
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of serum concentration and surface functionalization on the protein adsorption to mesoporous silica nanoparticles†
Chih-Yu Lina,
Chia-Min Yang *ab and
Mika Lindén*c
*ab and
Mika Lindén*c
aDepartment of Chemistry, National Tsing Hua University, Hsinchu 30013, Taiwan. E-mail: cmyang@mx.nthu.edu.tw
bFrontier Research Center on Fundamental and Applied Sciences of Matters, National Tsing Hua University, Hsinchu 30013, Taiwan
cInstitute of Inorganic Chemistry II, University of Ulm, Albert-Einstein-Allee 11, 89081 Ulm, Germany. E-mail: mika.linden@uni-ulm.de
First published on 22nd October 2019
Abstract
A study of a protein corona on mesoporous silica nanoparticles (MSNs) at in vitro and in vivo relevant serum concentrations is presented. Three MSNs different in terms of mesoscopic pore arrangement, surface chemistry, and surface roughness were studied. After incubation in either 10% or 100% serum, the hard protein corona–particle complexes were collected and analyzed by DLS, zeta-potential, and TGA, and the corona proteins were analyzed with SDS-PAGE. A good correlation between SDS-PAGE and TG results in terms of total amounts of proteins adsorbed was established. The results demonstrated that more proteins, especially apolipoproteins, were associated with the particles at higher serum concentration regardless of surface chemistry and morphological differences. Also, the mean molecular weight of the adsorbed proteins was clearly lower under full serum conditions modeling in vivo conditions as compared to under dilute conditions modeling in vitro conditions, but functionalization of the MSNs by carboxylic acid functionalities reduced protein adsorption. The influence of the structural characteristics of the MSNs on the protein adsorption was minor.
1. Introduction
Mesoporous silica nanoparticles (MSNs) have attracted considerable academic attention as both drug carriers, and contrast agents.1–10 Their attractive properties in this context include high surface areas and pore volumes that allow high drug loading levels to be achieved; tunable mesopore diameter and mesostructure allow accommodation of guest molecules spanning from molecular drugs to macromolecular proteins and DNA, and the rigidity and chemical inertness can prevent the cargoes from enzymatic degradation or inactivation. However, MSNs are not FDA approved yet, but non-porous silica in the form of fluorescent dye encapsulated (NCT02106598) and radioactive 89Zr-containing (NCT03465618) silica nanoparticles are currently undergoing clinical trials.11,12Despite the many attractive characteristics of MSNs for biomedical applications, they typically suffer from sub-optimal biodistribution,13 a common problem for many nanoscopic platforms. It is now well established that proteins adsorb to the nanoparticle surface immediately upon contact between nanoparticles and serum. This protein corona has a major influence on the fate of the nanoparticles in the body, as the physicochemical properties of the original NPs are changed,14,15 and can, in addition to the biodistribution, also influence targeting efficacy, cellular uptake, and toxicity.16–26 Studies on chemically related silicon NPs27–30 and MSNs31–33 have shown that protein adsorption can be affected by protein concentration,29,30 physicochemical properties such as particle size and porosity,31,32 and surface chemistry of the particles.27,28,33 Most of these studies have, however, been carried out under typical in vitro conditions, i.e. with serum concentrations in the range of 10% or in the presence of single proteins. Although such studies are very important for studying the influence of surface chemistry on protein adsorption or for investigating the relation between the protein corona and the in vitro behavior, the relevance of such results for describing in vivo conditions may be limited. Therefore, investigations of serum effects on MSNs are still highly desired for deeper understanding of in vitro and in vivo outcomes and the correlation between them.
This work aims at narrowing the aforementioned gap by studying protein adsorption on several MSNs. Three types of MSNs (i.e., MCM-41, MCM-48, and MCM-48) with a rough surface with moderate and comparable size (200–300 nm) were selected. In addition, their carboxylic acid- and dye (ATTO-647N)-functionalized derivatives were also examined due to the indispensability of MSNs surface modification for imaging and targeting. The studies were carried out in 10% and 100% fetal calf serum (FCS) which are relevant for in vitro and in vivo conditions, respectively. The analyses focused on the hard corona, i.e. strongly adsorbed proteins.16,17,20 The physicochemical properties of the protein–particle conjugates were monitored by dynamic light scattering (DLS) and zeta potential measurements, and the textural properties were studied using transmission electron microscopy (TEM) and gas sorption. Quantitative and qualitative analyses of the hard corona were performed by thermogravimetric analysis (TGA) and sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Our results demonstrated that serum concentration has much higher impact on the protein adsorption amount and relative protein composition than do MSNs type and surface chemistry. The present study provides insights into in vitro and in vivo extrapolation and serves as solid ground for further more detailed investigations.
2. Experimental section
2.1 Materials and characterization methods
For the synthesis and functionalization of MSNs, cetyltrimethylammonium bromide (CTAB, 99.2%), sodium hydroxide (ACS, Reag. Ph. Eur.), absolute ethanol (99.5%), and aqueous ammonium solution (28%) were purchased from VWR. F127 (Bioreagent), benzylcetyldimethylammonium chloride (BCDAC), diethylene glycol hexadecyl ether (C16E2), 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC), and N-hydroxysuccinimide (NHS) were obtained from Sigma-Aldrich. Tetraethoxysilane (TEOS, ≥99%) and 2-(4-(2-hydroxyethyl)-1-piperazi-nyl)-ethansulfonsäure (HEPES) were purchased from Merck, and carboxyethylsilanetriol di-sodium salt (25% in H2O) was provided by ABCR GmbH & Co. ATTO 647N-amine was purchased from ATTO-TEC GmbH, and dimethyl sulfoxide (DMSO) anhydrous was obtained from Invitrogen. For SDS-PAGE, acrylamide (≥98%), N-N′-methylene-bis-acrylamide (≥98%), tris(hydroxymethyl)aminomethane (Tris, ≥99%), ammonium persulfate (APS, ≥98%), tetramethylethylenediamine (TEMED, ≥99%), sodium dodecyl sulfate (SDS, ≥99.5%), dithiothreitol (DTT, ≥99.5%), glycine (≥99%) and sodium azide (≥99%) were purchased from Carl Roth. Glycerol (ACS, Reag. Ph. Eur.) and Coomassie brilliant blue G 250 (CBB, for microscopy) were obtained from Sigma-Aldrich, and hydrochloric acid (HCl, 32%) and bromophenol blue (ACS, Reag. Ph. Eur.) were obtained from Merck. PageRuler Broad Range Unstained Protein Ladder was purchased from Thermo Scientific. Dulbecco's Phosphate Buffered Saline (DPBS), Dulbecco's Modified Eagle Medium (DMEM), and Penicillin Streptomycin were purchased from Gibco, and FCS was obtained from HyClone.X-ray diffraction (XRD) patterns were recorded on X'Pert MPD PRO (PANalytical, Netherland) diffractometer using Cu Kα radiation. Nitrogen physisorption isotherms were measured at 77 K using QuadraSorb SI (Quantachrome Instruments, Odelzhausen, Germany). Transmission electron microscopy (TEM) images were taken by using an EM109 (Zeiss, Germany) electron microscope operated at 80 kV. Hydrodynamic size of the particles was analyzed by dynamic light scattering (DLS) (Zetasizer Nano-ZS ZEN 3600, Malvern Instruments, Herrenberg, Germany), and zeta potential was determined using the same equipment. Thermogravimetric analysis (TGA) was carried out in air using TG 209 F1 Libra (Netzsch Gerätebau GmbH, Selb, Germany). The fluorescence intensities of dye-labelled particles were determined by TECAN SPARK 10M reader (Tecan, Crailsheim, Germany).
2.2 Mesoporous silica nanoparticles synthesis
The synthesis procedures of MCM-41 particles were adopted from Zhao et al.34 A homogeneous solution containing CTAB (1.00 g), 2.00 M NaOH (3.50 mL) and H2O (480.00 mL) was heated to 80 °C, followed by dropwise addition of TEOS (5.00 mL) under vigorous stirring. The mixture was further stirred for 2 hours and collected via centrifugation (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm), repeatedly washed by H2O and ethanol, and then dried at 60 °C. MCM-48 particles were prepared according to the protocol from Bouchoucha et al.35 CTAB (1.00 g) and F127 (4.00 g) were mixed with ethanol (85.00 mL) and 28% aqueous ammonia solution (213.00 mL), and then TEOS (3.86 mL) was introduced to the solution under vigorous stirring for 1 minute. The solution was kept statically at room temperature under for 24 hours, collected by centrifugation (10000 rpm), washed repeatedly by H2O and ethanol, and then dried at 60 °C. Lastly, R-MCM-48 particles were synthesized by following the procedures we reported previously.36 BCDAC (0.74 g), C16 × 102 (0.26 g), 0.40 M NaOH (20.00 mL) and water (575.00 mL) were mixed in a polypropylene bottle and stirred at 35 °C for 12 hours. TEOS (5.98 mL) was then injected into the bottle at a rate of 7.50 mL h−1. The mixture was aged at 90 °C for 24 hours and the solid was filtrated, washed by H2O and ethanol and dried at 60 °C. For all the MSNs, the removal of surfactants was carried out via calcination at 550 °C for 6 hours.
000 rpm), repeatedly washed by H2O and ethanol, and then dried at 60 °C. MCM-48 particles were prepared according to the protocol from Bouchoucha et al.35 CTAB (1.00 g) and F127 (4.00 g) were mixed with ethanol (85.00 mL) and 28% aqueous ammonia solution (213.00 mL), and then TEOS (3.86 mL) was introduced to the solution under vigorous stirring for 1 minute. The solution was kept statically at room temperature under for 24 hours, collected by centrifugation (10000 rpm), washed repeatedly by H2O and ethanol, and then dried at 60 °C. Lastly, R-MCM-48 particles were synthesized by following the procedures we reported previously.36 BCDAC (0.74 g), C16 × 102 (0.26 g), 0.40 M NaOH (20.00 mL) and water (575.00 mL) were mixed in a polypropylene bottle and stirred at 35 °C for 12 hours. TEOS (5.98 mL) was then injected into the bottle at a rate of 7.50 mL h−1. The mixture was aged at 90 °C for 24 hours and the solid was filtrated, washed by H2O and ethanol and dried at 60 °C. For all the MSNs, the removal of surfactants was carried out via calcination at 550 °C for 6 hours.
2.3 Functionalization of mesoporous silica nanoparticles
Carboxylation of MSNs was conducted as the following: calcined MSNs (338.00 mg) were dispersed in HEPES (25 mM, pH 7.4) (45.00 mL) via sonication, carboxyethylsilanetriol di-sodium salt (25% in H2O) (0.76 mL) was then added to the solution. After 3 hours of rotation at room temperature, the resultant carboxylated MSNs (COOH-MSNs) were recovered after centrifugation (10000 rpm), washing repeatedly by H2O and ethanol, and drying at 60 °C. The preparation of fluorescent dye labelled MSNs (ATTO-MSNs) was described below: firstly, to activate carboxyl groups, COOH-MSNs (150.00 mg) were dispersed in HEPES (25 mM, pH 7.4) (30.00 mL) via sonication, and then EDC (44.63 mg) and NHS (26.76 mg) were introduced to the solution. After rotating at room temperature for 30 minutes, the activated COOH-MSNs were collected by centrifugation (10000 rpm) and washed twice by H2O to remove the excessive reagents. The activated COOH-MSNs were then again dispersed into HEPES (15.00 mL), to which ATTO-647N-amine/DMSO (1.00 mg mL−1) (0.15 mL) was added. The solution was rotated in the dark at room temperature for 1 hour, and the resultant ATTO-MSNs were obtained by centrifugation (10000 rpm), washing twice by ethanol, and drying in the dark under vacuum at 25 °C.
2.4 Protein adsorption and protein–particle complexes characterization
Particles were dispersed in PBS by Covaris S220 to obtain stock solutions (20.00 mg mL−1), from which 0.05 mL aliquot (containing 1.00 mg of particle) was withdrawn and added to 1.00 mL of DMEM (supplemented with 10% FCS and 1% antibiotics) or pure FCS, and the mixtures were rotated at 37 °C. At designated time-points, the protein–particle complexes were collected by centrifugation (13200 rpm), washed twice to remove the soft corona, and again dispersed in PBS before DLS and zeta potential measurement. For TGA, the washed complexes were dried at 60 °C overnight before measurement. For TEM, the washed complexes were dispersed in double distilled water (ddH2O) before dropping on carbon coated Cu grids.
2.5 SDS-PAGE
The washed complexes were treated with 100.00 μL 10% SDS buffer solution for 30 minutes to detach the hard protein corona, and the mixture was centrifuged (13200 rpm) to separate the particles. The protein containing supernatant (4.00 μL) was withdrawn, mixed with Laemmli buffer (1.20 mL Tris–HCl buffer (0.50 M, pH 6.8), 1.20 g SDS, 6.00 mg bromophenol blue, 5.92 mg glycerol, 2.90 mL ddH2O, and 0.93 g DTT) (20.00 μL), and heated for 5 minutes at 95 °C. Subsequently, 10.00 μL sample solution was loaded to the gel (12% separating gel, 5% stacking gel), and the proteins were separated in a PROTEAN® II XL Multi Cell (BioRad) electrophoresis chamber at constant 300 V for 3 hours. The gels were washed five times (10 minutes for each time) with ddH2O using orbital shaker, and the washed gels were stained with CBB staining solution for 90 minutes,37 followed by destaining in ddH2O overnight. The gels were documented using a ChemiDoc™ MP System (BioRad) and analyzed with the Image Lab software.
3. Results and discussion
3.1 Particle characterization
Three different types of MSNs (MCM-41, MCM-48, and R-MCM-48), exhibiting a comparable mean particle size distribution, mesopore dimensions (3.2–3.7 nm), BET surface areas (1018–1286 m2 g−1), and mesopore volumes but having different structural characteristics were used. Small-angle diffractograms, nitrogen sorption isotherms, and TEM images of the three particles are shown in Fig. 1, and derived key parameters are summarized in Table 1. The MCM-41 particles exhibit a characteristic XRD pattern of a 2-dimensional p6mm structure, while the MCM-48 and R-MCM-48 particles show reflections that can be indexed according to an Ia3d structure. The unit cell of the MCM-48 particles is smaller than that of the R-MCM-48 particles, as evidenced by the shift of the positions of the reflections observed for the MCM-48 particles towards higher angles as compared to those of R-MCM-48 (Fig. 1a). This is also mirrored in the nitrogen sorption isotherms, where the pore filling step for the MCM-41 and R-MCM-48 particles is centered at about 0.3p/po, while it is shifted towards lower relative pressures (about 0.22p/po) for the MCM-48 particles. In all cases the pore filling step is narrow, and the specific pore volumes of all particles are virtually identical. The mesopore size derived using the equilibrium NLDFT kernel developed for silica materials having cylindrical pores gives a mean pore diameter of 3.7 nm for the MCM-41 and the R-MCM-48 particles, while the mesopore size of the MCM-48 particles was 3.2 nm. Low-resolution TEM imaging was used for evaluating the mean particle size and the morphology of the particles, and the corresponding images are shown in Fig. 1c. The mean particle diameter was about 150 nm in all cases as determined based on particle diameter histograms including more than 200 particles in each case. The MCM-48 particles showed the narrowest particle size distribution and were spherical, while the particle size distribution of the MCM-41 was wider and the larger particles were slightly elongated. Both these particles had a smooth outer surface. The R-MCM-48 particle showed a completely different morphology, as they were clearly rougher although having a spherical shape. The roughness influence on the external surface area, as determined by V-t-de Boer method, was evident as expected, and the R-MCM-48 particles had a much higher external surface area (513 m2 g−1) than the MCM-41 (238 m2 g−1) and MCM-48 (227 m2 g−1) particles.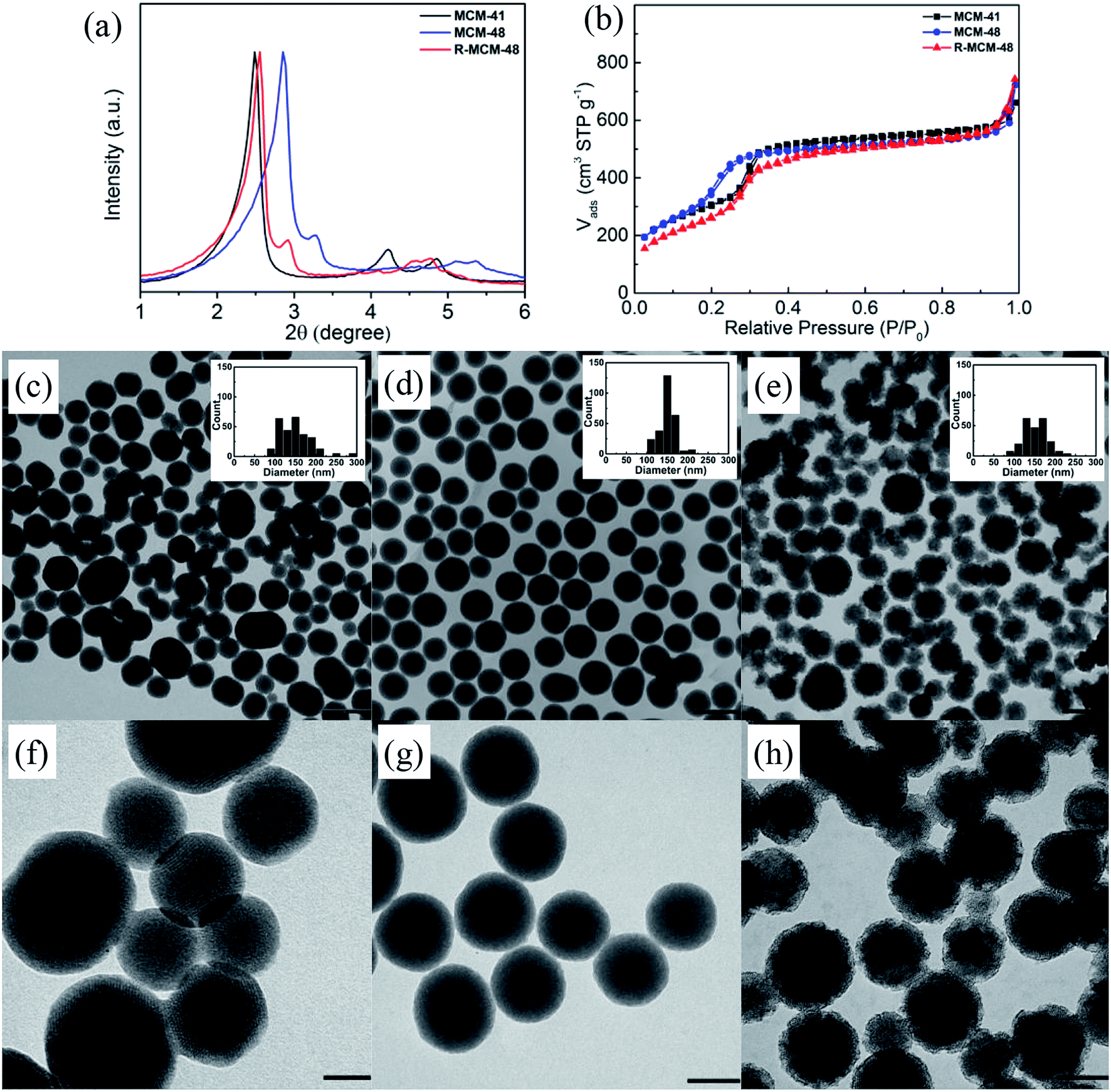 | ||
| Fig. 1 (a) XRD and (b) nitrogen sorption isotherms of pure-silica MSNs. TEM images of (c and f) MCM-41, (d and g) MCM-48, and (e and h) R-MCM-48. The scale bars indicate 250 nm in (c, d, and e) and 100 nm in (f, g, and h), respectively. | ||
| MSNs | DDFT (nm) | SBET (m2 g−1) | Sext (m2 g−1) | Vt (cm3 g−1) @ P/P0 ∼ 0.8 |
|---|---|---|---|---|
| MCM-41 | 3.7 | 1116 | 238 | 0.86 |
| MCM-48 | 3.2 | 1286 | 227 | 0.82 |
| R-MCM-48 | 3.7 | 1018 | 513 | 0.83 |
A portion of each particle batch was further surface functionalized by post-grafting in HEPES buffer using a functional silane carrying a carboxylic acid group at one end of the alkyl-function. The organic contents of three carboxylated particles were comparable (5.9–8.5 wt%, Table S1†), and each mesostructure was maintained, as indicated by XRD patterns and nitrogen sorption isotherms (Fig. S1†). Compared to the pure-silica MSNs, the pore size decreased slightly upon functionalization due to pore-resident functional group, and BET surface areas calculated per gram of silica were reduced by 9–20%, most probably as a result of partial silica dissolution during functionalization. As a further modification, a part of these batches of particles were functionalized by covalent linking of a red fluorescent ATTO-647N-amine dye to the carboxylate function by EDC/NHS coupling. Fluorescence spectroscopy analysis (Fig. S2†) suggested that the three different dye-labeled MSNs showed nearly identical dye loadings calculated as weight percent. No change in particle morphology was observed upon surface functionalization, as shown in Fig. S3 and S4.†
The particles were further characterized in the wet state by DLS and zeta-potential measurements in PBS buffer (pH = 7.4). The data are summarized in Table 2. The zeta-potential was similar for all particles being in the range −11 to −16 mV. The mean hydrodynamic diameter of the particles also measured in PBS was similar for the MCM-41 and MCM-48 particles, about 200 nm, while it was clearly higher for the R-MCM-48 particles, close to 300 nm, which probably can be ascribed to their rough outer surface. The rougher surface slows down the movement of these particles, and as the particle size in DLS is calculated based on the diffusion coefficient of the particles larger diameters are obtained.
| MSN | Time (h) | FCS (%) | Pure-silica MSNs | COOH-MSNs | ATTO-MSNs | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| ζ-Potential (mV) | DH (nm) | PDI | ζ-Potential (mV) | DH (nm) | PDI | ζ-Potential (mV) | DH (nm) | PDI | |||
| MCM-41 | 0 | 0 | −13.0 ± 0.3 | 224.2 ± 15.5 | 0.08 ± 0.02 | −14.9 ± 4.6 | 218.5 ± 9.6 | 0.18 ± 0.06 | −12.0 ± 1.1 | 257.6 ± 72.7 | 0.14 ± 0.14 |
| 0.5 | 10 | −10.6 ± 1.2 | 232.8 ± 6.1 | 0.10 ± 0.01 | −9.9 ± 0.2 | 232.1 ± 10.6 | 0.12 ± 0.03 | −9.6 ± 0.5 | 302.5 ± 97.0 | 0.15 ± 0.07 | |
| 5 | 10 | −10.5 ± 0.6 | 234.3 ± 3.3 | 0.07 ± 0.04 | −10.4 ± 0.1 | 234.8 ± 16.0 | 0.18 ± 0.12 | −9.7 ± 0.2 | 236.4 ± 43.0 | 0.23 ± 0.14 | |
| 0.5 | 100 | −8.7 ± 0.3 | 258.9 ± 7.9 | 0.10 ± 0.02 | −9.2 ± 1.3 | 251.0 ± 7.2 | 0.10 ± 0.02 | −8.7 ± 0.9 | 237.7 ± 7.7 | 0.17 ± 0.08 | |
| 5 | 100 | −8.5 ± 0.7 | 259.6 ± 6.8 | 0.08 ± 0.02 | −9.9 ± 1.3 | 254.6 ± 5.5 | 0.13 ± 0.03 | −9.1 ± 0.5 | 254.5 ± 15.2 | 0.06 ± 0.03 | |
| MCM-48 | 0 | 0 | −12.2 ± 0.4 | 199.8 ± 5.0 | 0.08 ± 0.02 | −12.2 ± 1.6 | 199.1 ± 5.5 | 0.10 ± 0.05 | −10.8 ± 1.0 | 198.4 ± 6.1 | 0.11 ± 0.06 |
| 0.5 | 10 | −10.1 ± 0.6 | 214.7 ± 5.4 | 0.06 ± 0.01 | −10.0 ± 0.6 | 225.0 ± 27.4 | 0.12 ± 0.01 | −9.7 ± 0.2 | 286.4 ± 73.1 | 0.19 ± 0.10 | |
| 5 | 10 | −10.1 ± 0.4 | 211.1 ± 3.0 | 0.07 ± 0.02 | −10.7 ± 0.7 | 215.8 ± 5.2 | 0.20 ± 0.16 | −10.2 ± 0.2 | 228.3 ± 30.9 | 0.23 ± 0.13 | |
| 0.5 | 100 | −8.3 ± 0.6 | 238.8 ± 4.7 | 0.08 ± 0.02 | −8.6 ± 0.3 | 227.5 ± 5.0 | 0.05 ± 0.02 | −8.9 ± 0.2 | 224.1 ± 2.0 | 0.07 ± 0.04 | |
| 5 | 100 | −8.4 ± 0.8 | 233.9 ± 2.0 | 0.08 ± 0.02 | −8.6 ± 0.2 | 231.1 ± 6.9 | 0.12 ± 0.06 | −8.6 ± 0.5 | 220.4 ± 0.7 | 0.06 ± 0.01 | |
| R-MCM-48 | 0 | 0 | −15.8 ± 0.2 | 287.0 ± 5.7 | 0.14 ± 0.05 | −12.7 ± 0.5 | 277.0 ± 20.6 | 0.18 ± 0.09 | −12.1 ± 0.4 | 334.5 ± 18.4 | 0.19 ± 0.05 |
| 0.5 | 10 | −10.2 ± 0.2 | 289.4 ± 9.1 | 0.10 ± 0.04 | −10.7 ± 0.4 | 320.3 ± 54.5 | 0.17 ± 0.05 | −10.6 ± 0.4 | 298.1 ± 8.5 | 0.12 ± 0.10 | |
| 5 | 10 | −10.5 ± 1.1 | 310.4 ± 5.3 | 0.14 ± 0.04 | −10.1 ± 0.5 | 310.7 ± 39.4 | 0.14 ± 0.07 | −10.1 ± 0.1 | 316.6 ± 42.4 | 0.13 ± 0.06 | |
| 0.5 | 100 | −9.3 ± 1.0 | 321.2 ± 15.0 | 0.13 ± 0.02 | −8.7 ± 0.8 | 303.6 ± 4.0 | 0.13 ± 0.08 | −9.1 ± 0.4 | 286.9 ± 14.4 | 0.06 ± 0.03 | |
| 5 | 100 | −8.9 ± 0.2 | 302.1 ± 9.2 | 0.12 ± 0.03 | −10.0 ± 0.3 | 299.1 ± 5.8 | 0.15 ± 0.09 | −9.4 ± 0.5 | 288.5 ± 8.5 | 0.17 ± 0.06 | |
3.2 Analyses of MSNs dispersed in serum
Dispersions of the above discussed particles were prepared in both 10% and 100% FCS (1.00 mg particles mL−1). The hydrodynamic diameters and the zeta-potentials of the particles, now carrying adsorbed proteins, were determined at two time-points, 0.5 and 5 h. The results are included in Table 2. The zeta potentials of all particles were nearly identical (−8.3 to −10.7 mV) at both times, indicating that the surface properties were determined by the adsorbed proteins. A serum concentration-dependent increase in the hydrodynamic diameter was evident in all cases, while the change in hydrodynamic radius was less sensitive to incubation time differences. An increase in the hydrodynamic diameter of the MCM-41 and MCM-48 particles was about 10–15 nm irrespective of incubation time at a serum concentration of 10% FCS. For the R-MCM-48 particles the hydrodynamic diameter measured after 0.5 h of incubation in a 10% FCS solution was virtually identical to that measured in PBS, while the value increased by 20 nm at the 5 h time-point. Although the pronounced roughness of the R-MCM-48 particles makes the evaluation of the hydrodynamic diameter more difficult, it appears that the protein corona thickness in 10% FCS was about 5–10 nm. In full serum (100% FCS), the protein corona thicknesses, as evaluated based on the increase in the hydrodynamic diameter (radius) of the particles, were clearly higher than that measured at 10% FCS, and mounted to about 15 nm, clearly indicating that more proteins were associated with the particles at higher FCS concentrations. The diameter increments were reasonable considering that hydrodynamic radii of plasma proteins range from 3 to 15 nm.38 The diameter increase determined in 100% FCS was close to the hard corona thickness (∼15 and ∼19 nm determined by TEM and DLS, respectively) formed in human serum on polystyrene NPs reported by Kokkinopoulou et al.39For the carboxylic acid-functionalized particles, similar protein corona thicknesses and serum concentration dependencies as those observed for the pure-silica particles were observed. For the dye-labeled particles rather a decrease in the hydrodynamic diameter was seen with increasing FCS concentration as compared to the values measured for the same particles dispersed in PBS. This indicates that the particles were partly aggregated in PBS, while the presence of serum proteins led to a better dispersability of the particles, as the values of the hydrodynamic diameters measured in full serum for the dye-labeled particles were almost identical to those measured for the other particles.
3.3 Analysis of the total amount of protein in the hard corona by SDS-PAGE and TG
In order to determine the total amounts of different proteins in hard corona, we first attempted to determine if SDS-PAGE analyses can be used for obtaining at least semi-quantitative results. This is not self-evident, as the staining efficiency can vary from gel to gel, and even within gels, and be different for different proteins. In order to minimize the contribution of gel-to-gel variations, we therefore, as a first step, integrated the total intensity of stained protein bands for each individual run in a gel, and normalized that intensity against the integrated intensity of the ladder measured on the same gel. In parallel, the same particles isolated after washing off the loosely bound proteins, but before desorbing the protein corona, were dried and the mass loss was measured by TGA. The SDS-PAGE gels used for the analyses are shown in Fig. 2. The correlation between the normalized total band intensities and the mass loss as determined by TGA for all types of particles is shown in Fig. 3. Here data was included for particles incubated in either 10% FCS or 100% FCS for either 0.5 h or 5 h. | ||
| Fig. 2 SDS-PAGE gel images of hard corona proteins isolated from (a) and (d) MSNs, (b) and (e) COOH-MSNs, and (c) and (f) ATTO-MSNs at 0.5 h (upper row) and 5.0 h (lower row). | ||
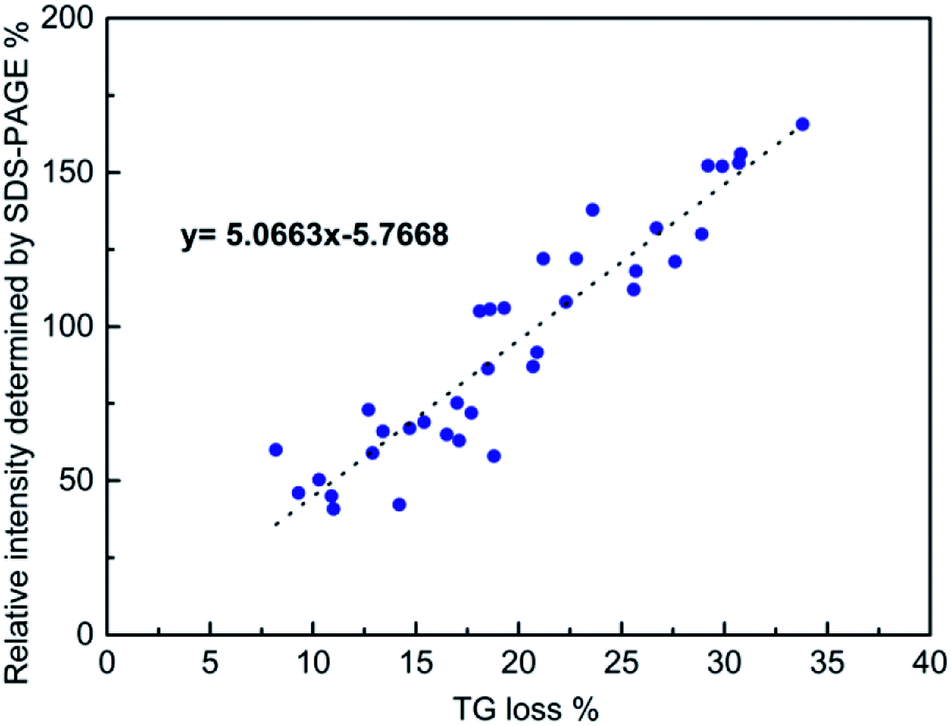 | ||
| Fig. 3 Correlation between the normalized total band intensity as determined by SDS-PAGE and the mass loss determined by TG. See text for details. | ||
Although there was some scatter in the data, there was a good correlation between the two sets of measurements, suggesting that normalized SDS-PAGE intensities can indeed be used to obtain at least semi-quantitative data on the total amount of proteins being part of the protein corona. As the protein adsorption patterns of the different particles was far from identical as seen in Fig. 2, normalized SDS-PAGE values most probably also could give a good description of the relative amounts of proteins of different molecular weights that constitute the inner, strongly bound portion of the protein corona. The protein corona contribution to the total mass loss ranged from about 8 to 34 wt% (see Table S2†), showing clear differences in the amounts of proteins adsorbed between the particles. The measured weight loss was within a reasonable range, considering that the density of the mesoporous silica particles and that of solid proteins both are about 1 g cm−3 and taking assuming a protein layer thickness in the range of 5–10 nm in the dehydrated state. The linear correlation did, however, not pass origin, and the TGA results indicated a higher degree of protein adsorption than did SDS-PAGE. This could reflect the inherent difficulty of identifying protein bands of low intensity in the SDS-PAGE integration, furthermore, as some silica would dissolve during the incubation time, this may also influence the results. However, as our focus was not a fully quantitative analysis but rather a comparative analysis of the protein corona composition as a function of particle type, serum concentration, and incubation time, the good correlation between the two completely different data sets gave us confidence that normalized SDS-PAGE data can be used for semi-quantitative analysis of the protein corona composition.
After having established a basis for semi-quantitative analysis of the SDS-PAGE results, we related the integrated intensities to the total mass of proteins adsorbed on the different particles as a function of incubation time and serum concentration. The results are shown in Fig. 4.
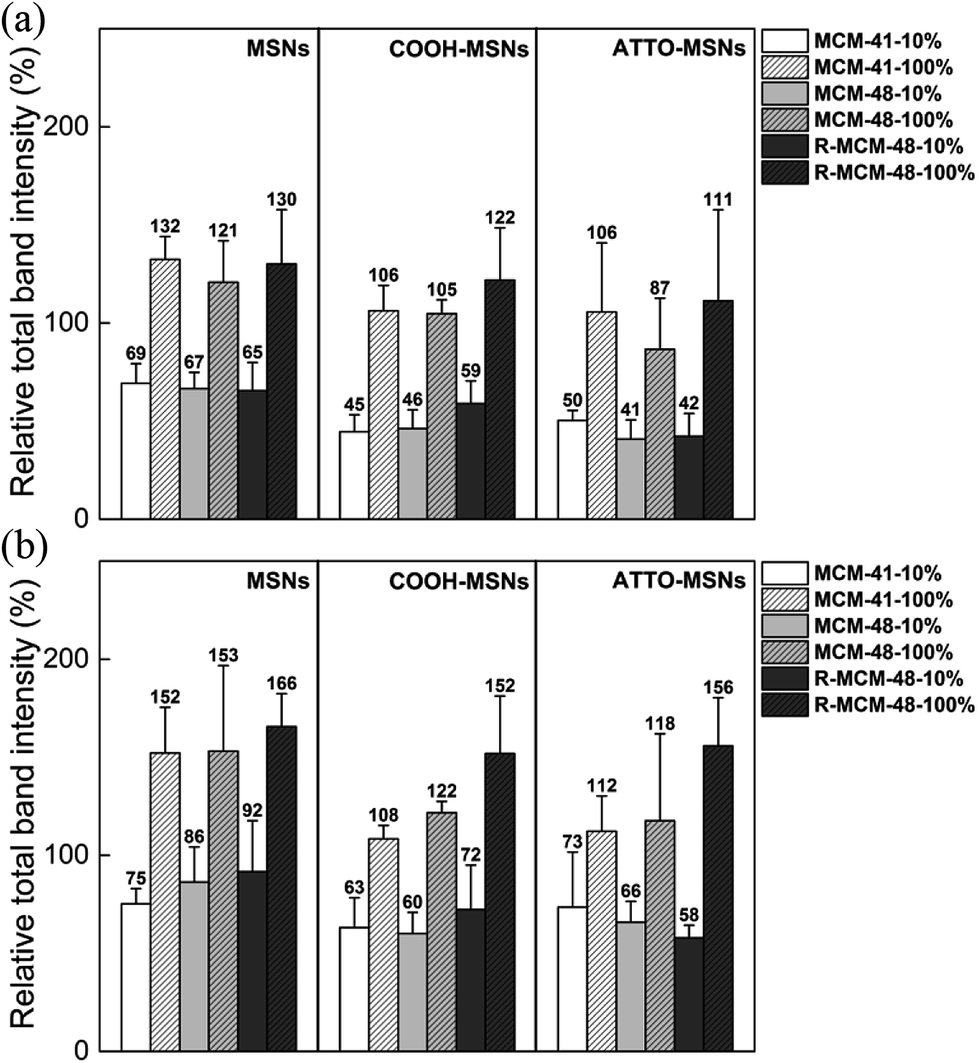 | ||
| Fig. 4 Histograms representing the total band intensity of hard corona on the particles at (a) 0.5 h and (b) 5 h. In each gel, the intensity of each particle lane is normalized against the intensity of ladder. The results were obtained from at least three independent replicates. | ||
The amount of proteins adsorbed increased by roughly a factor 1.5–2 when the serum concentration was increased from 10% to 100% irrespective of the particles. This is opposite to what was observed for non-porous 200 nm silica nanoparticles by Monopoli et al. for serum concentrations 3–80%.29 It is also evident from our results that the carboxy-functionalized MSNs adsorbed less proteins as compared to the all-silica MSNs. This was true also when taking the weight-contribution of the organic function into account (see Table S3†). These results are consistent with previous reports, where carboxy-functionalization was shown to decrease the extent of protein adsorption.25,33 Interestingly, the rough MCM-48 particles adsorbed similar, albeit slightly higher, amounts of proteins as the smooth MCM-41 and MSM-48 particles. Since most of the serum proteins are larger than the pore sizes of MSNs and adsorb mainly onto the external particle surface, the protein density on R-MCM-48 should be lower than MCM-41 and MCM-48, as the outer surface area was higher for the R-MCM-48 particles as compared to the other two studied particles. To sum up, despite that the variations were the sum of many factors, it was evident that the serum concentration is the most important parameter for protein adsorption, while surface functionality, particle type, pore size, surface roughness and incubation time had a lesser influence on the extent of protein adsorption to the particles studied.
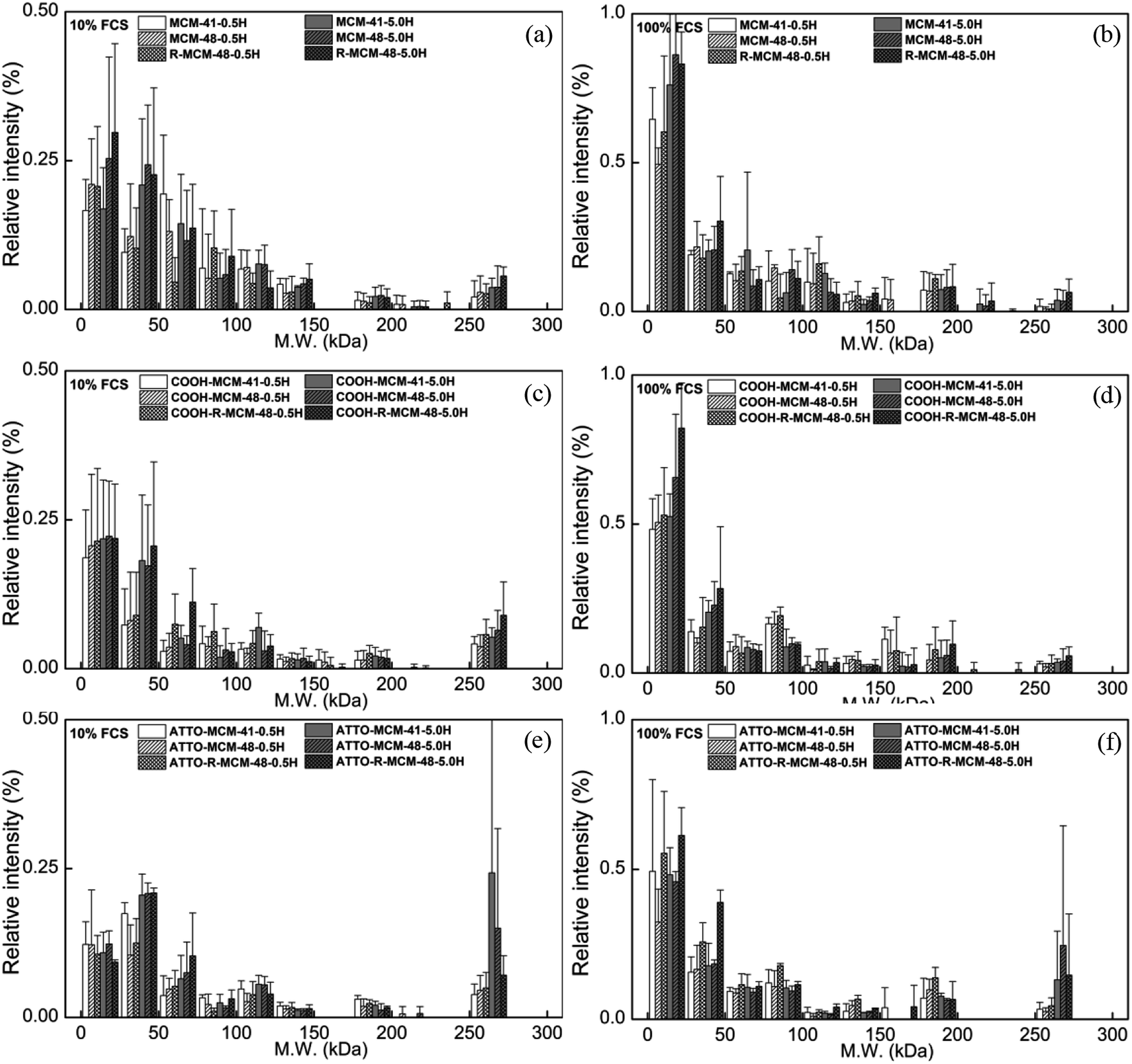 | ||
| Fig. 5 Compositions of hard corona on (a and b) pure-silica MSNs, (c and d) COOH-MSNs, and (e and f) ATTO-MSNs in 10% (left column) and 100% (right column) FCS. The intensity of each subgroup is normalized against the total intensity of the ladder lane next to FCS on each gel. | ||
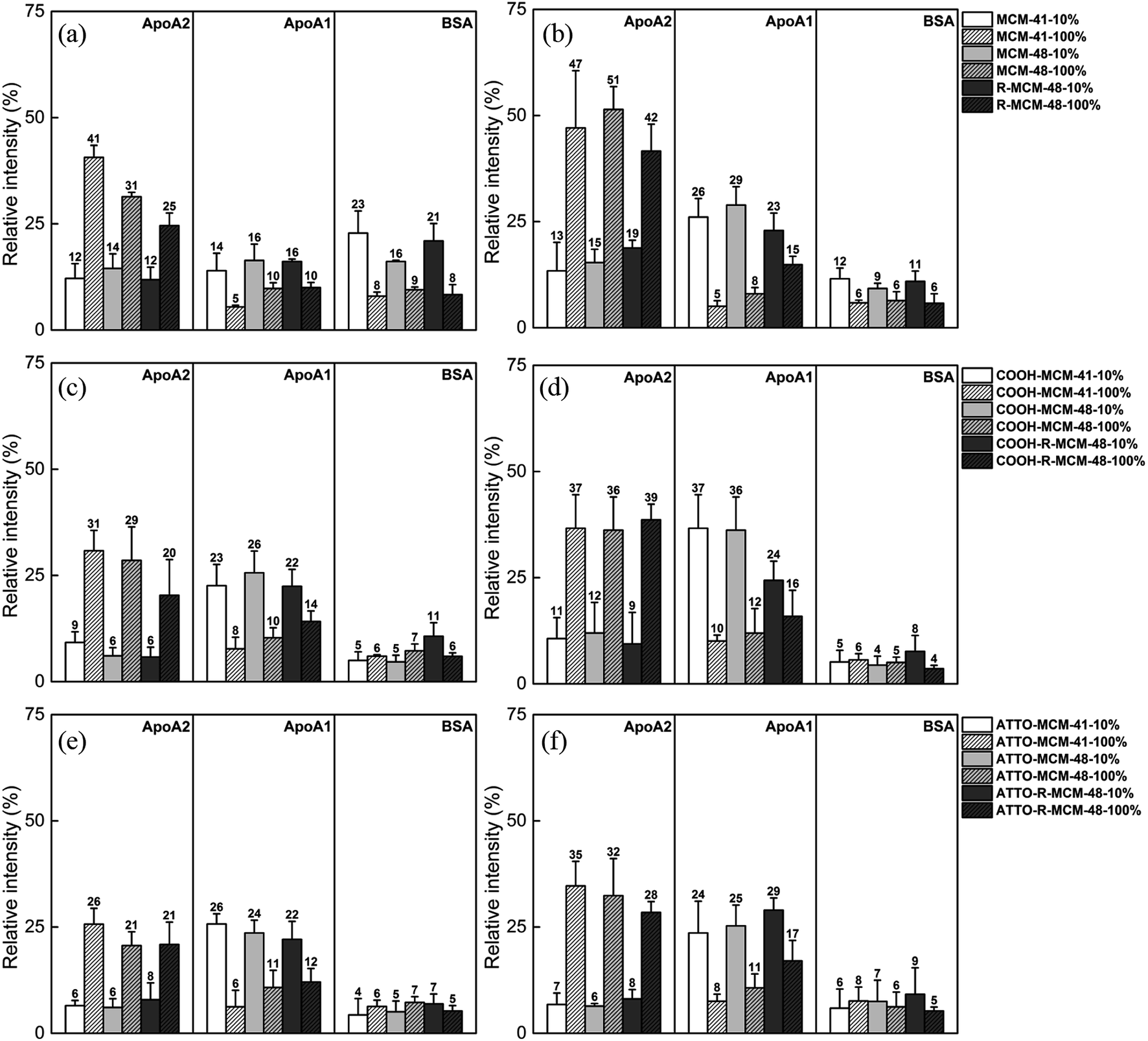 | ||
| Fig. 6 Relative intensity of selected proteins on (a and b) MSNs, (c and d) COOH-MSNs, and (e and f) ATTO-MSNs at 0.5 h (left column) and 5 h (right column). The intensity of each protein band is normalized against total intensity of its particle lane. | ||
As seen, the relative abundance of apolipoproteins was high with notably preferential adsorption of ApoA1 in 10% FCS and ApoA2 in 100% FCS, respectively, and their concentrations also increased with increasing incubation time. However, the particle surface chemistry had an impact on the relative extent of ApoA1 and ApoA2 adsorption, and the functionalized particles showed a higher ApoA1 adsorption and lower ApoA2 adsorption as compared to pure-silica particles. While the BSA adsorption on the functionalized particles remained low throughout, the initial BSA adsorption was relatively high on the pure-silica particles, but decreased with increasing incubation time. The results indicate that each protein is sensitive and responds differently to serum concentration, surface chemistry and time, but the serum concentration was still the most influential factor.
These results are of paramount importance when attempting to extrapolate in vitro data, typically obtained at FCS concentrations of 10–15%, to the behavior in vivo, where full serum conditions apply. Such clear differences in protein corona composition as a function of serum concentration may fully alter the biological response to the particles, and it is known that ApoA2 and ApoA1 both are critical components in high density lipoproteins (HDL) formation and responsible for cholesterol transport to liver. Their abundance in corona should be crucial to biological outcomes, especially biodistribution.
Some of our findings are in agreement with literature data typically obtained under dilute serum conditions (data summarized in Table S4†). The lower protein density (number of proteins per unit area) observed on the rough R-MCM-48 as compared to the smooth MCM-41 and MCM-48 particles is in agreement with the results reported by Clemments et al.31 and Saikia et al.32 Furthermore, the lower protein absorption in the carboxylic acid functionalized particles as compared to the pure-silica particles are in agreement with the results published by Beck et al.25 The high ApoA1 or ApoA2 and low BSA adsorption on pure-silica and carboxylated MSNs are consistent with the studies on 70 nm MSNs in 10% FCS by Clemments et al.,31,33 and in full serum for pure-silica particles reported by Pochert et al.41 An increasing adsorption of apolipoproteins and decreasing or constant BSA adsorption over time has also been observed in the study for magnetic MSNs by Pisani et al.42 A high ApoA1 and a low BSA adsorption was also observed by Monopoli et al.29 using human serum. An increase in the relative abundance of low molecular weight proteins in the corona with increasing serum concentration has also been observed for MSNs at low particle concentrations by Ghavami et al.30
4. Conclusions
We have studied the hard corona composition on pure-silica and functionalized MSNs formed under in vitro (10%) and in vivo (100%) relevant serum concentrations. Much more proteins were strongly adsorbed to all particles studied at higher serum concentrations, and although incubation time, surface roughness, and particle surface chemistry did have an influence on the amount of proteins adsorbed, the influence of these parameters remained small as compared to the serum concentration. Importantly, the relative amount of smaller proteins was clearly higher under full serum conditions as compared to 10% serum, and especially the amounts of apolipoproteins was increasing with increasing serum concentration. Our results thus highlight the necessity to carry out protein adsorption studies under full serum conditions if a link between the protein adsorption and the in vivo performance of nanoparticles is aimed for.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors (C.-Y. L. and C.-M. Y.) gratefully acknowledge the financial supports of the Ministry of Science and Technology, Taiwan under the contracts No. MOST 106-2113-M-007-025-MY3 and MOST 107-3017-F-007-002. This study was also partly supported by the Frontier Research Center on Fundamental and Applied Sciences of Matters from The Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) in Taiwan. The authors gratefully acknowledge the DFG funded CRC 1279 project “Exploiting the human peptidome for novel antimicrobial and anticancer agents” (M. L.) for financial support.References
- Q. He and J. Shi, J. Mater. Chem., 2011, 21, 5845–5855 RSC.
- R. R. Castillo, M. Colilla and M. Vallet-Regí, Expert Opin. Drug Delivery, 2017, 14, 229–243 CrossRef CAS.
- C. Argyo, V. Weiss, C. Bräuchle and T. Bein, Chem. Mater., 2014, 26, 435–451 CrossRef CAS.
- Z. Li, J. C. Barnes, A. Bosoy, J. F. Stoddart and J. I. Zink, Chem. Soc. Rev., 2012, 41, 2590–2605 RSC.
- S.-H. Wu, Y. Hung and C.-Y. Mou, Chem. Commun., 2011, 47, 9972–9985 RSC.
- M. Vallet-Regí, F. Balas and D. Arcos, Angew. Chem., Int. Ed., 2007, 46, 7548–7558 CrossRef.
- V. Mamaeva, C. Sahlgren and M. Lindén, Adv. Drug Delivery Rev., 2013, 65, 689–702 CrossRef CAS.
- I. T. Teng, Y.-J. Chang, L.-S. Wang, H.-Y. Lu, L.-C. Wu, C.-M. Yang, C.-C. Chiu, C.-H. Yang, S.-L. Hsu and J.-a. A. Ho, Biomaterials, 2013, 34, 7462–7470 CrossRef CAS.
- L.-S. Wang, L.-C. Wu, S.-Y. Lu, L.-L. Chang, I. T. Teng, C.-M. Yang and J.-a. A. Ho, ACS Nano, 2010, 4, 4371–4379 CrossRef CAS.
- Q. Jin, C.-Y. Lin, S.-T. Kang, Y.-C. Chang, H. Zheng, C.-M. Yang and C.-K. Yeh, Ultrason. Sonochem., 2017, 36, 262–269 CrossRef CAS.
- https://ClinicalTrials.gov/show/NCT03465618.
- https://ClinicalTrials.gov/show/NCT02106598.
- M. Lindén, in The Enzymes, ed. F. Tamanoi, Academic Press, 2018, vol. 43, pp. 155–180 Search PubMed.
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941 CrossRef CAS.
- S. Tenzer, D. Docter, J. Kuharev, A. Musyanovych, V. Fetz, R. Hecht, F. Schlenk, D. Fischer, K. Kiouptsi, C. Reinhardt, K. Landfester, H. Schild, M. Maskos, S. K. Knauer and R. H. Stauber, Nat. Nanotechnol., 2013, 8, 772 CrossRef CAS.
- C. Corbo, R. Molinaro, A. Parodi, N. E. Toledano Furman, F. Salvatore and E. Tasciotti, Nanomedicine, 2015, 11, 81–100 CrossRef.
- D. Walczyk, F. B. Bombelli, M. P. Monopoli, I. Lynch and K. A. Dawson, J. Am. Chem. Soc., 2010, 132, 5761–5768 CrossRef CAS.
- E. Frohlich, Int. J. Nanomed., 2012, 7, 5577–5591 CrossRef.
- J. Wolfram, Y. Yang, J. Shen, A. Moten, C. Chen, H. Shen, M. Ferrari and Y. Zhao, Colloids Surf., B, 2014, 124, 17–24 CrossRef CAS.
- C. D. Walkey and W. C. W. Chan, Chem. Soc. Rev., 2012, 41, 2780–2799 RSC.
- S. Ritz, S. Schöttler, N. Kotman, G. Baier, A. Musyanovych, J. Kuharev, K. Landfester, H. Schild, O. Jahn, S. Tenzer and V. Mailänder, Biomacromolecules, 2015, 16, 1311–1321 CrossRef CAS.
- P. Aggarwal, J. B. Hall, C. B. McLeland, M. A. Dobrovolskaia and S. E. McNeil, Adv. Drug Delivery Rev., 2009, 61, 428–437 CrossRef CAS.
- A. Lesniak, F. Fenaroli, M. P. Monopoli, C. Åberg, K. A. Dawson and A. Salvati, ACS Nano, 2012, 6, 5845–5857 CrossRef CAS.
- S. Shahabi, S. Döscher, T. Bollhorst, L. Treccani, M. Maas, R. Dringen and K. Rezwan, ACS Appl. Mater. Interfaces, 2015, 7, 26880–26891 CrossRef CAS.
- M. Beck, T. Mandal, C. Buske and M. Lindén, ACS Appl. Mater. Interfaces, 2017, 9, 18566–18574 CrossRef CAS PubMed.
- A. Salvati, A. S. Pitek, M. P. Monopoli, K. Prapainop, F. B. Bombelli, D. R. Hristov, P. M. Kelly, C. Åberg, E. Mahon and K. A. Dawson, Nat. Nanotechnol., 2013, 8, 137 CrossRef CAS.
- S. Shahabi, L. Treccani, R. Dringen and K. Rezwan, ACS Appl. Mater. Interfaces, 2015, 7, 13821–13833 CrossRef CAS.
- F. Catalano, G. Alberto, P. Ivanchenko, G. Dovbeshko and G. Martra, J. Phys. Chem. C, 2015, 119, 26493–26505 CrossRef CAS.
- M. P. Monopoli, D. Walczyk, A. Campbell, G. Elia, I. Lynch, F. Baldelli Bombelli and K. A. Dawson, J. Am. Chem. Soc., 2011, 133, 2525–2534 CrossRef CAS PubMed.
- M. Ghavami, S. Saffar, B. Abd Emamy, A. Peirovi, M. A. Shokrgozar, V. Serpooshan and M. Mahmoudi, RSC Adv., 2013, 3, 1119–1126 RSC.
- A. M. Clemments, P. Botella and C. C. Landry, ACS Appl. Mater. Interfaces, 2015, 7, 21682–21689 CrossRef CAS.
- J. Saikia, M. Yazdimamaghani, S. P. Hadipour Moghaddam and H. Ghandehari, ACS Appl. Mater. Interfaces, 2016, 8, 34820–34832 CrossRef CAS.
- A. M. Clemments, C. Muniesa, C. C. Landry and P. Botella, RSC Adv., 2014, 4, 29134–29138 RSC.
- Y. Zhao, X. Sun, G. Zhang, B. G. Trewyn, I. I. Slowing and V. S. Y. Lin, ACS Nano, 2011, 5, 1366–1375 CrossRef CAS.
- M. Bouchoucha, M.-F. Côté, R. C. Gaudreault, M.-A. Fortin and F. Kleitz, Chem. Mater., 2016, 28, 4243–4258 CrossRef CAS.
- N. Lai, C. Lin, P. Ku, L. Chang, K. Liao, W. Lin and C. J. N. R. Yang, Nano Res., 2014, 7, 1439–1448 CrossRef CAS.
- D. Kang, Y. S. Gho, M. Suh and C. Kang, Bull. Korean Chem. Soc., 2002, 23, 1511–1512 CrossRef CAS.
- N. Phogat, M. Kohl, I. Uddin and A. Jahan, in Precision Medicine, ed. H.-P. Deigner and M. Kohl, Academic Press, 2018, DOI:10.1016/b978-0-12-805364-5.00011-1, pp. 253–276.
- M. Kokkinopoulou, J. Simon, K. Landfester, V. Mailänder and I. Lieberwirth, Nanoscale, 2017, 9, 8858–8870 RSC.
- S. Tenzer, D. Docter, S. Rosfa, A. Wlodarski, J. Kuharev, A. Rekik, S. K. Knauer, C. Bantz, T. Nawroth, C. Bier, J. Sirirattanapan, W. Mann, L. Treuel, R. Zellner, M. Maskos, H. Schild and R. H. Stauber, ACS Nano, 2011, 5, 7155–7167 CrossRef CAS.
- A. Pochert, I. Vernikouskaya, F. Pascher, V. Rasche and M. Lindén, J. Colloid Interface Sci., 2017, 488, 1–9 CrossRef CAS.
- C. Pisani, J. C. Gaillard, M. Odorico, J. L. Nyalosaso, C. Charnay, Y. Guari, J. Chopineau, J. M. Devoisselle, J. Armengaud and O. Prat, Nanoscale, 2017, 9, 1840–1851 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra05585a |
| This journal is © The Royal Society of Chemistry 2019 |