
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAcetylene hydrochlorination over boron-doped Pd/HY zeolite catalysts†
Lu
Wang
 *ab,
Lizhen
Lian
a,
Haijun
Yan
a,
Feng
Wang
a,
Jide
Wang
*a,
Chao
Yang
a and
Lida
Ma
b
*ab,
Lizhen
Lian
a,
Haijun
Yan
a,
Feng
Wang
a,
Jide
Wang
*a,
Chao
Yang
a and
Lida
Ma
b
aKey Laboratory of Oil and Gas Fine Chemicals of Education, Xinjiang Uyghur Autonomous Region, College of Chemistry and Chemical Engineering, Xinjiang University, Urumqi, PR China 830046. E-mail: wanglu_4951@163.com; awangjd@126.com; Fax: +86 991 8581018; Tel: +86 991 8581018
bXinjiang De'an Environmental Protection Technology Co. Ltd, Urumqi, 830026, PR China
First published on 25th September 2019
Abstract
A novel boron-doped Pd/HY zeolite catalyst for acetylene hydrochlorination was prepared and exhibited an outstanding catalytic performance (the acetylene conversion was maintained at >95% for about 30 h). The boron species can stabilize catalytically active Pd2+ species and weaken carbon deposition and Pd2+ reduction during the reaction, thus improving the catalytic stability.
The coal-based acetylene hydrochlorination process is a major route for the production of vinyl chloride monomer (VCM) in China, which is essential for polyvinyl chloride (PVC) production. However, this process is restricted by the application of toxic and scarce mercury chloride, which is used as the active ingredient of catalysts in the industrial process.1,2 Hence, it is imperative to explore environmentally friendly catalysts for acetylene hydrochlorination.
Since Hutchings et al. suggested that the activities of metal catalysts were associated with the standard electrode potentials of the related metal ions,3 numerous studies have been carried out to develop non-mercury catalysts for efficient and sustainable hydrochlorination of acetylene in recent years, such as Au,4–11 Pd,12–19 Pt,20 Ru,21 Bi,22 Cu,23,24etc. Based on the detailed study by Hutchings' team,3 metal chloride/activated carbon catalysts (including Pd2+, Hg2+, Cu2+, Cu+, Ag+) had superior catalytic activity and the order of their activities was Pd2+ > Hg2+ > Cu2+ > Cu+ > Ag+. Therefore, palladium-based catalysts were reported to be active for acetylene hydrochlorination.12–19 Wang et al. prepared Pd-based/C catalysts via impregnation method and suggested that the Pd loss and the carbon deposition were main reasons for the catalyst deactivation, and the adding of K and La could improve the stability and regeneration of Pd/C catalysts.18 Mitchenko et al. reported that Pd(II) chloro complexes were the active sites of catalysts19 and the (NH4)2PdCl4 complex could stabilize the Pd species resulted in improving the catalytic performance over carbon-based materials.13 In our previous study, zeolites as new supports were prepared Pd/HY catalysts applied for acetylene hydrochlorination and the catalysts stabilities could be enhanced after modification with NH4F or polyaniline.14–17 While the carbon deposition and the active component reduction is the major obstacle to improve activity and stability of catalysts. It reported that heteroatom doping of support materials was effective for the improvement of the catalytic performance of metal catalysts25 and few reports were studied on the B-doped zeolite-based catalysts for acetylene hydrochlorination at present. In this work, a novel boron-doped Pd/HY zeolite-based catalyst for acetylene hydrochlorination was prepared and its outstanding catalytic performance was further studied.
The boron-doped HY zeolites supports were prepared using boron oxide (B2) as the boron source. The boron oxide aqueous solution was quantitatively (the weight percentage of B2 is 1.0%, 3.0%, 5.0%, 7.0%, 9.0%) added into HY zeolites (purchased by Nankai University, Si/Al = 9, 5.0 g) under stirring for 4 h, then washed with distiller water to pH about 7 and water was evaporated at 80 °C for 10 h, the B-doped HY zeolites supports were obtained. The Pd/B2-HY catalysts were prepared via ultrasonic-assisted impregnation using the B-doped HY or the original HY supports, as described in our previous work.14–17 Based on the weight percentage of the boron oxide, the obtained B-doped Pd/HY catalysts were named as Pd/1B2-HY, Pd/3B2-HY, Pd/5B2-HY, Pd/7B2-HY and Pd/9B2-HY, respectively. The Pd loading in all catalysts was 0.9 wt%.
X-ray diffraction (XRD) data were collected using a M18XHF22-SRA diffractometer with Cu–K irradiation at 50 kV and 100 mA in the scan range of 2 between 10°–80°. Scanning electron microscopy (SEM) and energy dispersive X-ray (EDX) analyses were performed using a LEO1450VP detector to determine the morphology. High-resolution transmission electron microscopy (HRTEM) experiment was performed using a JEM-2100F (JEOL, Japan) working at 200 kV. BET surface areas analysis was performed by JW-BK Brunauer–Emmett–Teller (BET) equipment. Thermogravimetric (TG) tests were detected by a NETZSCH SAT 449F3 multifunctional thermal analyzer in an air atmosphere at a flow rate of 50 mL min−1. The nature of carbon deposition was determined by an Agilent 7890A/5975C gas chromatography-mass spectrometry (GC-MS). Palladium contents were detected using an inductively coupled plasma (ICP-6300) instrument. X-ray photoelectron spectroscopy (XPS) data were recorded by AXIS ULTRA spectrometer (Kratos Analytical Ltd) and binding energies were referred to the C 1s line at 284.8 eV. Fourier transform infrared spectrometer (FTIR) was used EQUINOX-55 (Bruker Company, Germany) in transmittance (%) mode in the range 4000–400 cm−1 and temperature programmed decomposition (TPD) was determined using a TP-5080 adsorption instrument with ammonia over a temperature ramp of 0–900 °C, rate ramp of 10 °C min−1 and flow of 100 mL min−1.
The catalytic performances of catalysts were evaluated in a fixed bed with 10 mm-diameter a quartz tube micro reactor. The reactor temperature was calibrated by a CKW-110 temperature controller (Chaoyang automation instrument factory, Beijing, China) and the gas flow during the reaction was regulated by a D08-1F mass flow controller (Sevenstar Huachuang electronics. co. Ltd, Beijing, China). After N2 flow (20 mL min−1) purged, hydrogen chloride (12.6 mL min−1) and acetylene (10.1 mL min−1) were fed through a mixing vessel containing catalyst (3.0 g), giving a temperature at 160 °C, feed volume ratio VHCl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :VC2H2 = 1.25 and the C2H2 gas hourly space velocity (GHSV) of 110 h−1. The exit gas mixture from the reactor was passed through an absorption bottle filled with sodium hydroxide solution and then set into a gas chromatography (GC 2010, Shimadzu) to analyse the acetylene conversion and the VCM selectivity immediately.13–17
:VC2H2 = 1.25 and the C2H2 gas hourly space velocity (GHSV) of 110 h−1. The exit gas mixture from the reactor was passed through an absorption bottle filled with sodium hydroxide solution and then set into a gas chromatography (GC 2010, Shimadzu) to analyse the acetylene conversion and the VCM selectivity immediately.13–17
Fig. 1 and S1† display the catalytic performance of undoped and B-doped catalysts. The undoped Pd/HY catalyst exhibited a poor catalytic stability and its C2H2 conversion dropped dramatically from 93% to 13% in 3 h. After B doping, the C2H2 conversion was significantly enhance from 93% of the undoped Pd/HY catalyst to 96% of the B-doped Pd/B2-HY catalyst under the same conditions. It was clear that the Pd/7B2-HY catalyst displayed the optimal catalytic performance with the C2H2 conversion of 96% and the VCM selectivity of 98% after 10 h and its activity kept over 95% within 30 h (Fig. S2†). It is suggested that the appropriate amount of B doping can improve the activity and stability of Pd-based catalysts for acetylene hydrochlorination.
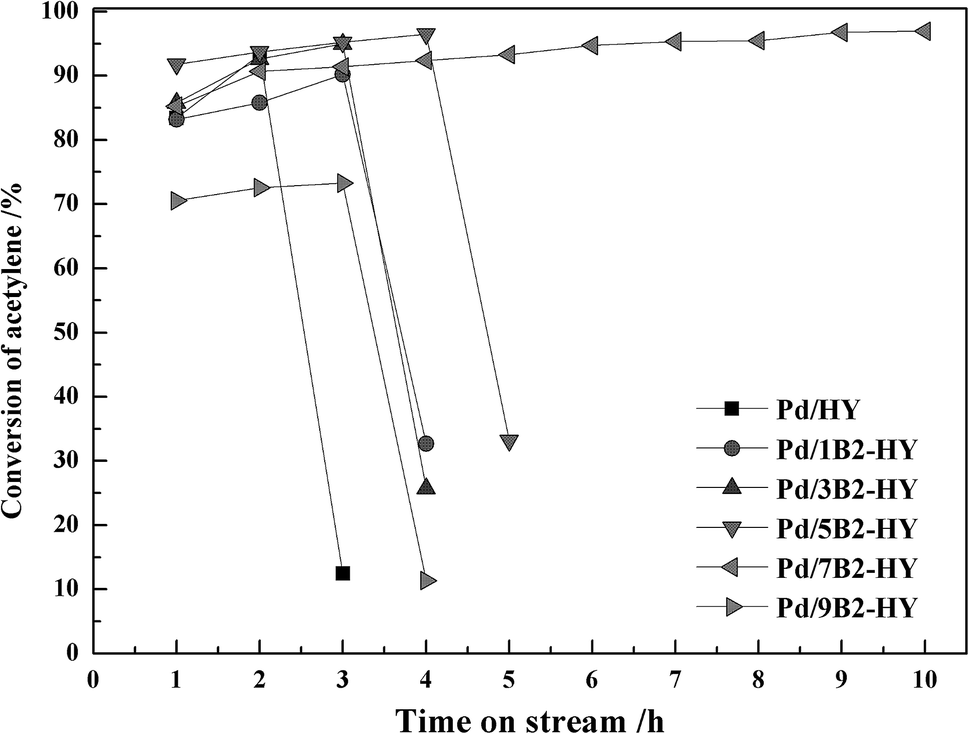 | ||
| Fig. 1 The catalytic performances of the B-doped Pd/B2-HY catalysts. | ||
It is obvious that no other diffraction peaks of Pd species were detected, except the strongest characteristic peak of HY zeolites, which could be attributed to the low concentration or high dispersion of Pd species26,27 (Fig. 2a). For the fresh Pd/7B2-HY catalyst, numerous Pd-based particles with the size about 2.81 nm were distributed uniformly on the HY support (Fig. 2b and d), which was slight bigger than that of the average pore width (by BET results, 2.49 nm) of the 7B2-HY zeolites, deducing that most Pd species involved in reaction might be on the support surface. In addition, the Pd species on the HY zeolite were visualized with well-defined fringes, which was ascribed to the (111) plane of face-centered cubic Pd with a d spacing of 0.23 nm (Fig. 2c).28 The characterization of HRTEM verifies that XRD pattern is reasonable and suggests that the Pd species are highly dispersed in the fresh Pd/7B2-HY catalyst (Table 1).
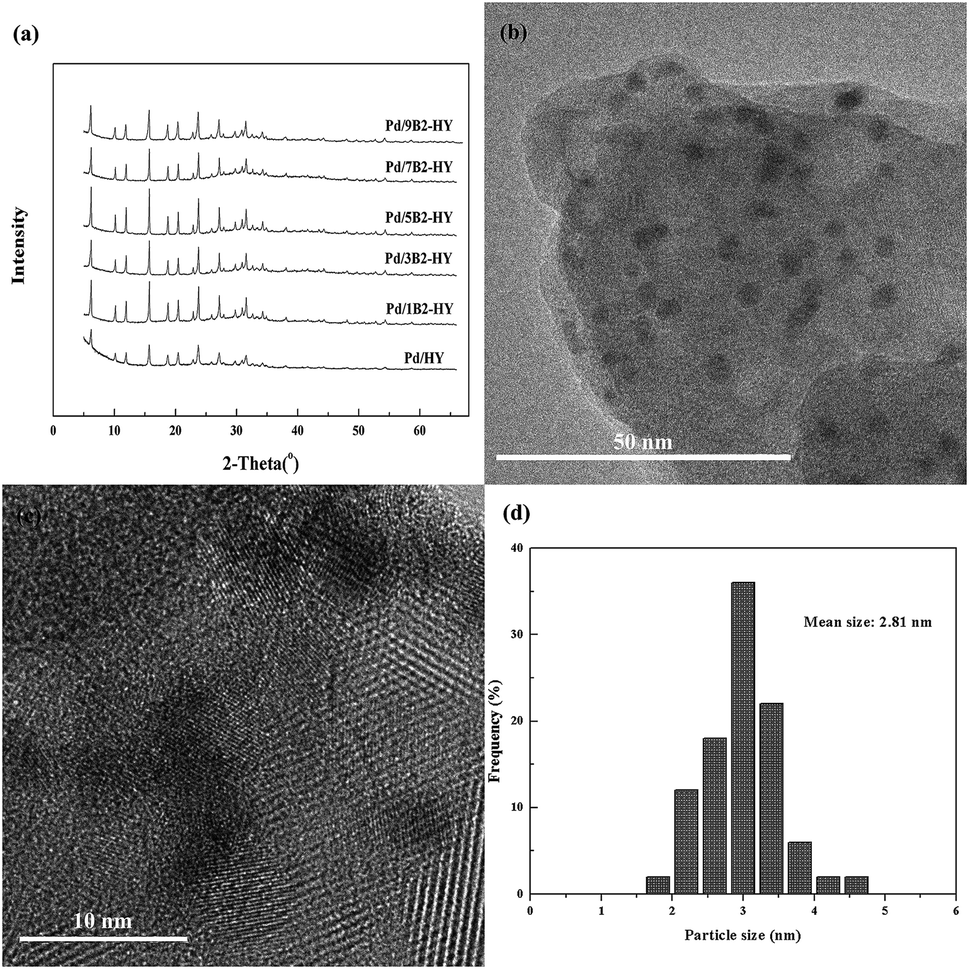 | ||
| Fig. 2 The characterization on the fresh catalysts: (a) XRD patterns of Pd-based catalysts. (b and c) HRTEM images of Pd/7B2-HY catalyst. (d) Pd particle size distribution of Pd/7B2-HY catalyst. | ||
After B doping, the 7B2-HY specific surface areas reduced and the decrease could result from the abundant presence of B species introduced on the surface (Table S1†), which possibly block the small pores in the zeolite-based support.29 Compared with the corresponding supports, the specific surface areas of the fresh catalysts showed a certain decrease because of the blocking of pores as a result of Pd addition.26 After reaction, the specific surface areas and total pore volumes of the catalysts were reduced and the decrease of catalysts BET surface area (ΔSBET%) order was Pd/HY (93.7%) > Pd/7B2-HY (91.7%). The carbon deposition, the agglomeration of Pd species and the collapse of zeolite framework are the possible reasons for clogging pores, covering the active sites and decreasing catalyst activity.30,31 SEM results (Fig. S3†) showed that the deactivated catalyst surface was aggregated more seriously and the carbon deposition was supposed to the main reason.32 To further testify the results, the TG analysis of the fresh and used Pd/HY and Pd/7B2-HY catalysts were showed in Fig. 3. The result indicated that the actual amount of carbon deposited on the Pd/7B2-HY catalyst could be calculated as 3.00%, which was about 29.41% less than that in the Pd/HY catalyst (4.25%). It is suggested that the B doping to Pd/HY catalysts can inhibit the formation of the carbon deposition to some extent, thus improving the catalytic stability of catalyst.33 In addition, the GC-MS results (Table S2†) showed that the carbon deposition is probably ascribed to some hydrocarbon compounds (such as benzene, 1,1-dichloroethane, chlorobenzene, 1-chlorobutylene and so on), which is also the main reason for the undesirable VCM selectivity of Pd/B2-HY catalysts.31,34
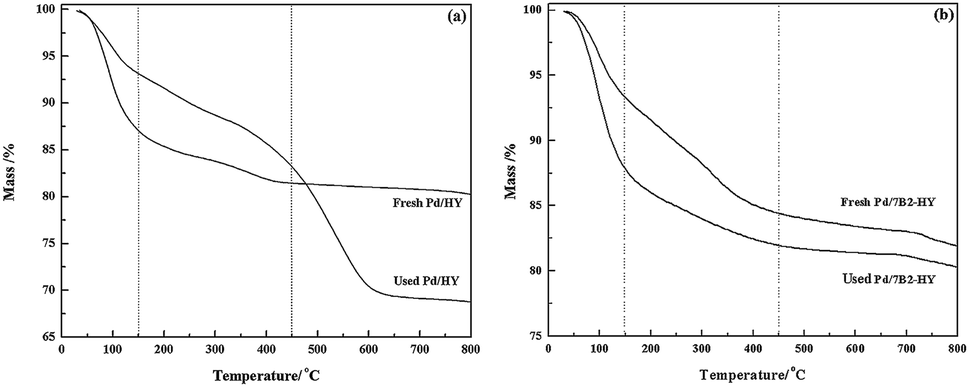 | ||
| Fig. 3 TG profiles of Pd-based catalysts. | ||
XPS analysis (Fig. 4) was carried out to investigate the valence state and relative amount of Pd species in the fresh and used Pd/HY and Pd/7B2-HY catalysts. It should be noted that all of the Pd (3d) signals had been divided into two components responding to metallic Pd0 (335.5 eV and 340.6 eV) and Pd2+ (337.1 eV and 342.3 eV) species35–37 and curve fitting were employed to analyse the ratio of Pd0 and Pd2+ species (the results were calculated in Table 2). The relative Pd2+ content is 42.5% presented in the fresh Pd/HY catalyst and the relative Pd2+ content of is decreased to 30.72% in the fresh Pd/7B2-HY catalyst. However, the relative Pd2+ content in the used Pd/HY and Pd/7B2-HY catalyst is 26.90% and 29.54%, respectively. It is established that Pd2+ was reduced into Pd0 during the reaction, contributing to the deactivation of Pd-based catalysts.13–17 Combination with the activity of catalysts (Fig. 1), the fresh Pd/7B2-HY catalyst with the enhanced catalytic performance is attributed to the presence of B (wide-scan spectra are shown in Fig. S4†), which can inhibit the less reduction of Pd2+ to Pd0 in the reaction process (Table 2). Moreover, only one B species (B3+) were detected in the fresh and used Pd/7B2-HY catalyst. The binding energy observed at 193.5 eV for the fresh Pd/7B2-HY catalyst was typically referred to as boron–oxygen bond, while the lower binding energy (193.2 eV) was detected in the used Pd/7B2-HY catalyst, indicating that the weakened interaction between boron and the support in the reaction. From ICP results (Table 2), the total Pd content in the used catalyst is much less than that in the fresh one. The loss of ratio of Pd in the Pd/HY catalyst is 71.2% and that in the Pd/7B2-HY catalyst is 40.0% after reaction. The less Pd loss in the Pd/7B2-HY catalyst also suggests that the B doping in the Pd-based catalysts can stabilize Pd species.
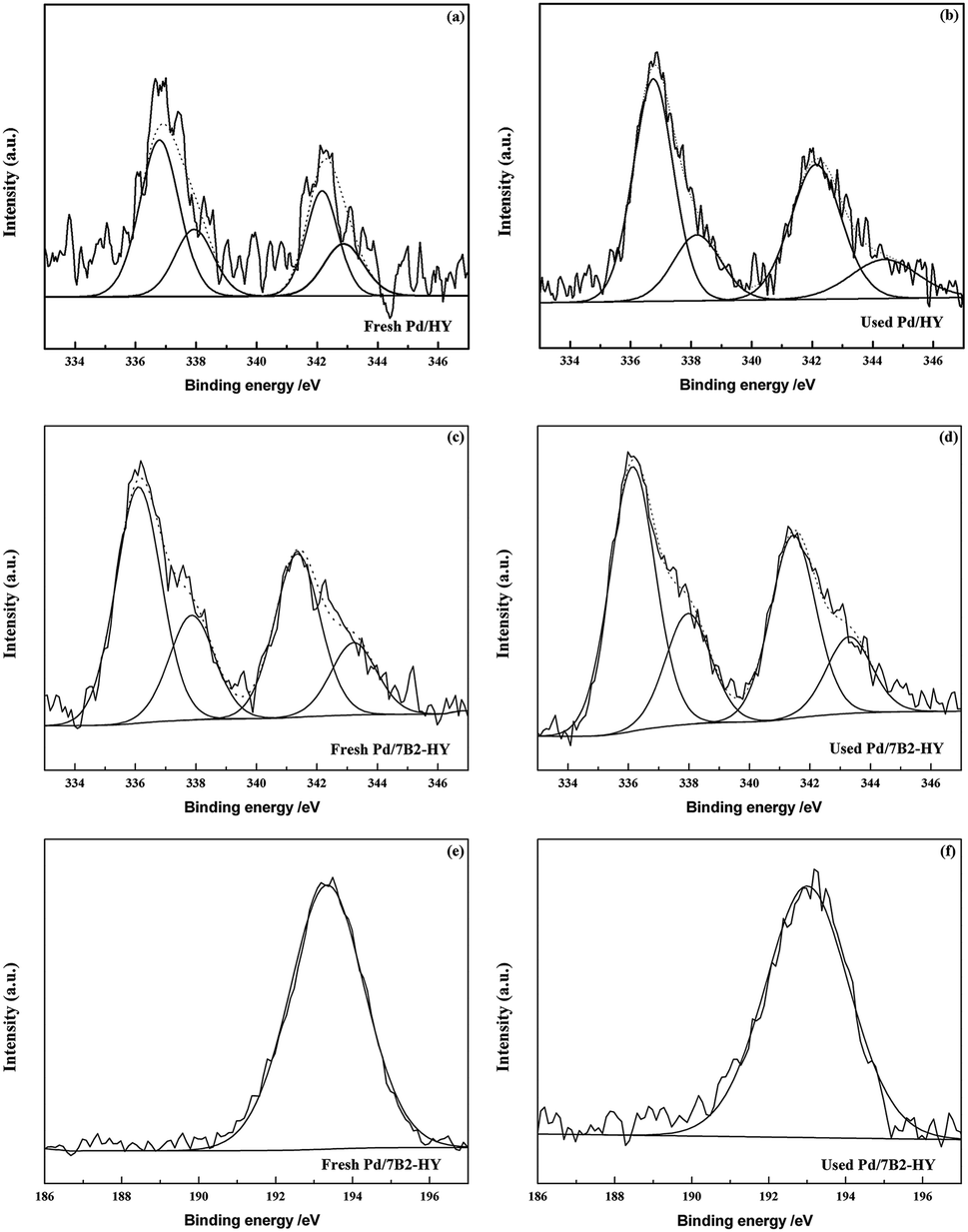 | ||
| Fig. 4 XPS Pd 3d profiles of the fresh and used Pd-based catalysts. | ||
| Catalysts | Pd0 (area%) | Pd2+ (area%) |
|---|---|---|
| Fresh Pd/HY | 57.50 | 42.50 |
| Used Pd/HY | 73.10 | 26.90 |
| Fresh Pd/7B2-HY | 69.28 | 30.72 |
| Used Pd/7B2-HY | 70.46 | 29.54 |
| Catalysts | Total Pd (wt%) | Loss ratio of Pd (%) | |
|---|---|---|---|
| Fresh | Used | ||
| Pd/HY | 0.59 | 0.17 | 71.2 |
| Pd/7B2-HY | 0.75 | 0.45 | 40.0 |
FT-IR (a) and NH3-TPD (b) results shown in Fig. 5 and S5† were investigated the surface functional groups and acidic properties of Pd-based catalysts. For the HY zeolites, the region from 3800 to 3000 cm−1 was attributed to the hydroxyl groups (including Si–OH, Al–OH and H-bonded hydroxyl groups) and the region from 1300 to 400 cm−1 was indicated the framework vibration of lattice cell (T-O-T unit: T is coordinated Si or Al atoms).38 Compared with the HY support, there was slight band position shift in the B-doped samples but they remained kept the zeolite framework. However, the band at 3550–3410 cm−1 (the hydroxyl groups) were disappeared in the used Pd/7B2-HY catalyst, which was suggested the existence of catalytic activity that originated from the acidic sites on Y zeolite. Moreover, NH3-TPD results could further provide evidence to analyse the changes of acidic properties on the fresh Pd/HY catalyst after B doping. Based on the desorption temperature, acid sites were identified to possess weak (∼115 °C), medium strength (300–500 °C) and strong strength (750–850 °C) in the Pd/HY catalyst, which could be ascribed to the original HY support.14–17 After B doping, the range of peak corresponds to weak acid sites shifted to the high temperature (∼196 °C) and no strong acid sites from the surface hydroxyl groups were observed, which was consistent with the FT-IR results discussed previously. Generally, the carbon deposition can produce in the strong acid sites39 and the Pd/7B2-HY catalyst had few strong acid sites, combination with the catalytic performance (Fig. 1) and TG characterizations (Fig. 3), it is suggested that the less carbon deposition occurred in the strong acidic sites on the Pd/7B2-HY catalyst surface was may be the possible reason for the enhancing the stability of catalyst.
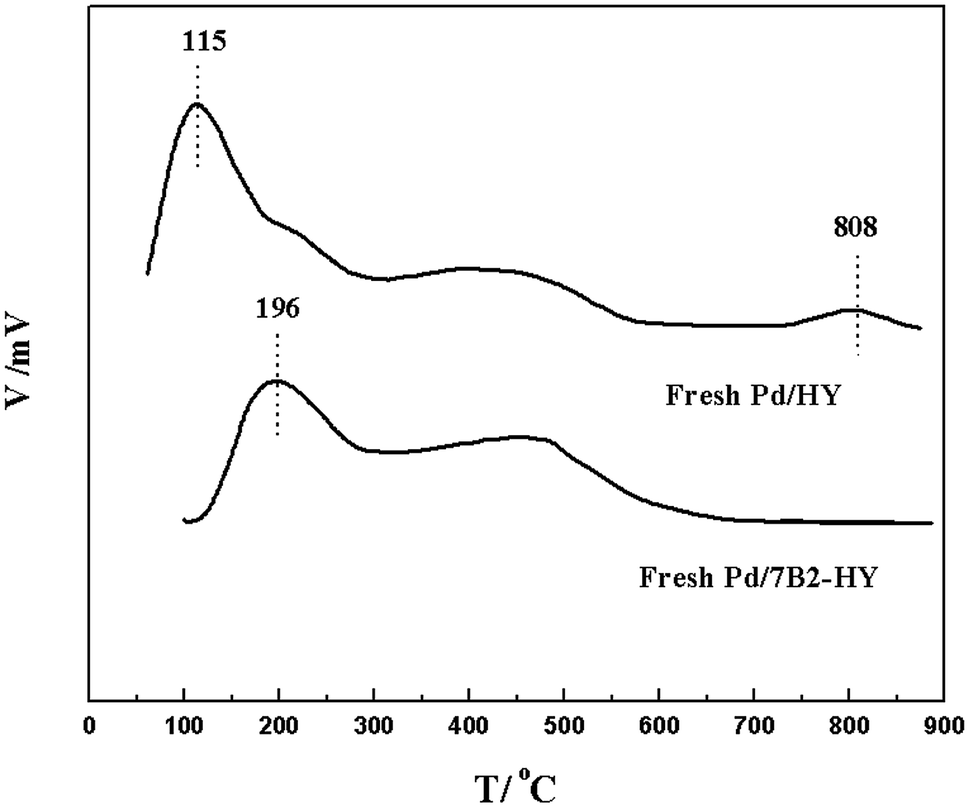 | ||
| Fig. 5 NH3-TPD profiles of the fresh Pd-based catalysts. | ||
Conclusions
The boron-doped Pd/HY catalyst was prepared and assessed for acetylene hydrochlorination reaction. It is exhibited that B additive can improve the activity and stability of Pd-based/HY catalysts during the reaction. The obtained Pd/7B2-HY catalyst shows the best catalytic performance with the acetylene conversion and the VCM selectivity over 95% more than 30 h. The results indicated that the presence of boron can make the Pd species dispersed well and partly weaken carbon deposition and Pd2+ reduction, thus improving the catalytic stability.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Natural Science Founds of General Programs of Xinjiang Uygur Autonomous Region (2017D01C034).Notes and references
- J. W. Zhong, Y. P. Xu and Z. M. Liu, Green Chem., 2018, 20, 2412–2427 RSC.
- Foreign Economic Cooperation Office, in R&D Progress of and Feasibility Study Report on Mercury-free Catalyst in China, Foreign Economic Cooperation Office, Ministry of Environmental Protection, People's Republic of China, 2011 Search PubMed.
- B. Nkosi, N. J. Coville and G. J. Hutchings, Appl. Catal., 1988, 43, 33–39 CrossRef CAS.
- G. Malta, S. A. Kondrat, S. J. Freakley, C. J. Davies, L. Lu, S. Dawson, A. Thetford, E. K. Gibson, D. J. Morgan, W. Jones, P. P. Wells, P. Johnston, C. R. A. Catlow, C. J. Kiely and G. J. Hutchings, Science, 2017, 355, 1399–1403 CrossRef CAS.
- G. Malta, S. J. Freakley, S. A. Kondrat and G. J. Hutchings, Chem. Commun., 2017, 53, 11733–11746 RSC.
- S. K. Kaiser, R. H. Lin, S. Mitchell, E. Fako, F. Krumeich, R. Hauert, O. V. Safonova, V. A. Kondratenko, E. V. Kondratenko, S. M. Collins, P. A. Midgley, N. López and J. P. Ramírez, Chem. Sci., 2019, 10, 359–369 RSC.
- L. Ye, X. P. Duan, S. Wu, T. S. Wu, Y. X. Zhao, A. W. Robertson, H. L. Chou, J. W. Zheng, T. Ayvalı, S. Day, C. Tang, Y. L. Soo, Y. Z. Yuan and S. C. E. Tsang, Nat. Commun., 2019, 10, 914–923 CrossRef PubMed.
- P. Johnston, N. Carthey and G. J. Hutchings, J. Am. Chem. Soc., 2015, 137(46), 14548–14557 CrossRef CAS.
- J. Wang, W. Q. Gong, M. Y. Zhu and B. Dai, Appl. Catal., A, 2018, 564, 72–78 CrossRef CAS.
- J. Zhao, S. C. Gu, X. L. Xu, T. T. Zhang, X. X. Di, Z. Y. Pan and X. N. Li, RSC Adv., 2015, 5, 101427–101436 RSC.
- J. Zhao, T. T. Zhang, X. X. Di, J. T. Xu, J. H. Xu, F. Feng, J. Ni and X. N. Li, RSC Adv., 2015, 5, 6925–6931 RSC.
- P. Li, M. Z. Ding, L. M. He, K. Tie, H. Ma, X. L. Pan and X. H. Bao, Sci. China: Chem., 2018, 61, 444–448 CrossRef CAS.
- H. H. He, J. Zhao, B. L. Wang, Y. X. Yue, G. F. Sheng, Q. T. Wang, L. Yu, Z. T. Hu and X. N. Li, RSC Adv., 2019, 9, 21557–21563 RSC.
- L. Wang, F. Wang, J. D. Wang, X. L. Tang, Y. L. Zhao, D. Yang, F. M. Jia and T. Hao, React. Kinet., Mech. Catal., 2013, 110, 187–194 CrossRef CAS.
- L. Wang, F. Wang and J. D. Wang, Catal. Commun., 2015, 65, 41–45 CrossRef CAS.
- L. Wang, F. Wang and J. D. Wang, New J. Chem., 2016, 40, 3019–3023 RSC.
- L. Wang, F. Wang and J. D. Wang, Catal. Commun., 2016, 74, 55–59 CrossRef CAS.
- S. J. Wang, B. X. Shen and Q. L. Song, Catal. Lett., 2010, 134, 102–109 CrossRef CAS.
- T. V. Krasnyakova, I. V. Zhikharev, R. S. Mitchenko, V. I. Burkhovetski, A. M. Korduban, T. V. Kryshchuk and S. A. Mitchenko, J. Catal., 2012, 288, 33–43 CrossRef CAS.
- S. A. Mitchenko, T. V. Krasnyakova, R. S. Mitchenko and A. N. Korduban, J. Mol. Catal. A: Chem., 2007, 275, 101–108 CrossRef CAS.
- S. K. Kaiser, R. Lin, F. Krumeich, O. V. Safonova and J. P. RamírezK, Angew. Chem., Int. Ed., 2019, 35, 12425–12432 CrossRef.
- K. Zhou, J. C. Jia, X. G. Li, X. D. Pang, C. H. Li, J. Zhou, G. H. Luo and F. Wei, Fuel Process. Technol., 2013, 108, 12–18 CrossRef CAS.
- X. M. Wang, M. Y. Zhu and B. Dai, ACS Sustainable Chem. Eng., 2019, 7, 6170–6177 CrossRef CAS.
- Y. F. Ren, B. T. Wu, F. M. Wang, H. Li, G. J. Lv, M. S. Sun and X. B. Zhang, Catal. Sci. Technol., 2019, 9, 2868–2878 RSC.
- J. Zhao, B. L. Wang, Y. X. Yue, C. F. Sheng, H. X. Lai, S. S. Wang, L. Yu, Q. F. Zhang, F. Feng, Z. T. Hu and X. N. Li, J. Catal., 2019, 373, 240–249 CrossRef CAS.
- X. Y. Li, Y. Wang, L. H. Kang, M. Y. Zhu and B. Dai, J. Catal., 2014, 311, 288–294 CrossRef CAS.
- X. L. Xu, J. Zhao, C. S. Lu, T. T. Zhang, X. X. Di, S. C. Gu and X. N. Li, Chin. Chem. Lett., 2016, 27, 822–826 CrossRef CAS.
- L. B. Ding, H. Yi, W. H. Zhang, R. You, T. Cao, J. L. Yang, J. L. Lu and W. X. Huang, ACS Catal., 2016, 6, 3700–3707 CrossRef CAS.
- J. Zhao, J. T. Xu, J. H. Xu, T. T. Zhang, X. X. Di, J. Ni and X. Li, Chem. Eng. J., 2015, 262, 1152–1160 CrossRef CAS.
- H. Y. Zhang, B. Dai, X. G. Wang, L. L. Xu and M. Y. Zhu, J. Ind. Eng. Chem., 2012, 18, 49–54 CrossRef CAS.
- H. Y. Zhang, B. Dai, X. G. Wang, W. Li, Y. Han, J. J. Gu and J. L. Zhang, Green Chem., 2013, 15, 829–836 RSC.
- J. G. Zhao, J. J. Zeng, X. G. Cheng, L. Wang, H. H. Yang and B. X. Shen, RSC Adv., 2015, 5, 16727–16734 RSC.
- Y. Jia, R. S. Hu, Q. H. Zhou, H. Y. Wang, X. Gao and J. Zhang, J. Catal., 2017, 348, 223–232 CrossRef CAS.
- C. Liu, C. H. Liu, J. H. Peng, L. B. Zhang, S. X. Wang and A. Y. Ma, Chin. J. Chem. Eng., 2018, 26, 364–372 CrossRef CAS.
- P. Weerachawanasak, G. J. Hutchings, J. K. Edwards, S. A. Kondrat, P. J. Miedziak, P. Prasertham and J. Panpranot, Catal. Today, 2015, 250, 218–225 CrossRef CAS.
- H. M. Chen, D. P. Huang, X. Y. Su, J. L. Huang, X. L. Jing, M. M. Du, D. H. Sun, L. S. Jia and Q. B. Li, Chem. Eng. J., 2015, 262, 356–363 CrossRef CAS.
- M. Jabłońska, A. Król, E. Kukulska-Zajac, K. Tarach, V. Girman, L. Chmielarz and K. Góra-MareK, Appl. Catal., B, 2015, 166–167, 353–365 CrossRef.
- T. K. Phung, M. M. Carnasciali, E. Finocchio and G. Busca, Appl. Catal., A, 2014, 470, 72–80 CrossRef CAS.
- X. P. Zhang, Q. D. Zhang, N. Tsubakic, Y. S. Tan and Y. Z. Han, Fuel, 2015, 147, 243–252 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra05576b |
| This journal is © The Royal Society of Chemistry 2019 |