
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of cancer immunotherapy based on PD-1/PD-L1 pathway blockade
Min Zhang a,
Kehai Liu*a and
Mingfu Wang*ab
a,
Kehai Liu*a and
Mingfu Wang*ab
aCollege of Food Science and Technology, Shanghai Ocean University, 999 Hucheng Ring Road, Shanghai 201306, China. E-mail: khliu@shou.edu.cn
bUniversity Hong Kong, School of Biological Sciences, Pokfulam Road, Hong Kong 999077, China
First published on 22nd October 2019
Abstract
Programmed death receptor 1 (PD-1)/programmed death ligand 1 (PD-L1) blockade therapy has achieved considerable success in various tumours. However, only a fraction of patients benefit from its clinical application, and some patients might be suffer from tumour resistance against PD-1/PD-L1 blockade therapy after the original response. In this review, we summarized the main reasons that caused the low response rate of PD-/PD-L1 blockade therapy: firstly, the off-target of PD-1/PD-L1 blocking agents, which is also the main factor of the side effect of autoimmune disorders; secondly, the insufficient infiltration of T cells in a tumour microenvironment; thirdly, the low immunogenicity of tumor cells; fourth, other immunosuppressive components impairing the therapeutic efficacy of the immunotherapy based on the PD-/PD-L1 blockade, and introducing some updated the delivery system of PD-1/PD-L1 blocking agents and the combination therapy based on PD-1/PD-L1 inhibitors and other therapeutics that can complement and promote each other to achieve improved immune response.
1. Introduction
Cancer immunotherapy aims to induce a durable human immune response in order to destroy tumour cells.1 Among them, immune checkpoint therapy (ICB), which targets the programmed death-1 (PD-1)/programmed death-ligand 1 (PD-L1) pathway, has achieved remarkable success in various types of tumours, such as nonsmall cell lung cancer, bladder cancer, melanoma and renal cell carcinoma, and has been regarded as a promising strategy for wiping out malignant cancer.2 PD-1 is a T-cell co-receptor and an important member of the immune regulatory molecule family. The activation of the PD-1 signal is to inhibit T cell-mediated immune responses.3 As its ligand, PD-L1 is expressed on a wide array of normal tissues and cells largely in response to inflammatory cytokines, and it can serve as a molecular shield to prevent the undesired damage caused by uncontrolled spread of immune responses to the normal tissue; ‘hijacking’ this ability seems to be a widespread mechanism for tumours to escape immune surveillance. Hence, scholars attempted to disrupt the engagement of PD-1 with its ligand PD-L1 to reactivate T cell functions and boost the immune system against tumour cells, and achieved remarkable success.However, PD-1/PD-L1 blockage therapy is not effective for all types of tumours, and still only a relatively small percentage of patients respond to that;4 What's more, some patients might be suffering from tumour resistance against PD-1/PD-L1 blockage therapy after original response.5 There are several possible explanations might be able to help answer and provide guidelines for potential enhancement. First of all, the off-target binding to normal tissues after the administration of PD-1/PD-L1 blocking agents may be one of the reasons why PD-1/PD-L1 blockade therapy shows a low response rate, and that is also the main factor on the side effects like autoimmune disorders. Second, insufficient infiltration of T cells in tumour microenvironment will also lead to a low response rate of PD-1/PD-L1 blockade therapy.6–8 Third, the tumour cells, which survive from immune surveillance and eventually develop into mature tumour tissue, generally have very low immunogenicity. In spite of the dissolve of immune-suppressive signaling on T cells by PD-1/PD-L1 blockade therapy, it is still not easy for the immune system to effectively recognize and kill tumour cells.9–12 Fourthly, the therapeutic efficiency of the immunotherapy is often influenced by the whole immunosuppressive network, while the PD-1/PD-L1 pathway is only one of the most important components of immunosuppressive networks.13,14 In this situation, PD-1/PD-L1 blockade therapy alone might not be able to achieve the evident anti-tumour effect. In addition to above-mentioned, recent studies discovered that some tumour cells could secrete a large proportion of their PD-L1 on exosomes instead of presenting the PD-L1 on their cell surface. Exosomal PD-L1 transmit immunosuppressive signals to draining lymph node to suppresses T cell function and inactivate immune cells at its source,15 and some tumour cells even secret a PD-L1 splicing variants, working as “decoys” of PD-L1 antibody, to induce the tumour resistance to PD-L1 blockade,16 which may also be responsible for the failures of PD-1/PD-L1 blockade therapy.
Thus, it has become a top priority to update the delivery system of PD-1/PD-L1 blocking agents, or develop combination therapy based on PD-1/PD-L1 inhibitors and other therapies that can complement and promote each other to achieve an improved immune response. This review mainly focuses on the recent advances in these two aspects.
2. Strategies to reduce side effects
The autoimmune-like toxicity caused by non-specific immunostimulation is one of the major problems that highly limit the clinical application of PD-1/PD-L1 blockade therapy. Although immune checkpoint inhibitors do not come without some risk of toxicity like traditional chemotherapy regimens, they can result in damage to normal cells by immune cells and affect almost pantosomatous organ function, including the skin, muscles, heart, lungs, liver, bowels, kidneys, eyes, endocrine tissues, and central nervous system.17 During the course of treatment, 60–77% of patients will suffer from an immune-related adverse event, and 10–42% of these patients will be severe (grade 3–4).18–20 It has been demonstrated that the local, cell-mediated delivery of immune checkpoint inhibitors from GM-CSF-secreting tumour cells immunotherapy activated potent anti-tumour responses in preclinical tumour mouse models, accompanied by the reducing evidence of systemic autoimmunity.21 And other research shows that the injection of subcutaneous slow-release delivery antibodies formulation in the tumour area exhibits as effective induction in tumour eradication as systemic delivery of antibody, as well as several orders of magnitude decrease in the serum antibody level, resulting in reduced adverse reactions and the risk of autoimmunity.22 These observations further support the call for the tumour specific delivery of immune checkpoint inhibitors.Nano-drug delivery system has emerged as a powerful weapon in tumour diagnosis and therapy due to its unique characteristic, such as the protective effect on payload in vivo, improving the targeting delivery, low side effect, etc.23,24 In order to elevate the accumulation and retention of PD-1/PD-L1 inhibitors in the target spots and decrease off-target effects, researchers are trying to incorporate nanotechnology into immunotherapy to enhance the immunotherapeutic responses against tumour cells (Fig. 1).
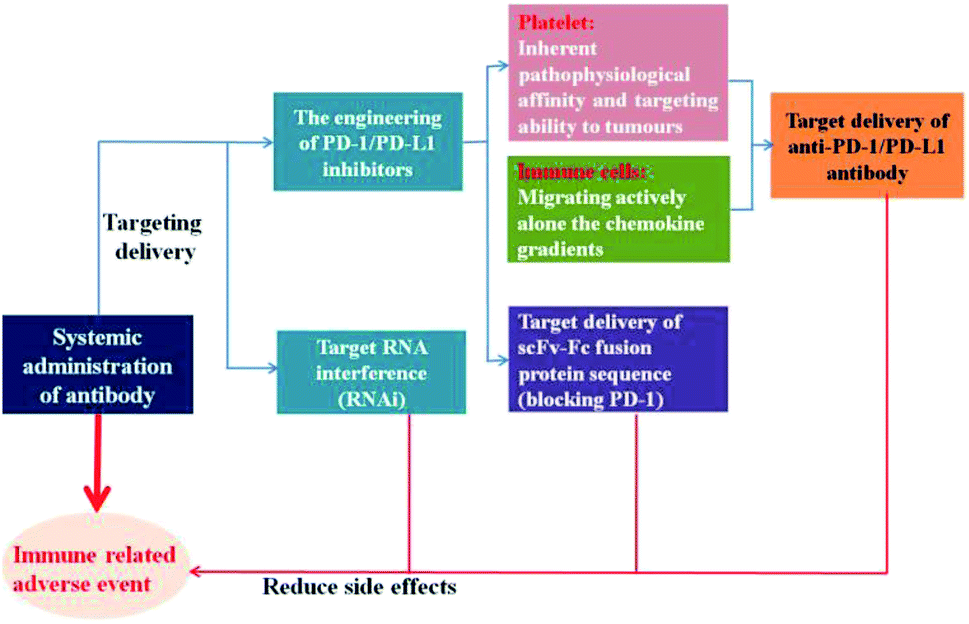 | ||
| Fig. 1 Strategies to reduce side effects. | ||
2.1 The engineering of PD-1/PD-L1 inhibitors
Platelets, as one of the most important bio-particulates in the body, play a great role in a variety of physiological activities, including thrombus formation, wound repair,25,26 maintain tumour blood vessel integrity and promote tumour metastasis.27,28 For instance, platelets can facilitate tumour metastasis by recognizing and interacting with circulating tumour cells shed from the primary tumour into the bloodstream.29 In view of the inherent pathophysiological affinity and targeting ability to tumours, platelets could be employed to conjugate with anti-PD-L1 antibody to reduce post-surgical tumour recurrence and metastasis.30 Through platelet activation, the anti-PD-L1 antibody could be effectively released via platelet-derived microparticles, and the administration of platelet-bound anti-PD-L1 antibody significantly reduced the risk of recurrence and metastasis and prolonged overall mouse survival after surgery (Fig. 2). In addition, the thrombin enrichment in B16F10 melanoma could lead to platelet aggregation, which could also be exploited to mediate the target delivery of anticancer cytokine.31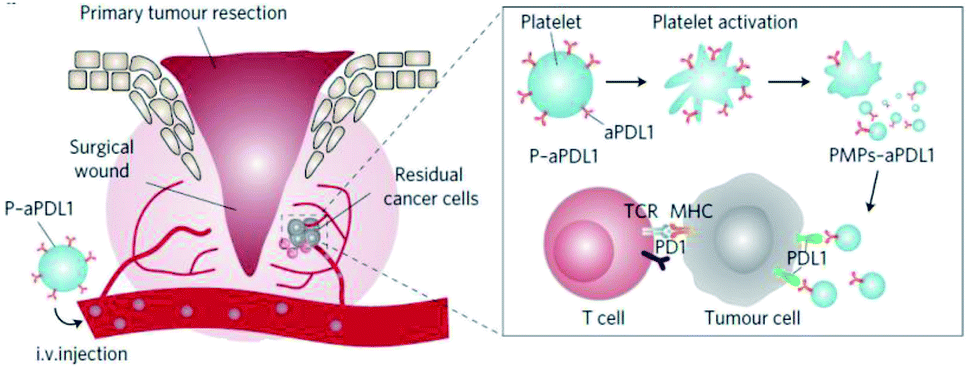 | ||
| Fig. 2 Schematic illustration of the delivery of aPDL1 to the primary-tumor resection site by platelets. Images were reproduced form ref. 30. | ||
Studies showed that the drug delivery system that directly targeted receptors on the surface of tumour cells, did not seem to work as expected. Most nanoparticles get into tumour tissues relying on enhanced permeability and retention effect, and their efficacy also has been highly pronounced in preclinical models of solid tumours equipped with leaky vasculature, however, which may not suitable for the tumours that develop over the course of years rather than days. It is noticeable that immune cells could migrate actively alone the chemokine gradients to sites of inflammation, like tumours. Instead of targeting tumours directly, Schmid et al.32 attempted to develop a PD-1 antibody-target nanoparticles that bind to CD8+ cells circulating in the blood or in the lymphoid tissues and tumours of mice, by means of which, immunomodulatory compounds and anti-PD-1 can be effectively delivered into tumour site better than systemic administration of free drug (Fig. 3).
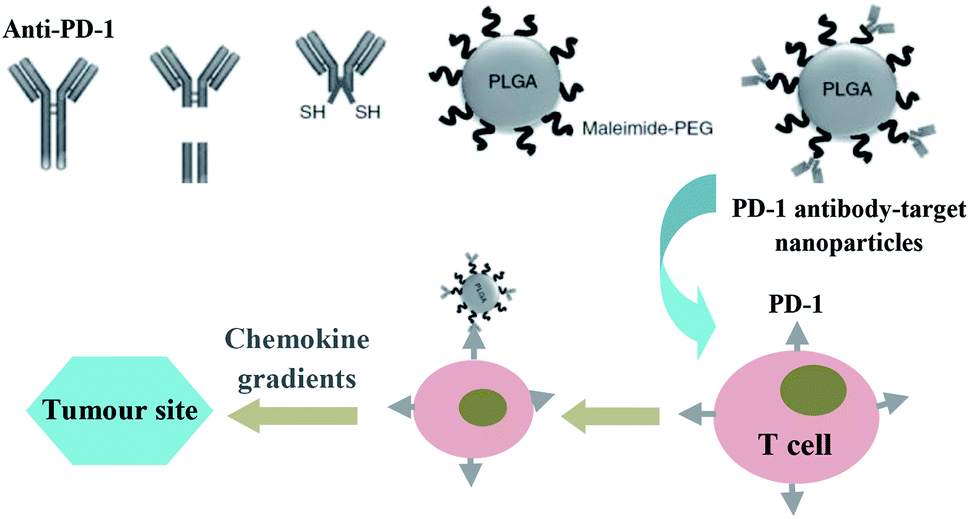 | ||
| Fig. 3 Schematic illustration of the preparation of PD-1 antibody-target nanoparticles, and its trafficking mechanism to the target spot. Images were partly reproduced form ref. 32. | ||
In addition to the direct target delivery of anti-PD-1/PD-L1 inhibitors aforementioned, researchers also tried to explore innovative strategies that aimed at transporting therapeutic proteins on a genetic level to accomplish the tumour-specific delivery of PD-1/PD-L1 inhibitors. Adeno-associated viral (AAV) vectors are one of the most promising vehicles for in vivo gene delivery. Reul et al.33 constructed a tumour-target Her2-AAV used as a vehicle for the coding sequence of an scFv-Fc fusion protein delivery that directed against mouse PD-1. Transduction of Her2/neu+ RENCA cells revealed that AAV-encoded aPD-1 could be readily detected and Her2-AAV also could mediate specific gene delivery into tumour lesions via intravenous administration in BALB/c mice bearing subcutaneous RENCA-Her2/neu tumours. Not only that, but AAV could also be levered to mediate extracellular domain of PD-1 (sPD-1) expression and disrupt the negative immunoregulatory signals provided by PD-1/PD-L1 pathway. The experimental results suggested that the expressed sPD-1 could block the PD-1/PD-L1interaction, and local gene transfer of sPD-1 in tumour site could potently inhibit tumour growth and prolong the survival of mice. These all provide proof of concept that tumour-targeted vectors can be used for the targeted delivery of PD-1/PD-L1 inhibitors on a genetic level to the tumour site.
2.2 Tumour-target PD-1/PD-L1 blockade by RNA interference
Although no detectable side effects have been reported following target administration of PD-1/PD-L1 inhibitors during short-term tumour experiments, gene knock-out mice exhibit that long-term interference with PD-1/PD-L1 during clinical maintenance therapies could lead to unexpected immune-related adverse responses.34 Alternatively, a more moderate approach that aims at targeted disturbing PD-1/PD-L1 signaling based on the utilization of short-hairpin double-stranded silencing RNA (siRNA), has demonstrated some potential in eliciting durable resistance against PD-1/PD-L1 pathway-mediated immunosuppressive signal. RNA interference (RNAi) is a promising strategy for tumour treatment by gene silencing.35 Through RNAi, targeted protein expression could be safely knocked down without introducing exogenous sequences into the genome.Based on the above theory, some researchers wondered whether this goal could be accomplished by means of retroviral siRNA delivery. The experimental results indicated that effective target siRNA sequences delivery reduced the surface PD-1 expression, which led to activated T-cell immune functions in response to PD-L1 positive melanoma cells.36 Some synthetic polymers, such as cyclodextrin-based polycations, polyethylenimine (PEI) and polyphosphates also have been used for siRNA delivery.37,38 After a series of functional modifications on these non-viral vector materials, it can successfully deliver siRNA to the target site against the complex in vivo environment and effectively release it for anti-tumour treatment. For example, the application of the adoptive T cell immunotherapy in tumour treatment is impressive. However, it also suffers from various immunosuppressive mechanisms exerted by tumour cells as well as host immune cells in the tumour microenvironment, including the PD-1/PD-L1 pathway. Hence, Teo et al. attempted to delivery PD-L1 siRNA utilizing various folic acid (FA)-functionalized PEI polymers to SKOV-3-Luc EOC cells, and studied the sensitization of the EOC cells to immunotherapy.39 The results indicated that all polymers induced 40% to 50% PD-L1 protein knockdown, and importantly, SKOV-3-Luc cells treated with the polymer/PD-L1 siRNA complexes showed up to twofold more sensitive to T cell immune response in comparison with scrambled siRNA treated controls.
These findings demonstrated that PD-L1 knockdown in PD-L1 positive tumour cells, via PD-L1 siRNA targeting delivery, are able to block the PD-1/PD-L1 pathway and enhance the anti-tumour immune response. More importantly, the PD-1/PD-L1 blocking by PD-L1 knockdown might be able to reduce the immune-related side effects and bypass the failure of PD-1/PD-L1 inhibitors caused by exosomal PD-L1 and “decoys” of PD-L1 antibody secreted on some tumour cells, as we mentioned before.
3. Immunosuppressive tumour microenvironment
It has been taken for granted that the recognition and elimination of tumour cells are the important missions of immune system. Nevertheless, along with gradual penetrating to tumour development research, scientists come to realize that the immune system can also facilitate the tumour progression by promoting chronic inflammation, lowing tumour immunogenicity and creating immunosuppressive tumour microenvironment. This dual functionality of the immune system on the tumour growth is termed as termed cancer immunoediting.40 At the beginning of cell carcinogenesis, innate and adaptive immunity can work together to eliminate cancer cells and prevent the occurrence of tumours. After the elimination phase, a small fraction tumour cell variant with very low immunogenicity can survive.9,41 When tumours' struggle to be free of immune recognition and/or destruction, they proceed into the escape phase and initiate combating with the immunosurveillance at the same time. The immunoedited tumour cells not only can evade the recognition of immune system by reducing their immunogenicity, but actively suppress the functions of immune cells by developing an arsenal of mechanisms, including overexpression of PD-L1, infiltration of immunosuppressive cells and secretion of immunosuppressive factors, such as transforming growth factor-β, interleukin-10 and etc.9,12,42,43 Furthermore, tumour cells also could send signals to these tumour-associated immune cells and modulate their function, resulting in the formation of immunosuppressive microenvironment that supports tumour survival, progression, especially contributes to tumour resistance to therapies (Fig. 4).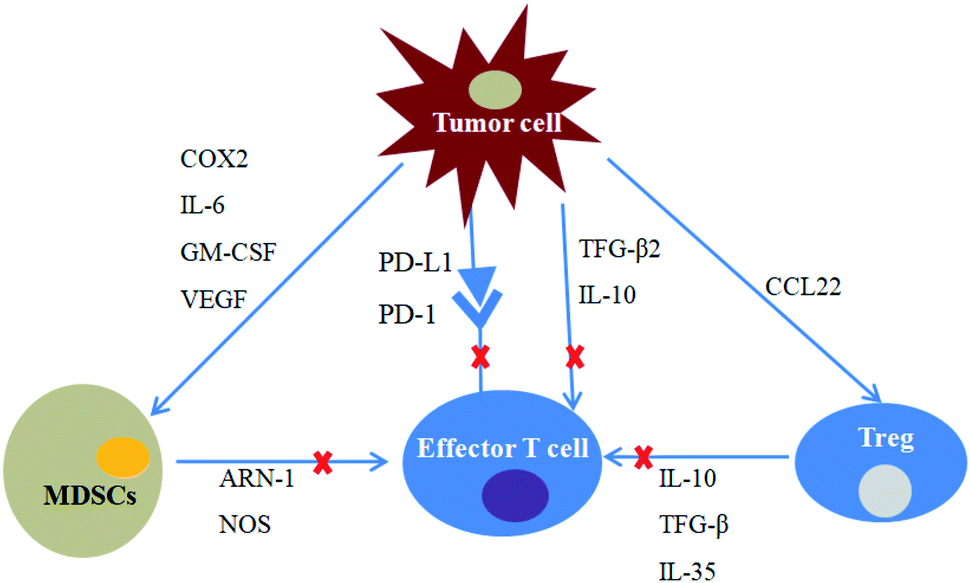 | ||
| Fig. 4 Immunosuppression in the tumour microenvironment. Tumour cells secrete immunosuppressive factors such as TGF-β2 and IL-10, all of which suppress the activity of effector T cells. PD-L1 expressed on its surface also suppresses effector T cells activity through PD-1/PD-L1 pathway. COX2, IL-6, GM-CSF and VEGF recruit MDSCs to the tumour microenvironment. Tumour cells secrete CCL22 to recruit Tregs and promote its proliferation. MDSCs and Tregs can suppress the activity of effector T cells via various cytokines. | ||
3.1 Combination treatment of PD-1/PD-L1 inhibitors and tumour vaccines
Active immunization mediated by tumour vaccines is considered as a promising strategy for effective anti-tumour response. Tumour vaccines aim at activating and amplifying tumour-specific cytotoxic T lymphocyte responses, inducing effector CD8+ and CD4+ cells to identify capture and expunge tumour cells.44–46 Various antigens including recombinant proteins, viral vectors, bacterial strains, tumour cell lysate, peptides and nucleic acids have been shown to deliver large amounts of immunogenic epitopes, and they have the potential to stimulate strong and durable immune response.44,45,47–50 However, in spite of the immune activation in T cells induced by tumour vaccines, there still exist some immune escape signals mediated by tumour immunosuppressive microenvironment that decrease the tumour-specific immune elimination,51 while PD-1/PD-L1 blockade therapy happens to remedy this deficiency.Tumour cell lysate, representing a spectrum of tumour-associated antigens, could be facilely processed into vaccines without further sequencing or antigen synthesis.52 However, it has been shown that tumour cell lysate based vaccination only induces weak tumour-specific T cells responses, and simulates slender therapeutic efficacy.53 To address this problem, Ochyl et al.54 reported a method for generating tumour cell lysate based PEGylated vaccine nanoparticles (PEG-NPs). This nano-vesicles PEG-NPs not only solved the problem for its own instability in vivo, but also improved the delivery efficiency of tumour vaccine. Results demonstrated that PEG-NPs vaccination elicited more antigen-specific T cell responses than standard freeze-thawed lysate vaccination by 3.7 times in tumour-bearing mice. Importantly, when combined with a PD-1 antibody, PEG-NP vaccination triggered 4.2 times stronger antigen-specific T cell responses, and resulted in 63% of tumour regression of tumour-bearing animals, while FT lysate and PD-1 antibody combination treatment displayed only 13% response rate. Besides, PEG-NP vaccination combined with PD-1 antibody immunotherapy could protect all survivors from further tumour cell re-challenge.
There were also documentaries demonstrated that whole tumour cell vaccines exhibited modest efficacy due to the similar antigens spectrum pattern between tumour cells and normal cells.55 Recent advances have examined closely the immunogenicity of various subcellular compartments of tumour cells, including cells membranes, cytosol, and nucleus, and the results showed that tumour-associated antigens were located on the tumour cells membranes in several types of tumour, which could be exploited as personalized therapeutic anti-tumour vaccines materials to irritate adaptive immune response.56 Therefore, the “artificial necroptotic cancer cell”, named αHSP70p-CM-CaP, was developed to deliver tumour membrane proteins plus additional boosting adjuvants.57 After the administration of αHSP70p-CM-CaP, effective lymph node trafficking and strong T cells response were detected in mice. Especially, when combined with a PD-1 antibody, αHSP70p-CM-CaP vaccination could result in the killing of tumour cells and mediate tumour regression in B16OVA melanoma mice.
In addition to these tumour vaccines, epigenetic modulators including hypomethylation agents (HMAs), were also used to enhance the immunogenicity of tumour cells via inducing tumour-associated antigen expression. Moreover, HMAs has been proved to relieve immunosuppressive tumour microenvironment by reducing the number of myeloid-derived suppressor cells (MDSCs), and not only that, HMAs also could mediate the upregulation of immunosuppressive ligands including PD-L1/PD-L2, increasing the sensitivity of tumours to PD-1/PD-L1 blockade therapy.58 The HMAs and PD-1 antibody combination treatment showed enhanced the immunogenicity of tumour cell, and reversed immunosuppressive tumour microenvironment to some extent, leading to the tumour regression and prolonged the survival time of mice.
Based on the above data, we could find that the personalized therapeutic anti-tumour vaccines eliciting endogenous cytotoxic T cells responses against tumour cells offer a promising strategy in working synergistically with PD-1/PD-L1 blockade therapy.
3.2 Combination treatment of PD-1/PD-L1 inhibitors and chemotherapy
Even though the significant tumour antigen-specific cytotoxic T cells responses initiated by tumour vaccines, their clinical applications are still highly limited due to the heterogeneity of cancer cells. Chemotherapy seems to be able to make up for this deficiency and provides the enhanced anti-tumour efficiency in coordination with PD-1/PD-L1 blockade therapy.59Chemotherapy is one of the three widely accepted conventional methods for tumour treatment that obtain anti-tumour effects by killing tumour cells using cytotoxic drugs.60 However, due to the poor selectivity, chemotherapy may cause a certain degree of toxic effects on the immune system. The traditional concept holds that there is an antagonistic effect between chemotherapy and immunotherapy, and these two treatment methods are difficult to use together. The situation has changed with an accumulation of data on the synergistic therapeutic effect obtained by a combination of immunotherapy and chemotherapy in multiple tumour treatments. Accumulating evidence suggests that chemotherapeutics induced cell death could produce plentiful of tumour debris in situ and mediate a large quantity of tumour-associated antigens releases, such as calreticulin (CRT), heat shock proteins (HSPs), high-mobility group box 1 protein (HMGB1) and adenosine triphosphate (ATP), which leads to the activation of tumour-specific cognate immune responses.61 This process is known as immunogenic cell death (ICD).62 In principle, ICD could establish an unbiased tumour antigen repertoire that covers all types of tumour antigens, and concomitantly trigger broad specific antitumour immunity. For example, Li et al.63 tried to subvert the GBM immunosuppressive microenvironment by DC-mediated delivery of doxorubicin–polyglycerol–nanodiamond composites (Nano-Dox). In vitro study on human cell models showed that Nano-DOX treated GC exhibited profuse DAMPs emission and antigen release.
In addition, chemotherapy could sensibilize tumour cells to cytotoxic T lymphocyte in vivo. For instance, paclitaxel (PTX), doxorubicin (DOX) and cisplatin were shown to increase the sensitivity of tumour cells to cytotoxic T lymphocytes specific killing effect due to the upregulation of mannose-6-phosphate receptors (M6PR) on cells and increase of the cells permeability to granzyme-B (GrzB) secreted by cytotoxic T lymphocytes.64 What's more, chemotherapy could also relieve the immunosuppression of tumour microenvironment by eliminating immunosuppressive cells, such as regulatory cells (tregs) and MDSCs.65 Immunotherapy, in turn, also could make a positive contribution to the efficiency of chemotherapy for tumour treatment (Fig. 5). For instance, immunotherapy performs an important role in rebuilding the human immune system and maintaining the immune balance of patients, and it also could effectively solve the patient's insensitivity to chemotherapy by enhancing the anti-tumour immune response, which can not only ensure the efficacy of chemotherapy but also improve the human immunity and reduce the toxic effects of chemotherapeutics.
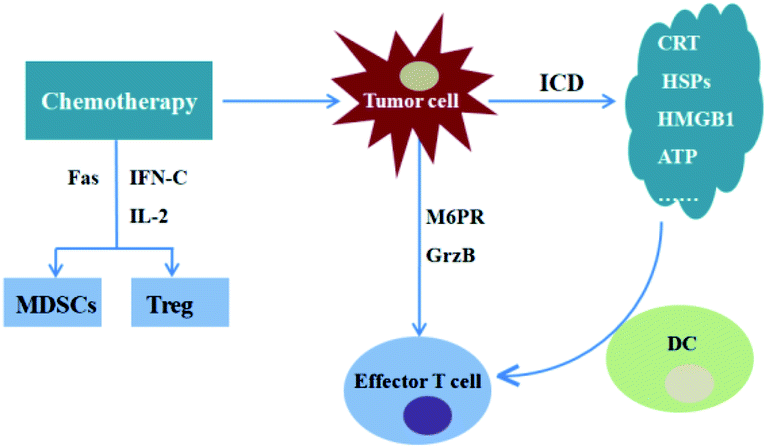 | ||
| Fig. 5 Combination treatments of chemotherapy and immunotherapy. Chemotherapeutics increase the sensitivity of tumour cells to cytotoxic T lymphocytes specific killing effect due to the upregulation of M6PR on cells and increase of the permeability of the cells to GrzB secreted by cytotoxic T lymphocytes. Chemotherapeutics induces ICD, resulting in tumour-specific immune response. Chemotherapeutics could decrease Treg and MDSCs levels. | ||
As mentioned above, under certain conditions, chemotherapeutics could promote anti-tumour immune responses in many aspects. However, indeed, systemic chemotherapy would damage the bone marrow and subsequently influence the number and activation state of resident immune cells,66 evoking concerns for potential antagonistic interactions between systemic chemotherapy and immunotherapy. One study showed that local chemotherapy could reduce the risk of immune cells damages and promote anti-tumour immune response, and when combined with anti-PD-1, it exhibited enhanced anti-tumour immune response and prolonged survival in glioblastoma. Besides, local chemotherapy-treated mice displayed increased infiltration of tumour-associated dendritic cells and proliferation of antigen-specific T effector cells, while systemic chemotherapy brought about systemic and intratumoural lymphodepletion, accompanied with decreased immune memory in long-term survivors. More importantly, adoptive transfer of CD8+ cells from local chemotherapy-treated mice partly rescued systemic chemotherapy-treated mice in rechallenge experiments.
In order to achieve improved anti-tumour efficiency and reduced side effects, Wang et al.67 engineered a therapeutic scaffold, which consisted of reactive oxygen species (ROS) degradable hydrogel that could release therapeutics in a programmed manner within the tumour microenvironment (TME) containing abundant ROS, aiming at achieving local release of gemcitabine (GEM) and anti-PD-L1 antibody (aPDL1). The experimental results showed that the aPDL1-GEM scaffold could elicit an immunogenic tumour phenotype and enhance an immune-mediated tumour regression, accompanied by prevention of tumour recurrence after primary surgical excision.
Based on above, we could find that the PD-1/PD-L1 blockade therapy combined with chemotherapy is expected to solve the problem of low response to PD-1/PD-L1 blocking agents caused by low tumour immunogenicity, and provide mutual promoted effects for tumour treatment at the same time. In addition to these chemotherapeutics, there are other tumour therapies that also could induce tumour ICD, including ionizing irradiation, photodynamic therapy, cardiac glycosides, cyclophosphamide, shikonin and oncolytic viruses. These tumour treatments also could be considered as a potential adjunctive treatment.
3.3 Combination treatment of PD-1/PD-L1 inhibitors and immune adjuvants
Immune adjuvants are immune promoters that can strengthen the body's immune response to immunogens. CpG-oligodeoxynucleotides (ODNs), a short single-stranded DNA molecule containing non-methylated, is a Toll-like receptor-9 agonist. By means of activation on antigen-presenting cells (APCs), including macrophages and dendritic cells, CpG ODNs can induce a signaling cascade, resulting in the release of pro-inflammatory cytokines such as interleukin-6 (IL-6), tumour necrosis factor (TNF), interleukin-12 (IL-12) and so on, and lead to initiation of effective immune response.68 As additional boosting adjuvants, CpG ODNs have been widely used to trigger innate immune system for synergistic anti-tumour immunotherapy.For example, scientists realized that the limited efficiency of PD-1/PD-L1 blockade therapy might be on account of costimulation deficiency in tumour microenvironment in the setting, at which the APCs encounter the tumour cells and T-cell. To address this issue, Wang et al.69 attempted to introduce the potent immunostimulatory effects of CpG ODNs into the checkpoint inhibition therapy and developed a novel stimuli-responsive delivery vector to realize the controlled release of anti-PD-1 antibody and CpG ODNs at tumour sites and exert synergistic antitumour activity. This innovative CpG ODNs-based drug delivery system not only served as a delivery intermediary for anti-PD-1 antibody but also could enhance anti-tumour efficiency after being fragmented. And the studies demonstrated that the bioresponsive controlled release of PD-1 antibody and CpG ODNs exerted more effective anti-tumour responses than either of them.
In addition, CpG-ODNs were also used as components of nanovaccine for tumour immunotherapy. Following CpG ODNs-based nanovaccine administration, the immunogenic antigen materials and CpG ODNs could be effectively co-delivered to APCs, boosting anti-tumour immunity. When combined with the anti-PD-1 antibody, CpG ODNs-based nanovaccine could mediate effective tumour regression in vivo.57,70 Altogether, these data demonstrated proof-of-concept evidence that CpG ODNs used as boosting adjuvants for triggering innate immune system also provide a potential strategy in working synergistically with PD-1/PD-L1 blockade therapy.
3.4 Combination treatment of PD-1/PD-L1 inhibitors and immunomodulators
Tumour immunosuppressive microenvironment plays a great role in the development of tumour. As above mentioned, taking cue from the tumour cells, the tumour-associated immune cells could modulate each other's functions, resulting in the formation of an immunosuppressive and relatively stable microenvironment which supports tumour survival, progression and especially contribute to tumour resistance to therapies, including overexpression of PD-L1, secretion of immunosuppressive factors and infiltration of immunosuppressive cells.9,12,41–43 Besides the above mechanism of immune suppression, there was a new finding suggested that complement receptors C3aR and C5aR expressed on CD8+ T cells also could be considered a novel class of immune checkpoints, which is independent of PD-1/PD-L1 immune checkpoint pathway. IL10 activates cytotoxic CD8+ T cells and stimulate their anti-tumour responses. Nevertheless, the IL10 production of cytotoxic CD8+ T cells could be inhibited by autocrine complement C3 through complement receptors C3aR and C5aR.71 As already mentioned, the immunosuppressive tumour microenvironment is composed of complex immunosuppression networks, and the PD-1/PD-L1 pathway is only one of the most important components of that. Thus, PD-1/PD-L1 blockade therapy alone is insufficient to resist the immunosuppressive effect of tumour microenvironment to achieve effective tumour killing effect. In these situations, co-inducing T-cell (re-)activation by blockade of other negative inhibitors or upregulation of stimulus signals might be able to work synergistically with PD-1/PD-L1 blockade therapy.72,73Type I interferons (IFNs) could inhibit tumour growth by promoting DC cross-priming to (re-)activate T cell,74 while the expression of IFNs in the tumour tissue is limited or restrained. In addition, although local delivery of IFNs can restore antigen presentation, it also upregulates the expression of PD-L1 and inhibits the following T cells activation. Liang et al.75 developed a conjugate based on anti-PD-L1 antibody and IFNα to create feedforward responses. It was shown that this conjugates could overcome both IFNs and PD-1/PD-L1 blockade therapy resistance and achieve a synergistic anti-tumour effect with the least side effects. Intriguingly, IFNα-mediated upregulation of PD-L1 increased the targeting distribution of the fusion protein. The antibody-cytokine fusions have been widely studied to conduct cytokine-based therapies for tumour. However, the size of these adducts severely decreased the tissue penetration and the subsequent concentration of cytokines at the right location. Alternatively, the heavy chain-only antibody fragments anti-PD-L1 antibody derived from alpaca was selected to construct antibody-cytokine fusion.76 Targeted delivery of antibody-cytokines conjugates in this manner significantly inhibited tumour growth.
Tumour or tumour-associated immune cells could also secrets immunosuppressive molecules to induce immune tolerance in the tumour microenvironment, such as indoleamine 2, 3-dioxygenase (IDO) and TGFβ. IDO, an important negative feedback protein overexpressed by tumour and IDO+ DCs, involves in the cell cycle arrest and apoptosis of effector T cells, and increasing the production of Tregs in tumour.77,78 In order to achieve better anti-tumour effect, researchers attempted to encapsulate IDO inhibitor into anti-PD-1 antibody delivery system for synergistically blocking immune tolerance signals in the tumour microenvironment, and exhibited significant tumour regression.79 Except for the directly delivery of immunosuppressive molecules inhibitor, it has been validated that intratumoural administration of mRNA encoding a fusion protein of the ectodomain of TGFβ receptor II and interferon-β showed therapeutic potential,80 and the anti-tumour efficacy could be improved when combined with blockade of PD-1/PD-L1 interactions. The ectonucleotidases CD39 and CD73, acting in unison to transform extracellular immune-stimulating ATP into adenosine, are two new drug targets. Hence, the inhibition of CD39 and CD73 may be able to promote the subversion of tumour immunosuppressive microenvironment and enhance the anti-tumour immune response.81
Macrophages play a crucial role in regulating tumour development and metastasis. Extensive researches suggested that tumours could constantly recruit M2 tumour associated macrophages (TAMs) into tumour tissue, and the TAMs density have a positive correlation with poor prognosis in various human tumours.82 Therefore, many scientists tried to set about manipulating the TAMs functions to improve the efficacy of immunotherapies, and also achieved certain positive effects.83,84
The tumour stroma is also one of the most important components of the immunosuppressive tumour microenvironment. Its highly fibrotic construction and structurally abnormal blood vessels pose a powerful physical barrier to CTL infiltration, leading to poor efficiency of PD-1/PD-L1 blockade therapy. A large portion of tumours has been examined to comprise high levels of hyaluronan (HA), which was an important component of the extracellular matrix (ECM) of tumours.85 Thus, in order to improve the intratumoural delivery of therapeutic molecules and enhance the CTL infiltration in tumour tissues, hyaluronidase (HAase) was used to digest the overexpressed HA, and achieved a positive effect.86 However, this approach is only suitable for patients with high levels of HA. For another, tumour vascular normalization has been gradually accepted as promising strategy for promoting drug delivery and encouraging immune cells infiltration.87 Sonic hedgehog (SHH) is generally upregulated, and plays an important role in the formation of tumour stroma in the majority of pancreatic ductal adenocarcinoma.88 Zhao et al.89 developed a nano-formulation of cyclopamine (CPA), a SHH inhibitor, used for tumour treatment. The preclinical results showed that the CPA nano-formulation increased tumour infiltration by CTLs and improved the susceptibility to anti-PD-1 antibody therapy in an orthotopic murine pancreatic ductal adenocarcinoma model. The lysophosphatidic acid receptor 4 (LPA4) is another therapeutic target of the stroma-modulating agent, and the activation of LPA4 induces fine vascular network formation in brain tumours. LPA treatment improved the delivery of anti-PD-1 antibody and lymphocyte infiltration into brain tumour tissues, resulting in the enhanced anti-tumour effect of PD-1 blockade therapy.90 These data demonstrated that co-delivery of a stroma-modulating agent and PD-1/PD-L1 pathway inhibitors is a promising approach to enhance the response rate of PD-1/PD-L1 blockade therapy.
4. Conclusion and prospects
PD-1/PD-L1 pathway-based immune checkpoint therapy has been regarded to initiating a revolution in tumour immunotherapy. Nowadays, an increasing number of PD-1/PD-L1pathway inhibitors have been developed and applied in preclinical trials. And the engineering of PD-1/PD-L1 inhibitors and its combination treatment with other therapeutics has preliminarily realized enhanced anti-tumour efficacy. However, despite that, this needs further study to achieve better therapeutic efficiency and eliminate or minimize the negative impact of side effects. For example, the biomaterials used for target delivery of PD-1/PD-L1 inhibitors, must possess safety, biocompatibility and low antigenicity, which can be potentially served as the carrier for target drug delivery in vivo. In addition, gene knockdown and chemical blockade have been proved to be efficient in inhibiting intracellular PD-L1 production, thereby disrupting signaling pathways, but they may be ineffective for serum PD-L1 or the PD-L1 that already expressed on the cell surface.91 As for the combination treatment, it is important to coordinate the different therapeutic agents for maximizing the anti-tumour efficiency because of the various anti-tumour mechanisms and different tolerance dose of each therapeutic agent.One more thing we can't ignore is that tumours are the living, dynamic changing organisms. Just as with the other organism, under the threat of adverse factors they are inclined to avoid danger or instinctively induce a series of resistance mechanisms to protect themselves. The most glaring examples are a range of drug-resistance mechanisms of tumours against chemotherapies. Recently, drug resistance phenomenon has also been observed in immunotherapy for tumours treatment. For example, PD-1 blockade therapy has been found to promote the expression of pro-tumour inflammatory cytokines that potentially counteract the anti-tumour effects of PD-1 blockade,92,93 and the anti-PD-L1 antibody “decoys” mechanism disables the anti-PD-L1 antibody blockade therapy;16 Most astonishingly, when the chimeric antigen receptors (CARs) T cells were used for tumour treatment, CARs could provoke reversible antigen loss via trogocytosis, and the target antigen could be transferred to T cells through this active process, thereby reducing target antigen density on tumour cells and inhibiting T cell activity by initiating fratricide T cell killing and promoting T cell exhaustion.94 Based on these understandings, we speculate that the future direction might lie in the combined therapy based on PD-1/PD-L1 blockade and personalized tumour vaccines. The activation of a specific and sustained anti-tumour immune response, rather than blindly suppressing the tumour using brute-force, is of great significance in tumour regression.
Conflicts of interest
There are no conflicts to declare.Abbreviations
| ICB | Immune checkpoint therapy |
| PD-1 | Programmed death-1 |
| PD-L1 | Programmed death-ligand 1 |
| AAV | Adeno-associated viral |
| sPD-1 | Extracellular domain of PD-1 |
| siRNA | Silencing RNA |
| RNAi | RNA interference |
| PEI | Polyethylenimine |
| FA | Folic acid |
| PEG-NPs | PEGylated vaccine nanoparticles |
| HMAs | Hypomethylation agents |
| MDSCs | Myeloid-derived suppressor cells |
| CRT | Calreticulin |
| HSPs | Heat shock proteins |
| HMGB1 | High-mobility group box 1 protein |
| ATP | Adenosine triphosphate |
| ICD | Immunogenic cell death |
| Nano-Dox | Doxorubicin–polyglycerol–nanodiamond composites |
| PTX | Paclitaxel |
| DOX | Doxorubicin |
| M6PR | Mannose-6-phosphate receptors |
| GrzB | Granzyme-B |
| Tregs | Regulatory cells |
| ROS | Reactive oxygen species |
| TME | Tumour microenvironment |
| GEM | Gemcitabine |
| aPDL1 | Anti-PD-L1 antibody |
| CpG ODNs | CpG-oligodeoxynucleotides |
| APCs | Antigen-presenting cells |
| IL-6 | Interleukin-6 |
| TNF | Tumour necrosis factor |
| IL-12 | Interleukin-12 |
| IFNs | Type I interferons |
| IDO | Indoleamine 2, 3-dioxygenase |
| TAMs | Tumour associated macrophages |
| HA | Hyaluronan |
| ECM | Extracellular matrix |
| HAase | Hyaluronidase |
| SHH | Sonic hedgehog |
| CPA | Cyclopamine |
| LPA4 | Lysophosphatidic acid receptor 4 |
| CARs | Chimeric antigen receptors |
Acknowledgements
This study was supported by the National Natural Science Foundation, China (grant no. 81572989) and the International Academic Cooperation Project of Science and Technology Committee of Shanghai, Shanghai, China (grant no. 18430721100).References
- M. Jacobson, L. Wang-Bishop, K. Becker and J. Wilson, Biomater. Sci., 2019, 7, 547–559 RSC.
- C. Robert, G. V. Long, B. Brady, C. Dutriaux, M. Maio, L. Mortier, J. C. Hassel, P. Rutkowski, C. McNeil, E. Kalinka-Warzocha, K. J. Savage, M. M. Hernberg, C. Lebbé, J. Charles, C. Mihalcioiu, V. Chiarion-Sileni, C. Mauch, F. Cognetti, A. Arance, H. Schmidt, D. Schadendorf, H. Gogas, L. Lundgren-Eriksson, C. Horak, B. Sharkey, I. M. Waxman, V. Atkinson and P. A. Ascierto, N. Engl. J. Med., 2015, 372, 311–319 CrossRef.
- K. S. Peggs and J. P. Allison, Br. J. Haematol., 2005, 130, 809–824 CrossRef CAS.
- W. Zou, J. D. Wolchok and L. Chen, Sci. Transl. Med., 2016, 8, 328 Search PubMed.
- P. Sharma, S. Hu-Lieskovan, J. A. Wargo and A. Ribas, Cell, 2017, 168, 707–723 CrossRef CAS.
- S. Spranger, R. M. Spaapen, Y. Zha, J. Williams, Y. Meng, T. T. Ha and T. F. Gajewski, Sci. Transl. Med., 2013, 200ra116 Search PubMed.
- D. T. Le, J. N. Uram, H. Wang, B. R. Bartlett, H. Kemberling, A. D. Eyring, A. D. Skora, B. S. Luber, N. S. Azad, D. Laheru, B. Biedrzycki, R. C. Donehower, A. Zaheer, G. A. Fisher, T. S. Crocenzi, J. J. Lee, S. M. Duffy, R. M. Goldberg, A. de la Chapelle, M. Koshiji, F. Bhaijee, T. Huebner, R. H. Hruban, L. D. Wood, N. Cuka, D. M. Pardoll, N. Papadopoulos, K. W. Kinzler, S. Zhou, T. C. Cornish, J. M. Taube, R. A. Anders, J. R. Eshleman, B. Vogelstein and L. A. Diaz Jr, N. Engl. J. Med., 2015, 372, 2509–2520 CrossRef CAS.
- C. Wang, Y. Ye, Q. Hu, A. Bellotti and Z. Gu, Adv. Mater., 2017, 29, 1606036 CrossRef.
- E. K. Nduom, M. Weller and A. B. Heimberger, Neuro-Oncology, 2015, 17, vii9–vii14 CrossRef CAS.
- D. F. Quail and J. A. Joyce, Cancer Cell, 2017, 31, 326–341 CrossRef CAS.
- A. Gieryng, D. Pszczolkowska, K. A. Walentynowicz, W. D. Rajan and B. Kaminska, Lab. Invest., 2017, 97, 498–518 CrossRef CAS.
- S. Chen and I. F. Parney, Glioma Cell Biology, 2014, 221–239 Search PubMed.
- W. Zou, Nat. Rev. Cancer, 2005, 5, 263–274 CrossRef CAS.
- T. J. Curiel, S. Wei and H. Dong, Nat. Med., 2003, 9, 562–567 CrossRef CAS.
- M. Poggio, T. Hu and C. Pai, Cell, 2019, 177, 414–427 CrossRef CAS.
- B. Gong, K. Kiyotani and S. Sakata, J. Exp. Med., 2019, 216, 982–1000 CrossRef CAS.
- G. De Velasco, Y. Je and D. Bosse, Cancer Immunol. Res., 2017, 5, 312–318 CrossRef CAS.
- F. S. Hodi, S. J. O'Day and D. F. McDermott, N. Engl. J. Med., 2010, 363, 711–723 CrossRef CAS PubMed.
- T. M. Beer, E. D. Kwon and C. G. Drake, J. Clin. Oncol., 2017, 35, 40–47 CrossRef CAS.
- A. M. Eggermont, V. Chiarion-Sileni and J. J. Grob, N. Engl. J. Med., 2016, 375, 1845–1855 CrossRef CAS PubMed.
- A. D. Simmons, M. Moskalenko, J. Creson, J. Fang, S. Yi, M. J. VanRoey and K. Jooss, Cancer Immunol. Immunother., 2008, 57, 1263–1270 CrossRef CAS.
- L. Van Hooren, L. C. Sandin and I. Moskalev, Eur. J. Immunol., 2016, 47, 385–393 CrossRef.
- M. Ferrari, Nat. Rev. Cancer, 2005, 5, 161–171 CrossRef CAS.
- D. Peer, J. M. Karp and S. Hong, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS PubMed.
- G. C. Gurtner, S. Werner and Y. Barrandon, Nature, 2008, 453, 314–321 CrossRef CAS PubMed.
- B. Furie and B. C. Furie, J. Clin. Invest., 2005, 115, 3355–3362 CrossRef CAS.
- B. Ho-Tin-Noé, T. Goerge and S. M. Cifuni, Cancer Res., 2008, 68, 6851–6858 CrossRef.
- L. J. Gay and B. Felding-Habermann, Nat. Rev. Cancer, 2011, 11, 123–134 CrossRef CAS.
- Q. Hu, W. J. Sun and C. J. Qian, Adv. Mater., 2015, 27, 7043–7050 CrossRef CAS.
- C. Wang, W. Sun and Y. Ye, Nat. Biomed. Eng., 2017, 1, 11 CrossRef.
- Y. T. Li, T. Nishikawa and Y. Kaneda, Sci. Rep., 2016, 6, 25077 CrossRef CAS.
- D. Schmid, C. G. Park and C. A. Hartl, Nat. Commun., 2017, 8, 1747 CrossRef.
- J. Reul, J. Frisch and C. Engeland, Frontiers in Oncology, 2019, 52 CrossRef.
- J. Weber, Cancer Immunol. Immunother., 2009, 58, 823–830 CrossRef CAS PubMed.
- D. Xie, J. Du, M. Bao, A. Zhou, C. Tian, L. Xue, C. Ju, J. Shen and C. Zhang, Biomater. Sci., 2019, 7, 901–913 RSC.
- L. Borkner, A. Kaiser and W. van de Kasteele, Cancer Immunol. Immunother., 2010, 59, 1173–1183 CrossRef CAS.
- Y. Ping, Q. Hu and G. Tang, Biomaterials, 2013, 34, 6482–6494 CrossRef CAS.
- C. Q. Mao, J. Z. Du and T. M. Sun, Biomaterials, 2011, 32, 3124–3133 CrossRef CAS.
- P. Y. Teo, C. Yang and L. M. Whilding, Adv. Healthcare Mater., 2015, 4, 1180–1189 CrossRef CAS.
- R. D. Schreiber, L. J. Old and M. J. Smyth, Science, 2011, 331, 1565–1570 CrossRef CAS PubMed.
- M. W. Teng, J. Galon and W. H. Fridman, J. Clin. Invest., 2015, 125, 3338–3346 CrossRef.
- D. F. Quail and J. A. Joyce, Cancer Cell, 2017, 326–341 CrossRef CAS.
- A. Gieryng, D. Pszczolkowska and K. A. Walentynowicz, Lab. Invest., 2017, 97, 498–518 CrossRef CAS.
- P. A. Ott, Z. Hu and D. B. Keskin, Nature, 2017, 547, 217–221 CrossRef CAS PubMed.
- U. Sahin, E. Derhovanessian and M. Miller, Nature, 2017, 547, 222–226 CrossRef CAS.
- K. C. Soares, A. A. Rucki and A. A. Wu, J. Immunother., 2015, 38, 1–11 CrossRef CAS.
- R. Kuai, L. J. Ochyl and K. S. Bahjat, Nat. Mater., 2017, 16, 489–496 CrossRef CAS.
- R. Kuai, X. Sun and W. Yuan, Bioconjugate Chem., 2018, 29, 771–775 CrossRef CAS.
- H. Song, P. Yang and P. Huang, Theranostics, 2019, 9, 2299–2314 CrossRef CAS.
- S. Kumar Gulla, B. Rama Rao, G. Moku, S. Jinka, N. Varma Nimmu, S. Khalid, C. Ranjan Patra and A. Chaudhuri, Biomater. Sci., 2019, 7, 773–788 RSC.
- D. E. Speiser, P. C. Ho and G. Verdeil, Nat. Rev. Immunol., 2016, 16, 599–611 CrossRef CAS.
- F. E. Gonzalez, A. Gleisner and F. Falcon-Beas, Hum. Vaccines Immunother., 2014, 10, 3261–3269 CrossRef.
- T. D. de Gruijl, A. J. M. van den Eertwegh and H. M. Pinedo, Cancer Immunol. Immunother., 2008, 57, 1569–1577 CrossRef.
- L. J. Ochyl, J. D. Bazzill and C. Park, Biomaterials, 2018, 182, 157–166 CrossRef CAS.
- I. Mellman, G. Coukos and G. Dranoff, Nature, 2011, 480, 480–489 CrossRef CAS.
- R. H. Fang, C. M. Hu and B. T. Luk, Nano Lett., 2014, 14, 2181–2188 CrossRef CAS.
- T. Kang, Y. Huang and Q. Zhu, Biomaterials, 2018, 164, 80–97 CrossRef CAS.
- H. Ruan, Q. Hu and D. Wen, Adv. Mater., 2019, 31, 1806957 CrossRef.
- Y. Fan, R. Kuai and Y. Xu, Nano Lett., 2017, 17, 7387–7393 CrossRef CAS PubMed.
- F. Raza, Y. Zhu, L. Chen, X. You, J. Zhang, A. Khan, M. Waseem Khan, M. Hasnat, H. Zafar, J. Wu and L. Ge, Biomater. Sci., 2019, 7, 2023–2036 RSC.
- G. Kroemer, L. Galluzzi and O. Kepp, Annu. Rev. Immunol., 2013, 31, 51–72 CrossRef CAS.
- O. Tacar, P. Sriamornsak and C. R. Dass, J. Pharm. Pharmacol., 2013, 65, 157–170 CrossRef CAS.
- T. F. Li, K. Li and Q. Zhang, Biomaterials, 2018, 181, 35–52 CrossRef CAS.
- R. Ramakrishnan, D. Assudani and S. Nagaraj, J. Clin. Invest., 2010, 120, 1111 CrossRef CAS PubMed.
- C. A. Chen, C. M. Ho and M. C. Chang, Mol. Ther., 2010, 18, 1233 CrossRef CAS.
- G. Lombardi, E. Rumiato and R. Bertorelle, Am. J. Clin. Oncol., 2013, 38, 514–519 CrossRef.
- C. Wang, J. Wang, X. Zhang, S. Yu, D. Wen, Q. Hu, Y. Ye, H. Bomba, X. Hu, Z. Liu, G. Dotti and Z. Gu, Sci. Transl. Med., 2018, 10, eaan3682 CrossRef.
- H. Zhang and X. D. Gao, Mater. Sci. Eng., C, 2017, 70, 935–946 CrossRef CAS.
- C. Wang, W. Sun and G. Wright, Adv. Mater., 2017, 29, 8912–8920 Search PubMed.
- X. Guan, J. Chen and Y. Hu, Biomaterials, 2018, 171, 198–206 CrossRef CAS.
- Y. Wang, S. N. Sun and Q. Liu, Cancer Discovery, 2016, 6, 1022–1035 CrossRef CAS.
- P. Sharma and J. P. Allison, Science, 2015, 348, 56–61 CrossRef CAS.
- D. S. Shin and A. Ribas, Curr. Opin. Immunol., 2015, 33, 23–35 CrossRef CAS.
- Z. Ren, J. Guo and J. Liao, Clin. Cancer Res., 2017, 23, 193–203 CrossRef CAS.
- Y. Liang, H. Tang, J. Guo, X. Qiu, Z. Yang, Z. Ren, Z. Sun, Y. Bian, L. Xu, H. Xu, J. Shen, Y. Han, H. Dong, H. Peng and Y. Fu, Nat. Commun., 2018, 9, 4586 CrossRef.
- M. Dougan, J. R. Ingram and H. J. Jeong, Cancer Immunol. Res., 2018, 6, 389–401 CrossRef CAS.
- J. R. Moffett and M. A. Namboodiri, Immunol. Cell Biol., 2003, 81, 247–265 CrossRef CAS.
- C. Uyttenhove, L. Pilotte and I. Théate, Nat. Med., 2003, 9, 1269–1274 CrossRef CAS.
- X. Zhang, C. Wang and J. Wang, Adv. Mater., 2018, 30, 1707112 CrossRef.
- K. Van der Jeught, P. T. Joe and L. Bialkowski, Oncotarget, 2014, 15, 10100–10113 Search PubMed.
- S. K. Abhishek, T. Tamara and K. Richard, J. Immunother. Cancer, 2019, 7, 1–12 CrossRef.
- X. Tang, Cancer Lett., 2013, 332, 3–10 CrossRef CAS.
- L. Yang, J. Sun and Q. Liu, Adv. Sci., 2019, 1802012 CrossRef.
- M. Daisuke, S. Naohiro and H. Tae, J. Clin. Invest., 2019, 129, 1278–1294 CrossRef PubMed.
- H. Gong, Y. Chao and J. Xiang, Nano Lett., 2016, 16, 2512–2521 CrossRef CAS.
- X. Guan, J. Chen and Y. Hu, Biomaterials, 2018, 171, 198–206 CrossRef CAS.
- R. K. Jain, Science, 2005, 307, 58–62 CrossRef CAS.
- R. L. Yauch, S. E. Gould and S. J. Scales, Nature, 2008, 455, 406–410 CrossRef CAS.
- J. Zhao, Z. Xiao and T. Li, ACS Nano, 2018, 12, 9881–9893 CrossRef CAS.
- D. Eino, Y. Tsukada and H. Naito, Cancer Res., 2018, 78, 6607–6620 CrossRef CAS.
- Y. Wu, W. Chen, Z. Xu and W. Gu, Front. Immunol., 2019, 10, 2022 CrossRef.
- M. A. Curran, W. Montalvo and H. Yagita, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4275–4280 CrossRef CAS.
- J. Dirks, A. Egli and U. Sester, Transplant Infect. Dis., 2013, 15, 79–89 CrossRef CAS.
- H. Mohamad, D. Anton and C. Annalisa, Nature, 2019, 568, 112–116 CrossRef.
| This journal is © The Royal Society of Chemistry 2019 |