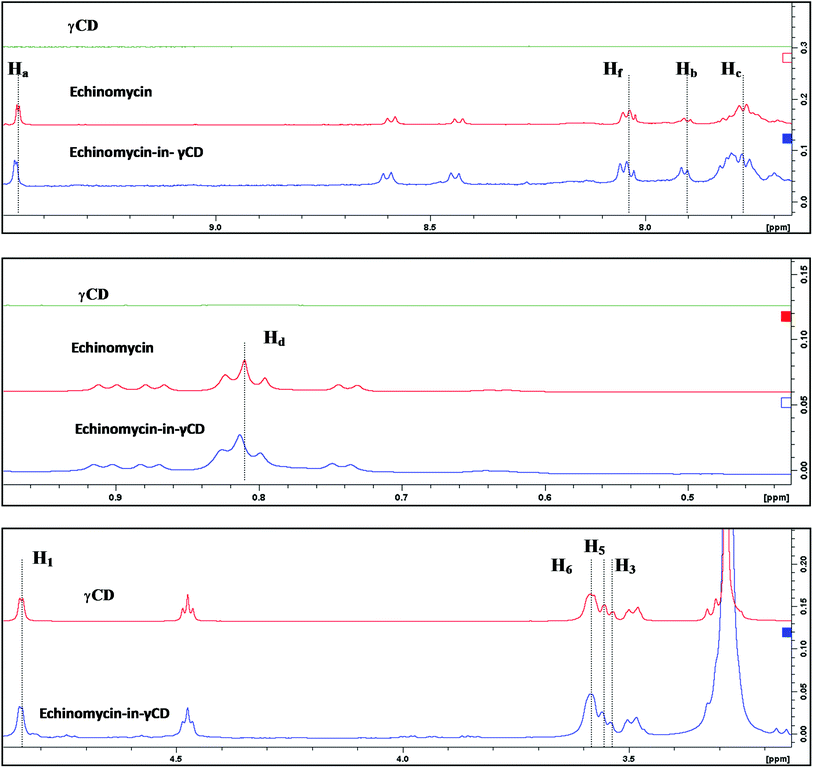 |
| | Fig. 3 Inclusion mechanism of echinomycin in γCD. 1H-NMR spectrums for γCD, echinomycin and echinomycin-in-γCD complex in DMSO. | |
Table 1 Chemical shifts (δ) of γ-CD, echinomycin (δo) and echinomycin-in-γCD complex (δC)
| |
Proton |
δ
o
|
δ
C
|
Δδ |
| γ-CD |
H1 |
4.8428 |
4.84423 |
0.00143 |
| H2 |
3.3086 |
3.3097 |
0.0011 |
| H3 |
3.5367 |
3.5413 |
0.0046 |
| H4 |
3.4912 |
3.4939 |
0.0027 |
| H5 |
3.5547 |
3.5602 |
0.0055 |
| H6 |
3.5853 |
3.5848 |
−0.0005 |
| Echinomycin |
Ha |
9.4594 |
9.4646 |
0.0052 |
| Hb |
7.902 |
7.909 |
0.0070 |
| Hc |
7.773 |
7.786 |
0.013 |
| Hd |
0.8099 |
0.8133 |
0.0034 |
| He |
3.278 |
3.2803 |
0.0023 |
| Hf |
8.037 |
8.044 |
0.0070 |
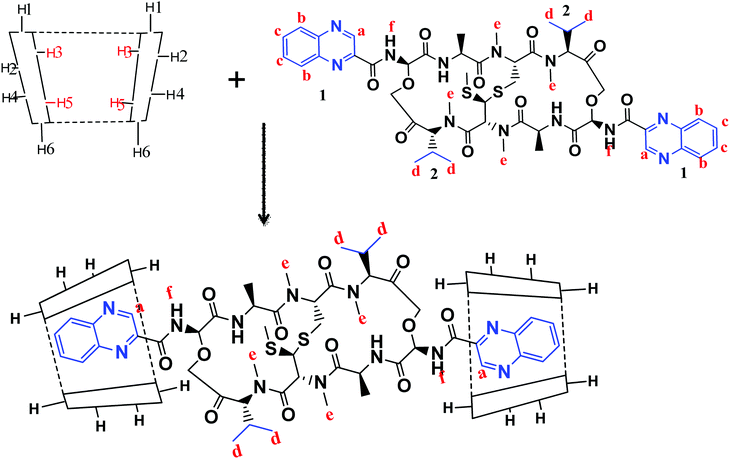 |
| | Fig. 4 The proposed model for echinomycin-in-γCD inclusion. Showing the inner and outer cavity hydrogen atoms of γCD and the chemical structure of echinomycin representing the major hydrophobic parts, (1) quinoxaline group and (2) isopropyl group. | |
3.3. Liposomes preparation and characterization
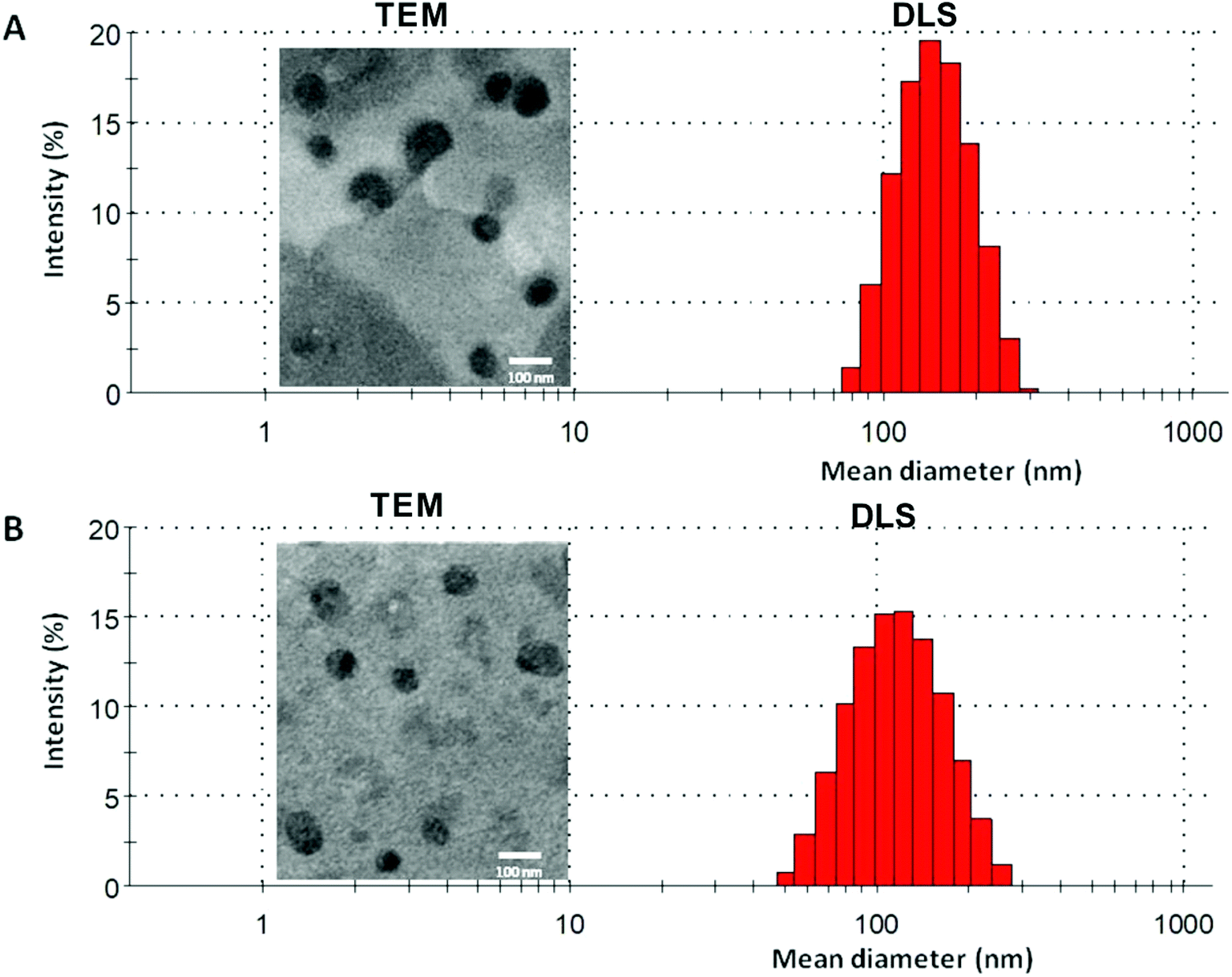
In the current work, echinomycin-in-γCD inclusion has been encapsulated into PEGylated liposomes formulation composed of DPPC:CHOL:DSPE-PEG with 62:35:3 mol% ratio. The elimination of liposomes from blood circulation is a key issue affecting its therapeutic efficiency. Therefore, PEGylated liposomes (Stealth® liposomes) approached to minimize the interaction with serum proteins and MPS.57 Moreover, previous reports have proven that the optimal formulations of PEGylated liposomes contain 3–7 mole% of PEG-DSPE2000.58 Several properties affect the biological interactions of liposomes,59 the size is one of the decisive parameters, especially for intravenous use. Size between 50–450 nm has an application in the clinical field.60 As shown in Table 2, blank liposomes (134 ± 10 nm) and echinomycin-in-γCD loaded liposomes (120 ± 16 nm) display no significant differences in the particle sizes. Both liposomes preparations showed similar polydispersity index (∼0.1) which demonstrates the presence of a monodisperse distribution. Moreover, the TEM of blank liposomes and echinomycin-in-γCD-in-liposomes formulations revealed that the blank and echinomycin loaded liposomes showed a homogeneous size and spherical shape as shown in Fig. 5A and B. Zeta potential of blank liposomes (−12 ± 2 mV) showed similar values to the one observed for echinomycin-in-γCD loaded liposomes (−15 ± 2 mV). The encapsulation efficiency of echinomycin was found to be around 5.1 ± 0.8% (molar ratio) which correspond to a drug loading of ∼0.025% (wt/wt). It worth to mention, the calculated MW of echinomycin-in-γCD (1:2) is 3.695 kDa which may occupy the available spaces and means one out of three encapsulated molecules is echinomycin.
Table 2 Blank and loaded liposomes characterization
| Treatment |
Mean hydrodynamic diameter (nm ± SD) |
Polydispersity Index (PDI) |
ζ-Potential (mV ± SD) |
Encapsulation efficacy (EE%) |
Drug loading, wt/wt (DL%) |
| Blank liposomes |
134 ± 10 |
0.11 ± 0.09 |
−12.3 ± 2.0 |
— |
— |
| Echinomycin-in-γCD-in-liposomes |
120 ± 16 |
0.10 ± 0.06 |
−15.2 ± 2.1 |
5.1 ± 0.8 |
0.025 ± 0.004 |
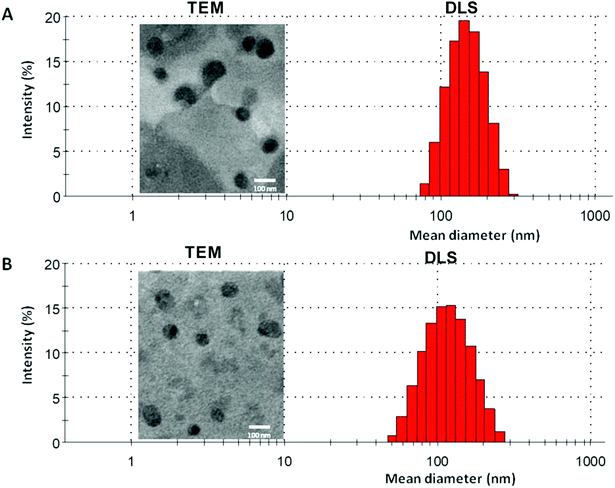 |
| | Fig. 5 A representative DLS and TEM graphs of (A) blank liposomes and (B) echinomycin-in-γCD-in-liposomes. | |
3.4.
In vitro stability and release
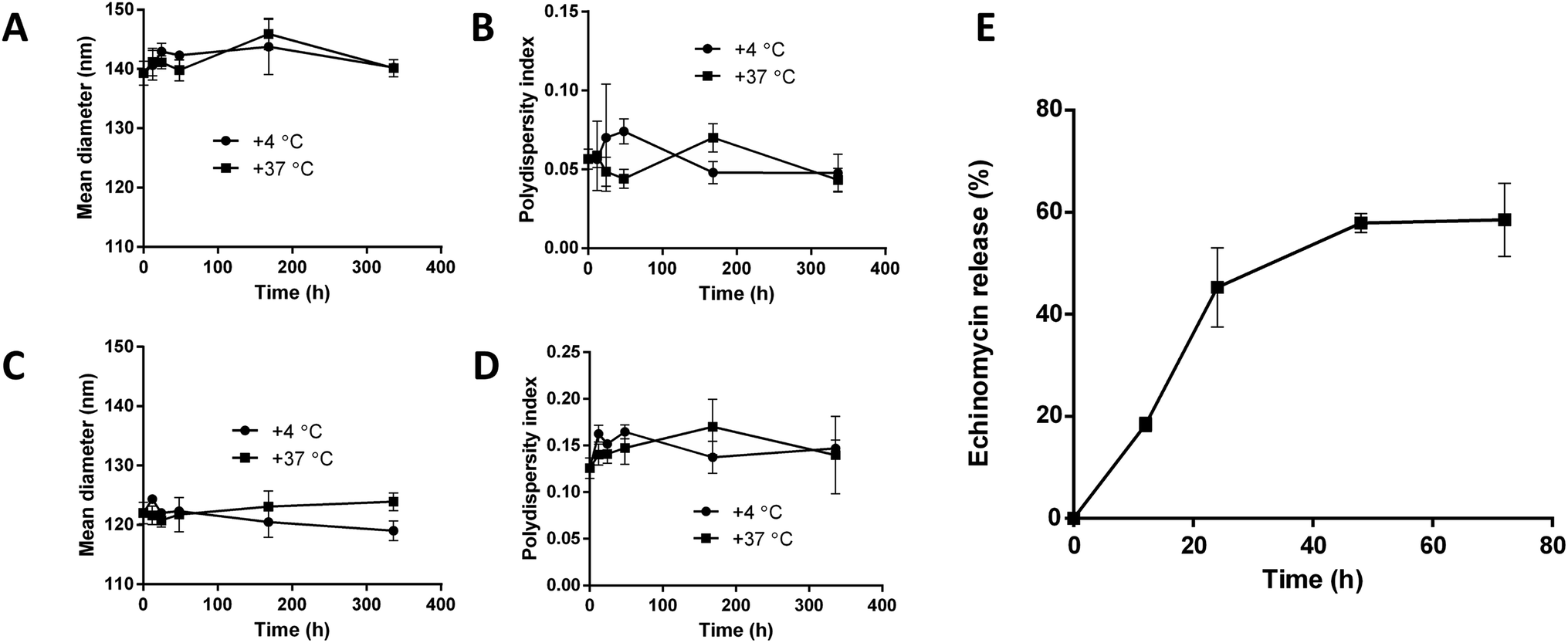
The stability of blank liposomes and echinomycin-in-γCD loaded liposomes were investigated by measuring the changes in the mean size and size distribution (PDI) over a period of 14 days in physiological buffer at 4 °C (storage temperature) and 37 °C (physiological temperature). The results showed good stability of both blank and loaded liposomes indicated by minimal changes in the mean size and PDI of blank liposomes and echinomycin-in-γCD loaded liposomes at tested temperatures (Fig. 6A–D). The in vitro release of echinomycin from echinomycin-in-γCD loaded liposomes was assessed in physiological buffer (PBS, pH 7.4) at 37 °C over a period of 72 h at different intervals. The results indicated that echinomycin showed a biphasic release from echinomycin-in-γCD loaded liposomes (Fig. 6E), with fast release at first 24 h (45.2%) followed by sustained release at 72 h (58.5%). The initial fast release in the biphasic behavior can be explained by the release of echinomycin trapped near liposomes membranes while the sustained release is due to the release of echinomycin trapped deep in the core of liposomes. These findings are in agreement previous studies described that the biphasic release drugs from liposomes.61–63
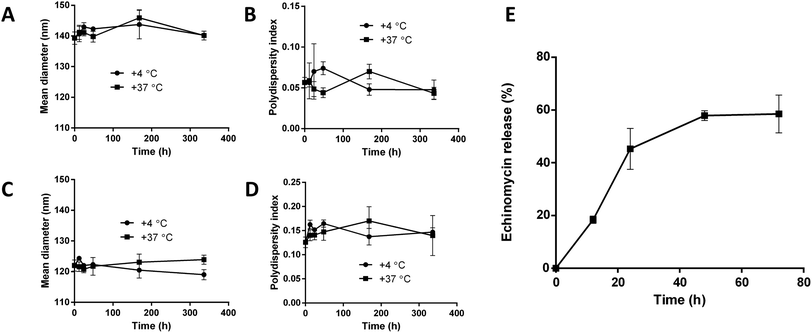 |
| | Fig. 6 Colloidal stability of blank liposomes and echinomycin-in-γCD-in-liposomes at 4 °C and 37 °C. (A) and (B) mean diameter (nm) and the polydispersity index (PDI) of blank liposomes. (C) and (D) mean diameter (nm) and the polydispersity index (PDI) of echinomycin-in-γCD-in-liposomes. (E) In vitro release of echinomycin from echinomycin-in-γCD-in-liposomes monitored over 72 h at 37 °C. All values represent the average ± SD of three independent experiments. | |
3.5. Cytotoxicity assessment by MTT
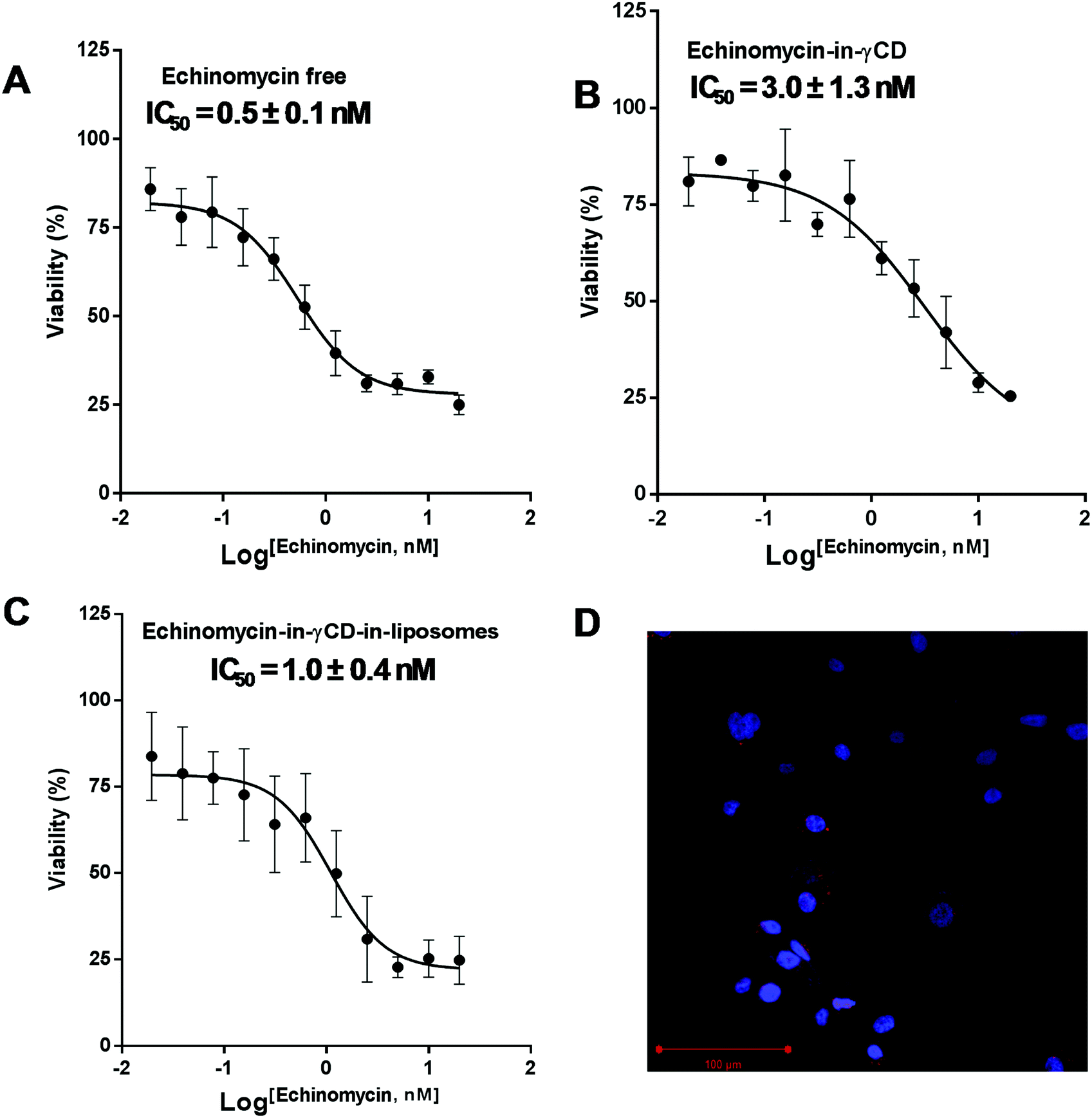
The cytotoxic activity of free echinomycin, echinomycin-in-γCD, and echinomycin-in-γCD-in-liposomes against U-87 MG cells was evaluated using cell viability assay (MTT) and the IC50 values were calculated. The IC50 values for free echinomycin, echinomycin-in-γCD, and echinomycin-in-γCD-in-liposomes were 0.5 ± 0.1 nM, 3.0 ± 1.3 nM, and 1.0 ± 0.4 nM, respectively (Fig. 7A–C). There was no significant toxicity related to blank liposomes. Moreover, the cellular uptake was performed by confocal laser scanning microscopy. Liposomes were fluorescently labeled by grafting of 0.5 mol% of with phosphatidylethanolamine Rhodamine-B (PE-Rhodamine). After 4 hours of incubation, liposomes can be seen within the cells, confirming the drug could be available to cells (Fig. 7D). The IC50 value of echinomycin-in-γCD was ∼6 folds higher compared to the free echinomycin, this can be explained by the fact that the echinomycin can be classified as water-insoluble and membrane permeable. On the other hand, the IC50 value of echinomycin-in-γCD-in-liposomes showed closed cytotoxicity effect compared to free echinomycin. Echinomycin has an extremely potent antitumor effect on different types of cancer cell lines (reviewed in47). Interestingly, Wang et al. showed that echinomycin can eradicate mouse lymphoma and transplanted human acute myeloid leukemia (AML) in the xenogeneic model by the elimination of cancer stem cells (CSCs).64 In a further study, echinomycin showed the ability to protect mice from relapsed AML without effect on hematopoietic stem cells.65
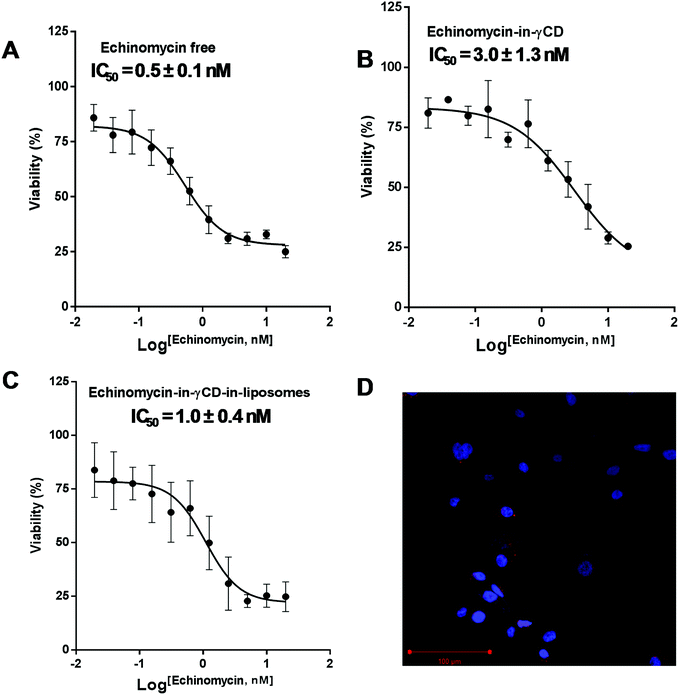 |
| | Fig. 7 IC50 values after treatment with different preparations of echinomycin. (A) The dose–response curve for U-87 MG cells treated with free echinomycin. (B) The dose–response curve for U-87 MG cells treated with echinomycin-in-γCD inclusion complexes. (C) The dose–response curve for U-87 MG cells treated with echinomycin-in-γCD-in-liposomes. (D) Confocal imaging microscopy treated with rhodamine labeled showing the localization of liposomes in the cytoplasm. All cytotoxicity values represent the average ± SD of three independent experiments. | |
3.6. Invasion assay
The 3D spheroid gel invasion assay is ideal in providing an indication about the interactions between the tumor cells and tumors microenvironments thereby representing the physiology of avascular solid tumors in vivo. Moreover, glioblastoma is an aggressive type of malignant brain tumors that can invade normal cells of the central nervous system. The spheroid gel invasion assay is characterized by its ability to distinguish invasive cancer cells from non-invasive ones. Therefore, this 3D-culture model is an excellent in vitro model that allow proper interpretation of cellular responses such as invasion to different therapeutics and drug delivery systems in a way that resembles the in vivo environments.66
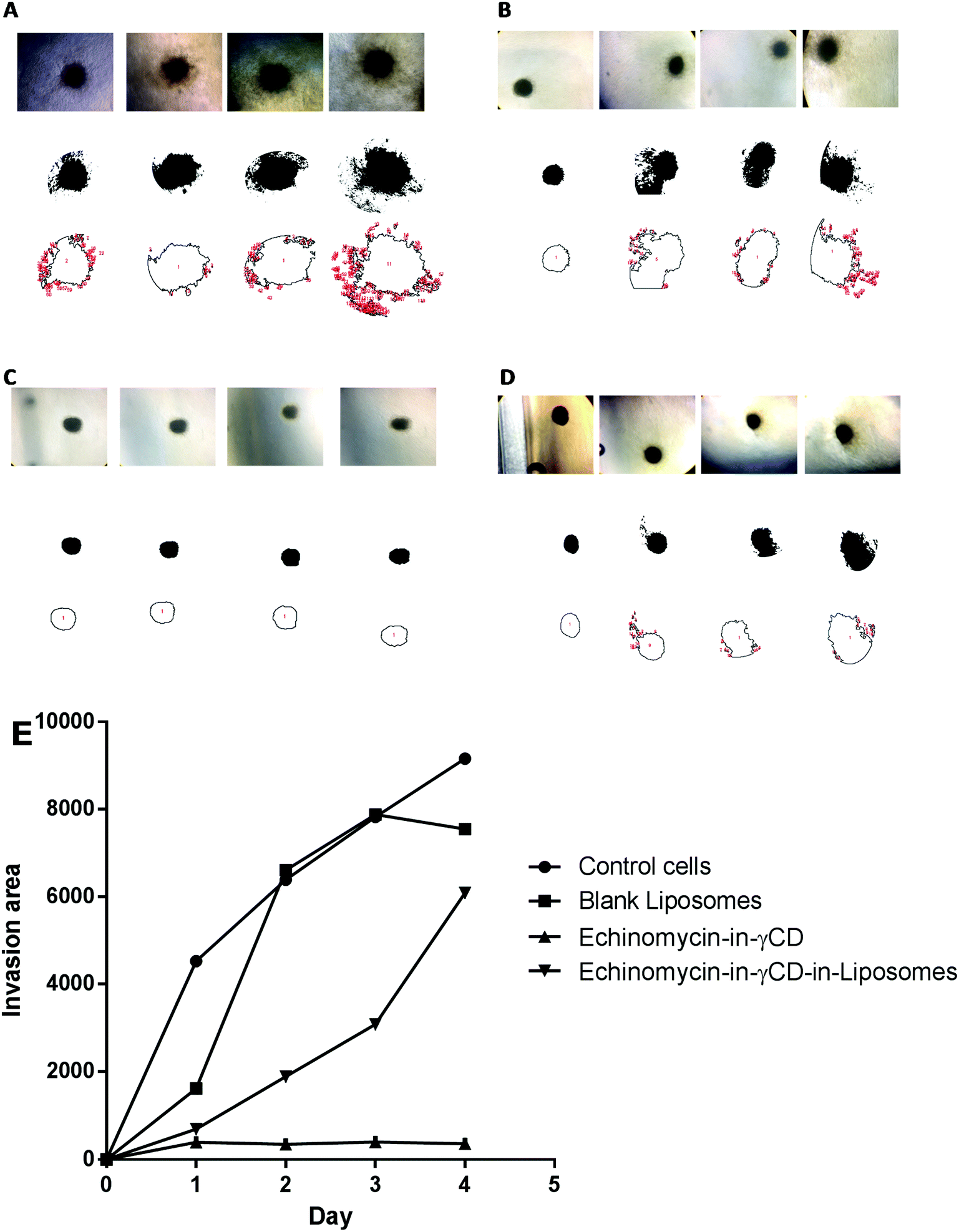
In the current study, spheroid gel invasion assay has been performed to investigate the anti-invasive effect of echinomycin-in-γCD-in-liposomes in comparison with the effect of echinomycin-in-γCD complexes, blank liposomes, and control untreated cells. Collagen type I was used as the matrix in this assay because it is known as the main interstitial matrix component in the solid tumors in vivo. Furthermore, the structural features of collagen that make it able to form gel-like architecture to get more realistic features of the cellular invasion process and making this assay more able to mimic the in vivo situation.67,68Fig. 8 shows that free echinomycin-in-γCD inclusion complexes echinomycin-in-γCD-in-liposomes was found to reduce the invasion of the U-87 MG spheroids in a time-dependent manner compared to the control and the blank liposomes treated spheroids. The echinomycin-in-γCD-in-liposomes was less able to reduce the invasion of the U-87 MG spheroids compared to the echinomycin-in-γCD inclusion complexes (Fig. 6E). This is likely related to the size of liposomes that cannot penetrate easily into the inner layers of the spheroids compared to the echinomycin-in-γCD inclusion complexes. The concentration of the echinomycin used was selected based on the IC50 values that were calculated through the MTT chemosensitivity assay. The 3D invasion assay is ideal in maintaining the structural and morphological properties of the cells in the spheres allowing real-time monitoring of the basic features of the cellular invasion during the running time of the assay. The invasive cells started to invade out from the embedded spheroids into the surrounding collagen as early as the first day after embedding. However, the non-invasive cancer cells grow as compact spheroids with clear borders from the surrounding matrix, with no signs of invasion even after 7 days of culture.
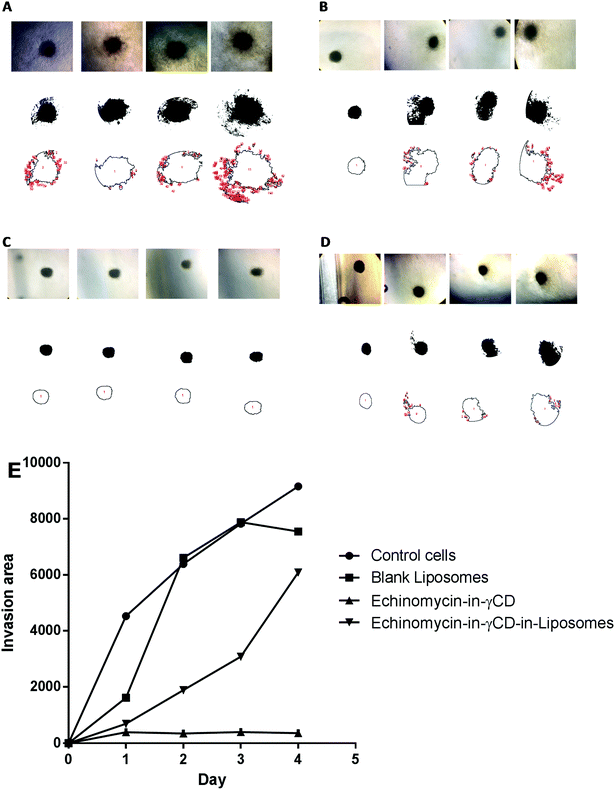 |
| | Fig. 8 The spheroid gel invasion assay. The invasion area of U-87 MG spheroids monitored until day 4 after treatment, (A) untreated U-87 MG spheroids, (B) U-87 MG spheroids treated with blank liposomes, (C) U-87 MG spheroids treated with echinomycin-in-γCD inclusion complexes, (D) U-87 MG spheroids treated echinomycin-in-γCD-in-liposomes. (E) The average invasion area from day 0 to day 4. All values represent the average ± SD of three independent experiments. | |
4. Conclusions
In this work, we show for the first time the complexation of echinomycin with γCD (echinomycin-in-γCD) and the loading into PEGylated liposomes (echinomycin-in-γCD-in-liposomes). The liposomes encapsulating echinomycin showed potent anti-proliferative and anti-invasive effect against U-87 MG glioblastoma cell line. Glioblastoma is an aggressive type of malignant brain tumors with less than 5% of patient survival after 5 years of diagnosis. The available therapies don't bear a higher survival rate in GBM patients due to the aggressive nature of glioblastoma that can invade normal cells of the central nervous system and the protection advantage by the neurovascular niche. Further animal studies should confirm the advantage of using the echinomycin-in-γCD-in-liposomes formulation as a promising alternative for the treatment of glioblastoma.
Contributions
M. Z., A. A., H. N., Z. L., D. A., E. A., T. A., A. A., S. W. carried out all experiments, data curation, software, and writing-original draft. W. A., M. Z., M. Z., and F. O. contributed to data investigation and writing-original draft. A. A., Y. B., and E. F. contributed to supervision, validation, and review-editing. W. A. contributed to conceptualization, methodology, investigation, project administration, data validation, and writing-original draft.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors would like to the thank Hamdi Mango Center for Scientific Research, The University of Jordan (Grant#4200053). Scientific Research Fund (Grant#MPH/2/8/2013). We also would like to thank Ms Hanan Azzam and Ms Rula Buqaien (The University of Jordan) for assistance on HPLC and TEM, respectively.
References
- J. A. Cuesta-Seijo and G. M. Sheldrick, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2005, 61, 442–448 CrossRef.
- B. J. Foster, K. Clagett-Carr, D. D. Shoemaker, M. Suffness, J. Plowman, L. A. Trissel, C. K. Grieshaber and B. Leyland-Jones, Invest. New Drugs, 1985, 3, 403–410 CrossRef CAS.
-
K. Katagiri, T. Yoshida and K. Sato, in Mechanism of action of antimicrobial and antitumor agents, Springer, 1975, pp. 234–251 Search PubMed.
- L. G. May, M. A. Madine and M. J. Waring, Nucleic Acids Res., 2004, 32, 65–72 CrossRef CAS.
- J. Yamaguchi, T. Tanaka, H. Saito, S. Nomura, H. Aburatani, H. Waki, T. Kadowaki and M. Nangaku, Sci. Rep., 2017, 7, 6516 CrossRef.
- Y. Wang, Y. Liu, F. Tang, K. M. Bernot, R. Schore, G. Marcucci, M. A. Caligiuri, P. Zheng and Y. Liu, Blood, 2014, 124, 1127–1135 CrossRef CAS.
- D. Kong, E. J. Park, A. G. Stephen, M. Calvani, J. H. Cardellina, A. Monks, R. J. Fisher, R. H. Shoemaker and G. Melillo, Cancer Res., 2005, 65, 9047–9055 CrossRef CAS.
- P.-C. Wu, S.-L. Tzeng, C.-K. Chang, Y.-F. Kao, M. J. Waring and M.-H. Hou, Nucleic Acids Res., 2018, 46, 7396–7404 CrossRef CAS.
- A. Y. Chang, K. Kim, H. Boucher, P. Bonomi, J. A. Stewart, D. D. Karp and R. H. Blum, Cancer, 1998, 82, 292–300 CrossRef CAS.
- W. J. Gradishar, N. J. Vogelzang, L. J. Kilton, S. J. Leibach, A. W. Rademaker, S. French and A. B. Benson 3rd, Invest. New Drugs, 1995, 13, 171–174 CrossRef CAS.
- W. J. Gradishar, N. J. Vogelzang, L. J. Kilton, S. J. Leibach, A. W. Rademaker, S. French and A. B. Benson, Invest. New Drugs, 1995, 13, 171–174 CrossRef CAS.
- S. Wadler, L. Tenteromano, L. Cazenave, J. A. Sparano, E. S. Greenwald, A. Rozenblit, R. Kaleya and P. H. Wiernik, Cancer Chemother. Pharmacol., 1994, 34, 266–269 CrossRef CAS.
- D. H. Shevrin, T. E. Lad, P. Guinan, L. J. Kilton, A. Greenburg, P. Johnson, R. R. Blough and H. Hoyer, Invest. New Drugs, 1994, 12, 65–66 CrossRef CAS.
- A. Y. Chang, Z. N. Tu, G. T. Bryan, J. M. Kirkwood, M. M. Oken and D. L. Trump, Invest. New Drugs, 1994, 12, 151–153 CrossRef CAS.
- H. B. Muss, J. A. Blessing and B. DuBeshter, Am. J. Clin. Oncol., 1993, 16, 492–493 CrossRef CAS.
- M. E. Marshall, M. K. Wolf, E. D. Crawford, S. Taylor, B. Blumenstein, R. Flanigan, F. J. Meyers, H. E. Hynes, B. Barlogie and M. Eisenberger, Invest. New Drugs, 1993, 11, 207–209 CrossRef CAS.
- S. A. Taylor, J. Crowley, J. Townsend, F. S. Vogel, H. Eyre, J. J. Braun and J. W. Goodwin, J. Neuro-Oncol., 1993, 15, 181–184 CrossRef CAS.
- H. B. Muss, J. A. Blessing, P. Hanjani, J. H. Malfetano, G. M. Kemp and K. Webster, Am. J. Clin. Oncol., 1992, 15, 363–364 CrossRef CAS.
- H. B. Muss, J. A. Blessing, G. L. Eddy and R. McGehee, Invest. New Drugs, 1992, 10, 25–26 CrossRef CAS.
- R. L. Schilsky, D. Faraggi, A. Korzun, N. Vogelzang, J. Ellerton, W. Wood and I. C. Henderson, Invest. New Drugs, 1991, 9, 269–272 CrossRef CAS.
- J. G. Kuhn, D. D. Von Hoff, M. Hersh, T. Melink, G. M. Clark, G. R. Weiss and C. A. Coltman, Eur. J. Cancer Clin. Oncol., 1989, 25, 797–803 CrossRef CAS.
- M. E. Davis, Clin. J. Oncol. Nurs., 2016, 20, S2 CrossRef.
- F. Hanif, K. Muzaffar, K. Perveen, S. M. Malhi and S. U. Simjee, Asian Pac. J. Cancer Prev., 2017, 18, 3 Search PubMed.
- S. Y. Lee, Genes Dis., 2016, 3, 198–210 CrossRef CAS PubMed.
- Z. Huang, L. Cheng, O. A. Guryanova, Q. Wu and S. Bao, Protein Cell, 2010, 1, 638–655 CrossRef CAS.
- J. Shi, P. W. Kantoff, R. Wooster and O. C. Farokhzad, Nat. Rev. Cancer, 2017, 17, 20 CrossRef CAS.
- A. Wicki, D. Witzigmann, V. Balasubramanian and J. Huwyler, J. Controlled Release, 2015, 200, 138–157 CrossRef CAS PubMed.
- U. Bulbake, S. Doppalapudi, N. Kommineni and W. Khan, Pharmaceutics, 2017, 9, 12 CrossRef.
- M. Voinea and M. Simionescu, J. Cell. Mol. Med., 2002, 6, 465–474 CrossRef CAS.
- S. Tran, P.-J. DeGiovanni, B. Piel and P. Rai, Clin. Transl. Med., 2017, 6, 44 CrossRef.
- G. Bangale, K. Rajesh and G. Shinde, Int. J. Pharma Sci. Res., 2014, 5, 750–759 Search PubMed.
- B. Gidwani and A. Vyas, BioMed Res. Int., 2015, 2015, 198268 Search PubMed.
- T. Higuchi, Adv. Anal. Chem. Instrum., 1965, 4, 117–211 CAS.
- T. Loftsson, D. Hreinsdottir and M. Masson, Int. J. Pharm., 2005, 302, 18–28 CrossRef CAS.
- S. Sur, A. C. Fries, K. W. Kinzler, S. Zhou and B. Vogelstein, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 2283–2288 CrossRef CAS.
- V. Chaudhary and J. Patel, Int. J. Pharma Sci. Res., 2013, 4, 68–76 CrossRef CAS.
- A. D. Bangham and R. W. Horne, J. Mol. Biol., 1964, 8, 660 CrossRef CAS.
- M. Placzek and M. Kosela, Acta Pharm., 2016, 66, 1–22 CAS.
- G. L. Amidon, H. Lennernäs, V. P. Shah and J. R. Crison, Pharm. Res., 1995, 12, 413–420 CrossRef CAS.
- B. Gidwani and A. Vyas, BioMed Res. Int., 2015, 2015, 198268 Search PubMed.
- A. Lodagekar, R. M. Borkar, S. Thatikonda, R. B. Chavan, V. G. M. Naidu, N. R. Shastri, R. Srinivas and N. Chella, Carbohydr. Polym., 2019, 212, 252–259 CrossRef CAS.
- N. Li, N. Wang, T. Wu, C. Qiu, X. Wang, S. Jiang, Z. Zhang, T. Liu, C. Wei and T. Wang, Drug Dev. Ind. Pharm., 2018, 44, 1966–1974 CrossRef CAS PubMed.
- P. Saokham, C. Muankaew, P. Jansook and T. Loftsson, Molecules, 2018, 23, 1161 CrossRef.
- L. Kai-Hang, S. Mengying, T. Guping and H. U. Xiurong, Zhejiang Daxue Xuebao, Yixueban, 2017, 46, 151–159 Search PubMed.
- A. R. Prado, F. Yokaichiya, M. Franco, C. Silva, L. Oliveira-Nascimento, M. Franz-Montan, M. C. Volpato, L. F. Cabeca and E. de Paula, J. Pharm. Pharmacol., 2017, 69, 652–662 CrossRef CAS.
- S. Charumanee, S. Okonogi, J. Sirithunyalug, P. Wolschann and H. Viernstein, Sci. Pharm., 2016, 84, 694–704 CrossRef CAS.
- Y.-S. Park, W.-S. Shin, C.-S. Kim, C. M. Ahn, X.-F. Qi and S.-K. Kim, Mol. Cell. Toxicol., 2018, 14, 9–18 CrossRef CAS.
- R. Pfoh, J. A. Cuesta-Seijo and G. M. Sheldrick, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2009, 65, 660–664 CrossRef CAS.
- A. M. Socha, K. L. Laplante, D. J. Russell and D. C. Rowley, Bioorg. Med. Chem. Lett., 2009, 19, 1504–1507 CrossRef CAS PubMed.
- Y. S. Park, W. S. Shin and S. K. Kim, J. Antimicrob. Chemother., 2008, 61, 163–168 CrossRef CAS.
- J. A. Cuesta-Seijo and G. M. Sheldrick, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2005, 61, 442–448 CrossRef.
- T. Mihajlovic, K. Kachrimanis, A. Graovac, Z. Djuric and S. Ibric, AAPS PharmSciTech, 2012, 13, 623–631 CrossRef CAS.
- P. Jansook and T. Loftsson, Int. J. Pharm., 2008, 363, 217–219 CrossRef CAS.
- K. Srinivasan, T. Stalin and K. Sivakumar, Spectrochim. Acta, Part A, 2012, 94, 89–100 CrossRef CAS.
- L. Sbârcea, L. Udrescu, L. Drăgan, C. Trandafirescu, Z. Szabadai and M. Bojiţă, Farmacia, 2010, 58, 478–484 Search PubMed.
- B. Cwiertnia, T. Hladon and M. Stobiecki, J. Pharm. Pharmacol., 1999, 51, 1213–1218 CrossRef CAS PubMed.
- W. Zhao and X.-R. Q. Song Zhuang, Int. J. Nanomed., 2011, 6, 3087 CAS.
-
S. Zalipsky, J. Gittelman, N. Mullah, M. M. Qazen and J. A. Harding, in Targeting of Drugs 6, Springer, 1998, pp. 131–138 Search PubMed.
- B. Sabeti, M. I. Noordin, S. Mohd, R. Hashim, A. Dahlan and H. Akbari Javar, BioMed Res. Int., 2014, 2014, 765426 Search PubMed.
- G. Bozzuto and A. Molinari, Int. J. Nanomed., 2015, 10, 975 CrossRef CAS.
- L. R. Tefas, B. Sylvester, I. Tomuta, A. Sesarman, E. Licarete, M. Banciu and A. Porfire, Drug Des., Dev. Ther., 2017, 11, 1605–1621 CrossRef CAS.
- T. Yang, F. D. Cui, M. K. Choi, J. W. Cho, S. J. Chung, C. K. Shim and D. D. Kim, Int. J. Pharm., 2007, 338, 317–326 CrossRef CAS.
- P. Panwar, B. Pandey, P. C. Lakhera and K. P. Singh, Int. J. Nanomed., 2010, 5, 101–108 CAS.
- Y. Wang, Y. Liu, S. N. Malek, P. Zheng and Y. Liu, Cell Stem Cell, 2011, 8, 399–411 CrossRef CAS.
- Y. Wang, Y. Liu, F. Tang, K. M. Bernot, R. Schore, G. Marcucci, M. A. Caligiuri, P. Zheng and Y. Liu, Blood, 2014, 124, 1127–1135 CrossRef CAS.
- R. Edmondson, J. J. Broglie, A. F. Adcock and L. Yang, Assay Drug Dev. Technol., 2014, 12, 207–218 CrossRef CAS.
- F. Bruyere, L. Melen-Lamalle, S. Blacher, G. Roland, M. Thiry, L. Moons, F. Frankenne, P. Carmeliet, K. Alitalo, C. Libert, J. P. Sleeman, J. M. Foidart and A. Noel, Nat. Methods, 2008, 5, 431–437 CrossRef CAS.
- F. Sabeh, R. Shimizu-Hirota and S. J. Weiss, J. Cell Biol., 2009, 185, 11–19 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra05636j |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Manar
Zraikat‡
b,
Amer
Amer
c,
Hamdi
Nsairat
d,
Zainab
Lafi
c,
Dana A.
Alqudah
a,
Enas
Al Qadi
b,
Tasneem
Alsheleh
b,
Fadwa
Odeh
d,
Arwa
Alkaraki
ab,
Malek
Zihlif
b,
Yasser
Bustanji
ce,
Elias
Fattal
*a,
Manar
Zraikat‡
b,
Amer
Amer
c,
Hamdi
Nsairat
d,
Zainab
Lafi
c,
Dana A.
Alqudah
a,
Enas
Al Qadi
b,
Tasneem
Alsheleh
b,
Fadwa
Odeh
d,
Arwa
Alkaraki
ab,
Malek
Zihlif
b,
Yasser
Bustanji
ce,
Elias
Fattal