
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVOX supported on TiO2–Ce0.9Zr0.1O2 core–shell structure catalyst for NH3-SCR of NO
Lvesheng
Sun
 a,
Shunxin
Cao
a,
Yun
Huang
a,
Yiming
Zhang
a,
Youhong
Xiao
b,
Guojun
Dong
*a and
Yu
Su
*c
a,
Shunxin
Cao
a,
Yun
Huang
a,
Yiming
Zhang
a,
Youhong
Xiao
b,
Guojun
Dong
*a and
Yu
Su
*c
aCollege of Materials Science and Chemical Engineering, Key Laboratory of Superlight Materials and Surface Technology of Education Ministry, Harbin Engineering University, 150001 Harbin, Heilongjiang, P. R. China. E-mail: dgj1129@163.com
bCollege of Power and Energy Engineering of Harbin Engineering University, 150001 Harbin, Heilongjiang, P. R. China
cSchool of Chemistry, Chemical Engineering and Materials of Heilongjiang University, 150080, Harbin, Heilongjiang, P. R. China. E-mail: 2003086@hlju.edu.cn
First published on 25th September 2019
Abstract
In this experiment, a TiO2–Ce0.9Zr0.1O2 support with core–shell structure was successfully prepared by a precipitation method and VOX/TiO2–Ce0.9Zr0.1O2 catalyst was prepared by an impregnation method, and the catalyst was used to catalyze the NH3-SCR of NO. Based on the results of HRTEM, XRD, BET, H2-TPR, NH3-TPD, XPS, Py-IR, it was speculated that due to the interaction between TiO2 and Ce0.9Zr0.1O2, more oxygen vacancies and Ce3+ are generated, which are beneficial to the existence of low-valence V by electron transfer between high valence state V and Ce3+and increase the acidic sites on the catalyst surface. The catalytic activity (>97%) of the VOX/TiO2–Ce0.9Zr0.1O2 catalyst is superior to the current commercial catalyst (V2O5–WO3/TiO2) and has a higher N2 selectivity (>97.5%) at 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1 GHSV and 250–400 °C.
000 h−1 GHSV and 250–400 °C.
Introduction
Nitrogen oxides (NOX) cause serious environmental pollution. The technology of selective catalytic reduction (SCR) of NOX with NH3 has been widely used to reduce pollution caused by NOX. A lot of research work on SCR catalysts has been carried out.1–4 But the most widely used catalyst is still V2O5–WO3(MoO3)/TiO2 catalyst.5,6 The wide application of this catalytic system benefits from its reasonable cost and high NO conversion. However, these catalysts still have some disadvantages such as narrow temperature windows of activity (300–400 °C) and poor catalytic activity in low temperature. Although many known SCR catalysts have relatively high NO conversion in a wider temperature range,7,8 their selectivity, stability, and resistance to water and sulfur are not excellent. On the other hand, the production costs of catalysts with the above advantages may be greatly increased, making it difficult to achieve industrial production.For two decades, CeO2 has attracted more attention in the catalytic field due to its excellent ability to store and release oxygen as a result of the efficient change of Ce valence between +3 and +4.9 The earliest application of CeO2 in cleaning flue gases is mainly to deal with automobile exhaust as the three-way catalyst (TWC) in the presence of noble metals such as Rh, Pt and Pd. In order to further expand the application of CeO2, a large number of modification studies have been performed. Using the isovalent metal cations of smaller diameter to prepare solid solution by co-precipitation is one of the most common methods. Studies show that these Ce-based solid solutions have great potential for deNOX.10–14 Thanh Huyen Vuong and co-workers have investigated the relations between structural properties and catalytic performance in VOX/CeXTi1−XO2 and VOX/CeXZr1−XO2 catalysts, respectively.15,16 The results indicates that VOX sites effectively attach to the support surface and the redox properties of the materials governs catalytic activity rather than Lewis and Brønsted surface acidic sites in this catalytic system. All above mentioned researches reveal the importance of oxygen mobility in catalytic reactions. Li et al.17 prepared CeO2–WO3/TiO2 by impregnation method and confirmed that CeO2 is favorable to keep more acid sites and enhanced NO reduction in NH3-SCR reaction compared with V2O5–WO3/TiO2. Li et al.18 prepared a CeO2–MnOX catalyst with a core–shell structure and it exhibits relatively good NO conversion in the presence of SO2 on account of the fact that CeO2 shell prevents the active sites of MnOX from being poisoned.
The SCR catalytic activity of V2O5–WO3(MoO3)/TiO2 mainly stems from the adsorption of NH3 by acidic sites possessed by highly dispersed V species on the TiO2 surface, forming Bronsted-NH3 and further reacting with NO in gas phase.19,20 Differently, the Mars-van Krevelen mechanism of Ce-based solid solution catalyst has been clearly clarified.15,16 We expect that adding this structure to traditional catalysts in another way can bring different effects and further optimize this catalytic system. In this work, the core–shell structure carrier of TiO2 and Ce0.9Zr0.1O2 was prepared by precipitation method, and loaded VOX on the carrier by impregnation method. Combined with the results of XRD, BET, H2-TPR, NH3-TPD, Py-IR, HRTEM and XPS, the reasons for the high activity and thermal stability of the catalyst are explained.
Experimental
Chemical reagents
Ce(NO3)3·6H2O (Analytical Reagent) was purchased from Tianjin Guangfu Fine Chemical Research Institute. Zr(NO3)4·5H2O (Analytical Reagent) was purchased from Tianjin Fuchen Chemical Reagents Factory. Nano titanium dioxide (Analytical Reagent) was purchased from Tianjin Guangfo Materials Co., Ltd. Ammonium metavanadate (Analytical Reagent) was purchased from Tianjin Zhiyuan Chemical Reagent Co., Ltd. Citric acid (Analytical Reagent) was purchased from Tianjin Dongli District Tianda Chemical Reagent Factory.Catalysts preparation
TiO2–Ce0.9Zr0.1O2 support were prepared by precipitation method. 0.0225 M of cerium nitrate (Ce(NO3)3·6H2O) and 0.0025 M of zirconium nitrate (Zr(NO3)4·5H2O) were completely dissolved in an appropriate amount of deionized water, respectively. Then, the resulting solutions were mixed at room temperature for 10 min. 0.025 M TiO2 powder was subsequently suspended in the mixed solution under vigorous stirring and the obtained mixture kept stirring for 1 h at room temperature, then heated to 80 °C to evaporate part of the water. When the mixture was slightly viscous, the ammonia water was added under stirring until pH = 10, and metal ions were precipitated on the surface of TiO2. After the mixture was stirred at room temperature for 1 h, the obtained solution was aged at 70 °C for 5 h in the blast drying oven. Then, the precipitate was filtered and washed with deionized water. Finally, the obtained product was dried at 120 °C for 12 h and subsequently calcined in air at 500 °C for 4 h.VOX/TiO2–Ce0.9Zr0.1O2 (VOX/TiO2, VOX/Ce0.9Zr0.1O2) catalyst with 3 wt% V2O5 loaded was prepared by wet impregnation. 0.195 g of the ammonium metavanadate (NH4VO3) was dissolved in 60 mL citric acid solution (10 wt% C6H8O7·H2O). 4.85 g of the TiO2–Ce0.9Zr0.1O2 (TiO2, Ce0.9Zr0.1O2) powder was suspended in vanadium-containing solution and the obtained mixture kept stirring for 1 h then heated to 100 °C to evaporate the excess water completely. The obtained product was further dried at 120 °C for 12 h and subsequently calcined in air at 400 °C for 3 h.
NH3-SCR activity test
The SCR activity tests were conducted in a fixed-bed quartz reactor with 250 mm length and 8 mm internal diameter 0.3 mL (400 mg) catalyst with 40–60 mesh size were used for all tests. The typical reactant gas contained: 1000 ppm NO, 1000 ppm NH3, 3%O2 and balance Ar. The total gas flow rate was 200 mL min−1 and regulated by mass flow controllers (Sevenstar D08 series Flow Readout Boxed, Beijing Sevenstar Electronics Co., Ltd., Beijing, China), corresponding to the gas hourly space velocity (GHSV) of about 40000 h−1. NO, N2, NH3, NO2 and N2O outlet concentration were analyzed by the ThermoStar Gas Analysis System GSD320 analyzer (Pfeiffer Vacuum GmbH, Berlin, Germany). Activity data was recorded with a temperature gradient of 50 °C in the range of 100 °C −450 °C. The NO conversion (CNO) and N2 selectivity (SN2) are calculated as following equations: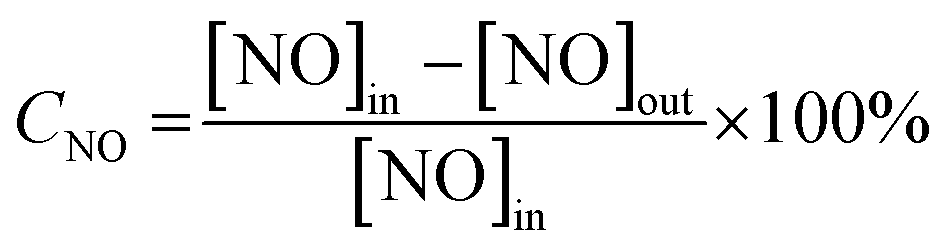
where [NO]in and [NH3]in represent the inlet concentration of NO and NH3. The [NO]out, [NH3]out, [NO2]out and [N2O]out represent the outlet concentration of NO, NH3, NO2 and N2O.
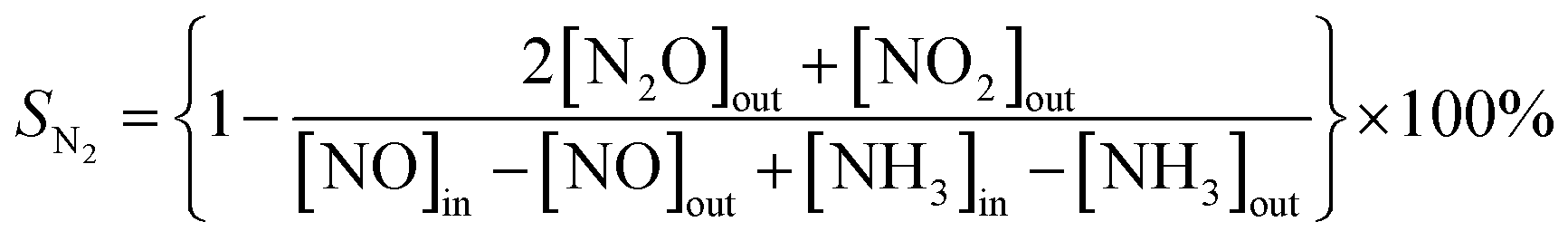
Catalyst characterization
X-ray diffraction (XRD) powder patterns were recorded by a D/MAX-3A Auto X-ray diffractometer (Rigaku Corporation, Tokyo, Japan) with Cu Kα radiation (λ = 1.5418 Å, 40 kV, 40 mA). The diffraction patterns were taken in the 2θ range of 10–80° at a scan speed of 10° min−1.HRTEM were recorded by FEI Talos F200X G2 electron microscope (PHILIPS, Amsterdam, The Netherlands).
The pore structure and specific surface area (SSA) of the catalyst were measured using a SSA-6000 pore and surface area analyzer manufactured by Beijing Builder Electronic Instrument Co., Ltd., and calculated by the Brunauer–Emmett–Teller (BET) and the Barrett–Joyner–Halenda (BJH) method. The sample was pretreated under vacuum at 200 °C for 2 h, the adsorption medium was N2 with He was carrier. The inlet pressure of the system was 0.3 MPa.
In situ Py-IR was performed on a Fourier transform infrared spectrometer (Nicolet 6700, Thermo Fisher Scientific, Waltham, MA, America) equipped with a smart collector and MCT detector cooled by liquid N2, collecting 32 scans with a resolution of 4 cm−1. The catalysts were firstly pretreated in Ar at 250 °C for 40 min, then cooled down to 20 °C. Then, the background spectrum was recorded with the flowing of Ar and subtracted from the sample spectrum. Subsequently, the gaseous pyridine were introduced to the catalyst for 30 min, and then flushed with Ar for 10 min. The spectra were normally collected at temperature ranging from room temperature to 350 °C.
H2-TPR was performed by PCA-1200 chemical adsorption analyzer and 0.05 g sample was pretreated in Ar (30 mL min−1) at 250 °C for 40 min and then heated up to 800 °C at a rate of 10 °C min−1 under 5 vol% H2/Ar.
NH3-TPD was performed by PCA-1200 chemical adsorption analyzer and 0.05 g sample was pretreated in Ar (30 mL min−1) at 250 °C for 40 min and then the NH3 was adsorbed at room temperature for 10 min. It was purged in Ar at 100 °C for 1 h, then cooled down to the room temperature and finally heated to 650 °C at a rate of 10 °C min−1 under Ar.
X-ray photoelectron spectra were obtained with K-Alpha spectrometer (Thermo Fisher Scientific, Waltham, MA, America) using Al Kα (1486.7 eV) radiation as the excitation source with a precision of ±0.3 eV. All binding energies were referenced to the C 1s line at 284.8 eV.
Results and discussion
The HRTEM analysis
High-resolution transmission electron microscopy (HRTEM) was used to characterize the VOX/TiO2–Ce0.9Zr0.1O2 catalysts (Fig. 1). The catalyst was well dispersed and a relatively obvious core–shell structure was observed (Fig. 1a and b). The 3.5 nm Ce0.9Zr0.1O2 shell is coated on the outside of the TiO2 core the magnified portion of the core–shell structure in Fig. 1c was shown in Fig. 1d. The lattice fringes of VOX/TiO2–Ce0.9Zr0.1O2 display inter planar spacings of 0.312 nm and 0.189 nm in the particle, which match well with those of the (111) plane of cubic Ce0.9Zr0.1O2 (CeO2) and (200) plane of anatase TiO2, respectively.21 It needs to note that no lattice fringes attributed to VOX were observed. In order to verify the distribution of the elements, the catalyst was subjected to mapping (Fig. 1e), and the distribution of the V element is shown in Fig. 1f. The V element is evenly distributed on the support surface, also indicating its existence. | ||
| Fig. 1 HRTEM images (a–d) and V element mapping of VOX/TiO2–Ce0.9Zr0.1O2 (e and f). | ||
XRD and BET analysis
Fig. 2 is the XRD comparison of the support and the VOX supported catalyst. The diffraction pattern for catalyst containing TiO2 has six peaks at 25.28°, 37.80°, 48.05°, 53.89°, 55.06° and 62.69° corresponding to (101), (004), (200), (105), (211), and (204) crystal faces of anatase TiO2 (PDF#21-1272), respectively.22 The diffraction peak at 28.55°, 33.08°, 47.48°, and 56.34° of the catalyst containing Ce0.9Zr0.1O2 corresponds to (111), (200), (220), (311) of cubic CeO2 (PDF#43-1002), respectively.23,24 It is worth noting that in the XRD spectrum, no diffraction peaks attributed to ZrO2 were observed, which indicates that the size of Zr ion (0.084 nm) is significantly smaller than that of Ce ion (0.097 nm), the addition of Zr4+ does not destroy the lattice structure of CeO2 itself meaning that the formation of a CeO2–ZrO2 solid solution.25,26 This is consistent with the result that the lattice fringes belonging to the (111) crystal plane having a crystal face spacing of 0.312 nm in CeO2 were observed in HRTEM.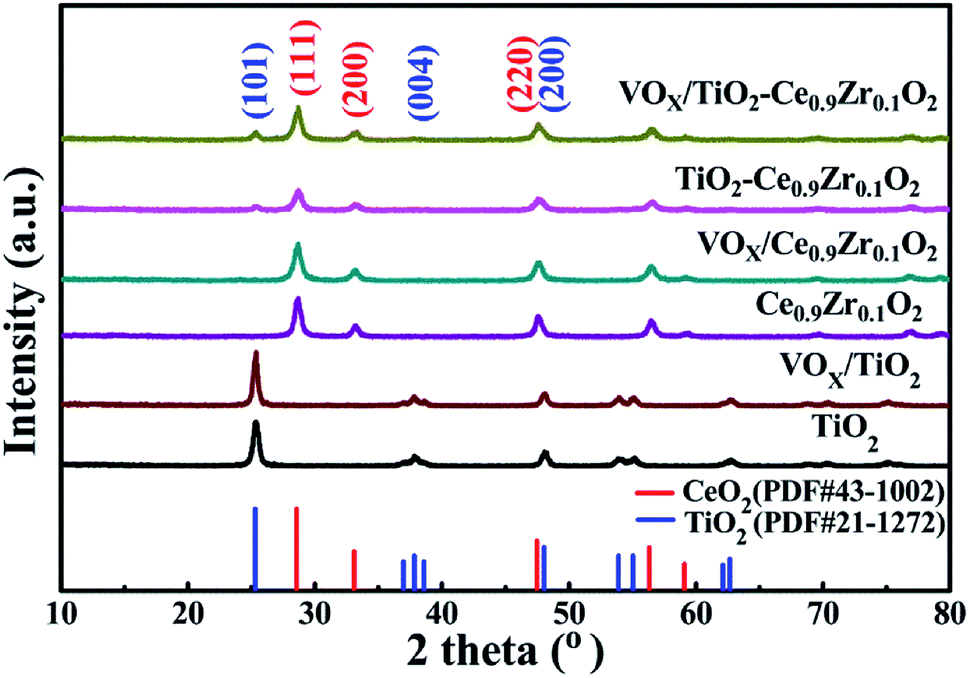 | ||
| Fig. 2 XRD spectrum of VOX supported catalysts and carriers. | ||
For TiO2–Ce0.9Zr0.1O2 and VOX/TiO2–Ce0.9Zr0.1O2, although TiO2 has a larger specific surface area (SSA), the diffraction peaks attributed to Ce0.9Zr0.1O2 and TiO2 with a molar ratio of 1:1 indicate, that Ce0.9Zr0.1O2 is not simply dispersed on TiO2 surface.27 Instead, it crystallizes on the surface of TiO2 core to form a shell structure or forms incomplete coating on the surface of TiO2 core. Interestingly, due to the load of VOX, the diffraction peak intensity of the VOX/TiO2–Ce0.9Zr0.1O2 catalyst is enhanced, and the peak shape becomes sharper, which may be that the loading of VOX weakens the interaction between Ce0.9Zr0.1O2 and TiO2, and increases the degree of crystallization of the TiO2–Ce0.9Zr0.1O2 core–shell structure. However, compared with Ce0.9Zr0.1O2 and VOX/Ce0.9Zr0.1O2, the loaded VOX has no significant effect on Ce0.9Zr0.1O2. From the partial comparison of XRD, we speculate that for TiO2–Ce0.9Zr0.1O2, the loaded VOX complements the oxygen vacancies in Ce0.9Zr0.1O2, and the cell of Ce0.9Zr0.1O2 is more complete, and the crystallinity of the sample is higher.28,29 For VOX/Ce0.9Zr0.1O2, the oxygen vacancies cannot be replenished due to the lack of interaction between TiO2 and Ce0.9Zr0.1O2, and the crystallinity and unit cell distortion are not significantly improved. For the VOX-loaded catalyst, the VOX loading mass percentage is small, the diffraction peak attributed to VOX cannot be observed, indicating that the VOX is highly dispersed on the core–shell structure carrier or in an amorphous form.30,31 This is consistent with the fact that the lattice fringe attributed to VOX was not observed in the HRTEM of VOX/TiO2–Ce0.9Zr0.1O2.
It is worth mentioned that TiO2 has the largest specific surface area in all samples (Table 1), the loading of VOX or the formation of core–shell structure with Ce0.9Zr0.1O2 will cause the specific surface area to decrease. However, for Ce0.9Zr0.1O2, the loading of VOX and the formation of a core–shell structure with TiO2 will increase the specific surface area. This results in a core–shell structure having a specific surface area between Ce0.9Zr0.1O2 and TiO2. Combining the pore volume (PV) and the pore size (PS) information, we can speculate that VOX and Ce0.9Zr0.1O2 enter the pores of TiO2 during the formation process of VOX/TiO2 and TiO2–Ce0.9Zr0.1O2, resulting in a decrease in specific surface area, pore volume and pore size. The cerium–zirconium solid solution constituting the core–shell structure is different from pure Ce0.9Zr0.1O2 in terms of specific surface, pore volume and pore size. It is a shell formed by spreading on the surface of TiO2. The specific surface area is relatively large compared with the simple Ce0.9Zr0.1O2, and this can provide more acidic sites. Due to the interaction between TiO2 and Ce0.9Zr0.1O2, the pores formed by Ce0.9Zr0.1O2 also change. When VOX is loaded, it is arranged on the surface of the pure Ce0.9Zr0.1O2 sample, and some special pores may be formed to increase the specific surface area. For TiO2–Ce0.9Zr0.1O2, VOX is not only arranged on the surface, but also may enter certain special channels. Therefore, the specific surface area, pore volume and pore size are reduced.
| Samples | SSA (cm2 g−1) | PV (cm3 g−1) | PS (nm) |
|---|---|---|---|
| TiO2 | 114 | 0.42 | 9 |
| VOX/TiO2 | 72 | 0.04 | 1 |
| Ce0.9Zr0.1O2 | 56 | 0.03 | 1 |
| VOX/Ce0.9Zr0.1O2 | 57 | 0.26 | 9 |
| TiO2–Ce0.9Zr0.1O2 | 74 | 0.04 | 1 |
| VOX/TiO2–Ce0.9Zr0.1O2 | 65 | 0.03 | 1 |
H2-TPR analysis
The redox performance of several catalysts was analyzed by H2-TPR technique, and the spectrums are illustrated in Fig. 3. According to Chen's report, the reduction ability of the anatase TiO2 phase is weak. There is no obvious reduction peak in the range of 50–800 °C, and the reduction performance of V2O5 is strong, and there is obvious reduction peak in the range of 50–800 °C.32 All samples contained only one reduction peak at 400–650 °C. The reduction peak of VOX/TiO2 at the temperature of 482 °C corresponds to the transition of V5+ to V4+ and V4+ to V3+, while in other samples, no reduction peak is observed at the corresponding position.30 The reduction temperature of the cerium zirconium solid solution is 537 °C, which is caused by the conversion of Ce4+ to Ce3+.33 For the VOX-loaded cerium–zirconium solid solution, the reduction temperature is slightly higher, and the peak temperature is at 541 °C. It illustrates that there is a certain interaction between VOX and Ce0.9Zr0.1O2 inhibiting the conversion of V5+ to V4+ and a small amount of V4+ to V3+, result in the shift to the higher temperature of the reduction peak. The reduction peak area is increased compared with Ce0.9Zr0.1O2, which can be attributed to the effect of superposition of V5+, V4+ and Ce4+ reduction peaks.34 The reduction peak of TiO2–Ce0.9Zr0.1O2 is higher at 544 °C than that of Ce0.9Zr0.1O2 alone, indicating the interaction between Ce0.9Zr0.1O2 and TiO2. While the reduction peak of the catalyst supported by TiO2–Ce0.9Zr0.1O2 is at 566 °C, which is higher than that of other catalysts and carriers. Similarly, the presence of the Ce0.9Zr0.1O2 coating interacts strongly with the surface VOX of the support, inhibiting the reduction process of VOX. The larger reduction peak is the result of the superposition of Ce4+ to Ce3+, V5+ to V4+ and V4+ to V3+. | ||
| Fig. 3 H2-TPR curve of catalysts and carriers. | ||
The hydrogen consumption of the samples are listed in Table 2. Since only Ce4+ on the surface is reduced, Ce of the internal bulk phase cannot be reduced in this temperature range of 400–650 °C, so the hydrogen consumption of Ce0.9Zr0.1O2 is the least. For TiO2–Ce0.9Zr0.1O2, the specific surface area of Ce0.9Zr0.1O2 is increased due to the formation of the core–shell structure, and more Ce is exposed and reduced, and the hydrogen consumption is significantly increased. TiO2 is not reduced in the range of 50–800 °C, and all the hydrogen consumption of VOX/TiO2 belongs to the reduction of VOX. By comparing the hydrogen consumption between the VOX-loaded catalyst and the support, we can confirm our previous inference about the superposition of the reduction peaks.
| Samples | Hydrogen consumption (μmol g−1) |
|---|---|
| VOX/TiO2–Ce0.9Zr0.1O2 | 163.5 |
| TiO2–Ce0.9Zr0.1O2 | 87.8 |
| VOX/Ce0.9Zr0.1O2 | 136.5 |
| Ce0.9Zr0.1O2 | 50.0 |
| VOX/TiO2 | 96.2 |
NH3-TPD and Py-IR Analysis
In order to compare the number of surface acidic sites of several samples, we analyzed them with NH3-TPD and showed the test results in Fig. 4. The desorption peak of NH3 adsorbed physically on the surface of the catalyst is before 100 °C, and the desorption range at 100–200 °C, 200–400 °C and 400–500 °C correspond to weak acidic sites, medium strong acidic sites and strong acidic sites on the catalyst surface, respectively. TiO2 and VOX/TiO2 mainly provide weak acid sites and a small amount of medium acid sites, Ce0.9Zr0.1O2, VOX/Ce0.9Zr0.1O2 and TiO2–Ce0.9Zr0.1O2 can provide a small amount of strong acidic sites. The VOX/TiO2–Ce0.9Zr0.1O2 catalyst has desorption peaks throughout the temperature range, indicating that the catalyst can provide three types of acidic sites.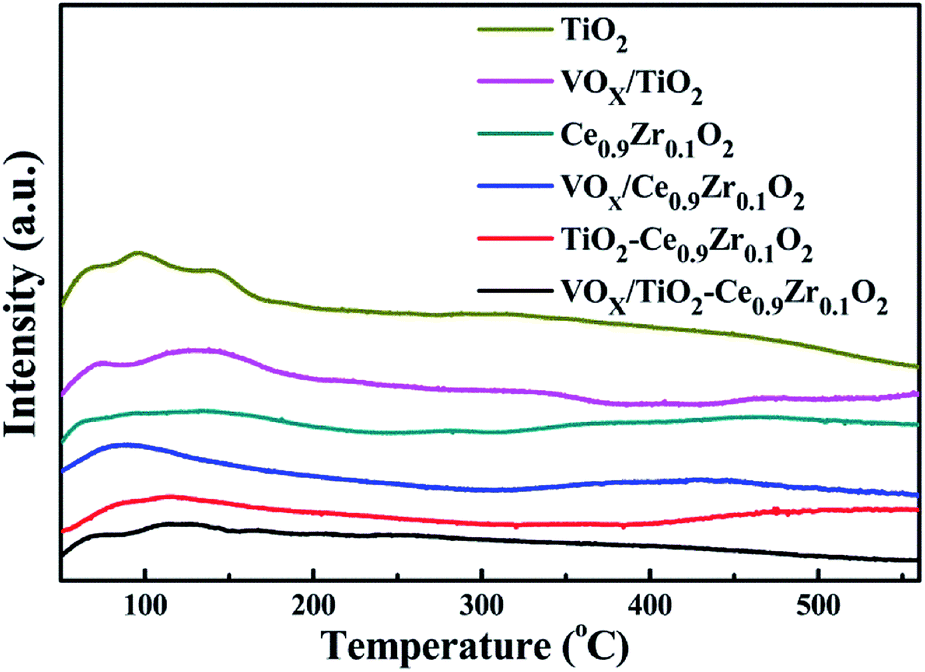 | ||
| Fig. 4 NH3-TPD curves of different samples. | ||
VOX/TiO2–Ce0.9Zr0.1O2 has the largest amount of NH3 desorption (Table 3), indicating that the sample has the best ability to adsorb NH3. This region can be thought of as VOX and TiO2–Ce0.9Zr0.1O2 together to provide weak, medium and strong acidic sites. The catalyst has a prominent ability to adsorb NH3 and has more medium and strong acidic sites, thus the catalyst can exhibit the best catalytic activity and thermal stability.
| Samples | B/L | Amount of NH3 desorption (μmol g−1) |
|---|---|---|
| VOX/TiO2–Ce0.9Zr0.1O2 | 0.53 | 462.4 |
| TiO2–Ce0.9Zr0.1O2 | 0.86 | 244.5 |
| VOX/Ce0.9Zr0.1O2 | 0.47 | 122.7 |
| Ce0.9Zr0.1O2 | 0.88 | 148.3 |
| VOX/TiO2 | 0.76 | 401.3 |
| TiO2 | — | 350.4 |
Acid site type analysis was carried out at different temperatures for different catalysts and carriers. The Py-IR spectrum is shown in Fig. 5. The wave number corresponds to Lewis acid at 1437 cm−1 and 1597 cm−1, and 1580 cm−1 and 1633 cm−1 correspond to Brønsted acid and the ratio of Brønsted acid to Lewis acid (B/L) is listed in Table 3.35–37 According to studies by Li and Liu et al., anatase TiO2 can only provide L acidic sites.38,39 Compared with other samples containing TiO2, the peak area of TiO2 is smaller and the wave number is slightly higher. It can be considered that the peak of VOX/TiO2 is a result of superposition of VOX and TiO2 peaks. Based on the special structure formed by Ce0.9Zr0.1O2 and TiO2, the TiO2 core is mainly encapsulated in the Ce0.9Zr0.1O2 shell. Although the medium acidic sites and strong acidic sites on the surface of Ce0.9Zr0.1O2 are increased, it does not provide too much weak acidic sites (Fig. 4). Therefore, the properties of providing weak acidic sites are not apparent in the samples of the core–shell structure. The largest B/L ratio is 0.88 that belongs to Ce0.9Zr0.1O2. Ce0.9Zr0.1O2 forms a core–shell structure with TiO2, it also has a large B/L ratio. However, due to the loading of VOX, the ratio of B/L is significantly reduced, indicating that the loading of VOX can provide more L acid sites. VOX/TiO2–Ce0.9Zr0.1O2 catalyst has the largest acid area. There is interaction between the core–shell structure and the active component, which increases the number of acidic sites on the surface and is more conducive to the adsorption of NH3 resulting in better catalytic activity of the catalyst.
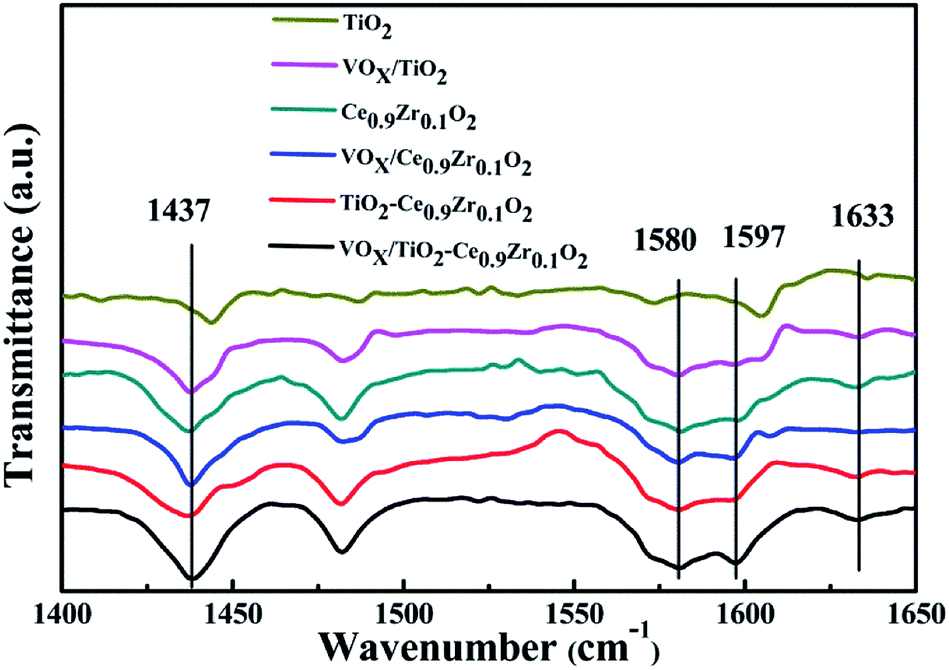 | ||
| Fig. 5 Py-IR spectra of different samples. | ||
In order to specifically explore the acid site composition of the surface of VOX/TiO2–Ce0.9Zr0.1O2 catalyst, we performed Py-IR desorption analysis at different temperatures, and the spectrum is shown in Fig. 6. With the increase of the temperature desorption of weakly acidic sites combined with pyridine, and the characteristic peak area of acidic sites is decreasing. Due to the temperature rise, the increase of the vibration frequency of the in-plane ring deformation of pyridine combined with the medium acid sites and the strong acidic sites causes the characteristic peak to continuously move toward the high wavenumber. When the temperature reaches 150 °C, the adsorption peak attributed to the B acidic site disappears completely, and when the temperature reaches 300 °C, the characteristic adsorption peak of the L acidic site disappears, indicating that the L acidic site is the main role in the reaction process. For medium acid and strong acid sites, the temperature at which they desorb pyridine is relatively high. The presence of a medium acid and a strong acid site is a major factor in the relatively stable activity of the catalyst at higher temperatures.
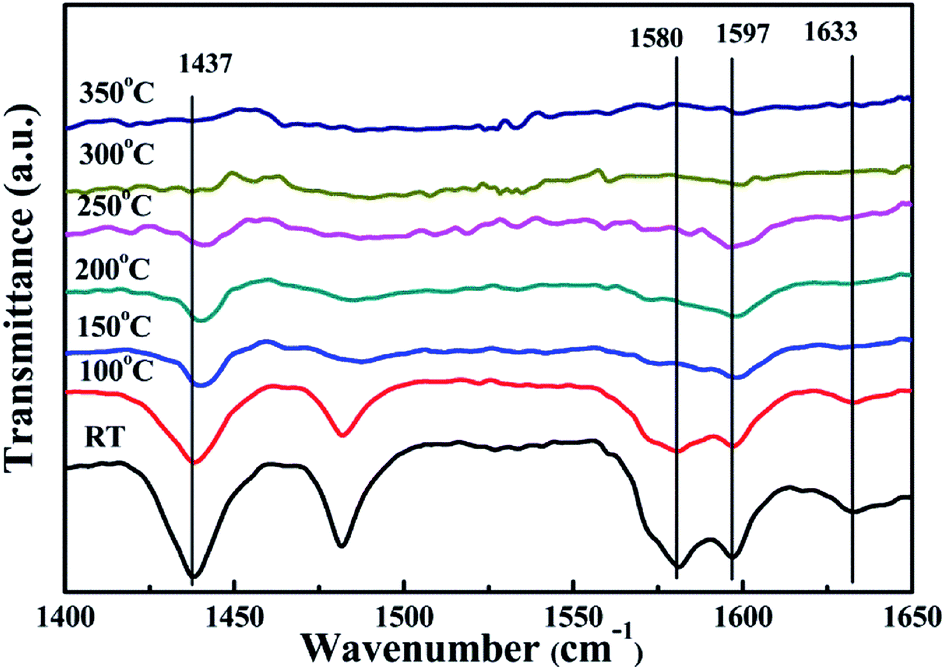 | ||
| Fig. 6 Py-IR spectra of VOX/TiO2–Ce0.9Zr0.1O2 at different temperatures. | ||
XPS analysis
To investigate the chemical states of the elements in the near surface region of the catalyst samples, X-ray photoelectron spectroscopy (XPS) analysis was performed. All binding energies were referenced to the C 1s line at 284.8 eV. Table 4 is the calculated value of bulk compositions and the surface atomic ratios of different samples, for samples with a core–shell structure, the shell structure is relatively thin, and/or the incomplete coating, while the photoelectrons excite the electrons of the Ce0.9Zr0.1O2 shell structure, some of the electrons of Ti are also excited, the ratio of Ce and Zr to Ti is close to 1.5:1. It is quite different from the calculated ratio in the core–shell structure, indicating that the solid solution is not simply mixed with TiO2. The Zr atom has a relatively stable atomic ratio, which is consistent with the study by Yao et al.40 Although we could not confirm the existence of V element in XRD and HRTEM, the existence of V atom was found in XPS detection, which indicates that V element is supported on the carrier. It should be noted that for VOX-loaded catalysts, the atomic ratio of the surface V element is much higher than that of the bulk composition, because the V element is mainly supported on the surface of the carrier, and the high content of the V element on the surface is favorable for the NH3-SCR reaction of NO.
| Catalysts | Calculated value of bulk compositions (%) | Surface atomic ratio (%) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| V | Ce | Ti | Zr | V | O | Ce | Ti | Zr | |
| VOX/TiO2–Ce0.9Zr0.1O2 | 4.03 | 43.19 | 47.98 | 4.80 | 21.00 | 59.12 | 8.87 | 8.07 | 2.94 |
| TiO2–Ce0.9Zr0.1O2 | — | 45.00 | 50.00 | 5.00 | — | 76.01 | 11.56 | 9.13 | 3.30 |
| VOX/Ce0.9Zr0.1O2 | 5.38 | 85.16 | — | 9.46 | 20.95 | 63.21 | 12.68 | — | 3.16 |
| Ce0.9Zr0.1O2 | — | 90.00 | — | 10.00 | — | 78.49 | 17.92 | — | 3.59 |
| VOX/TiO2 | 2.65 | — | 97.35 | — | 21.21 | 56.02 | — | 22.77 | — |
| TiO2 | — | — | 100 | — | — | 70.79 | – | 29.21 | — |
The spectra of V 2p, O 1s, Ce 3d, Ti 2p and Zr 3d are given in Fig. 7, and the relative contents of different valence elements are listed in Table 5. Fig. 7a shows the V 2p photoelectron spectroscopy of different catalyst surfaces. The vanadium element is mainly present on the surface of the catalyst in the valence state of V5+ (2p3/2) and V4+ (2p3/2), and the corresponding binding energy is 517.3 eV and 516.3 eV, respectively.41 The binding energy of V3+ (2p3/2) is 515.5 eV.42,43 The content of V3+ of Ce0.9Zr0.1O2 and TiO2 as carrier is small, while the content of V3+ in TiO2–Ce0.9Zr0.1O2 carrier is increased, and the content of V4+ is increased at the same time. Combined with the results of the H2-TPR test, we speculate that this result is attributed to the interaction between Ce0.9Zr0.1O2 and TiO2, which increases the oxygen vacancies in the carrier, and the V element is supported on the surface of the core–shell structure carrier in a lower valence state. Due to the higher proportion of the V element on the surface and the presence of more low valence states V element, the catalyst has a higher catalytic activity.29,31
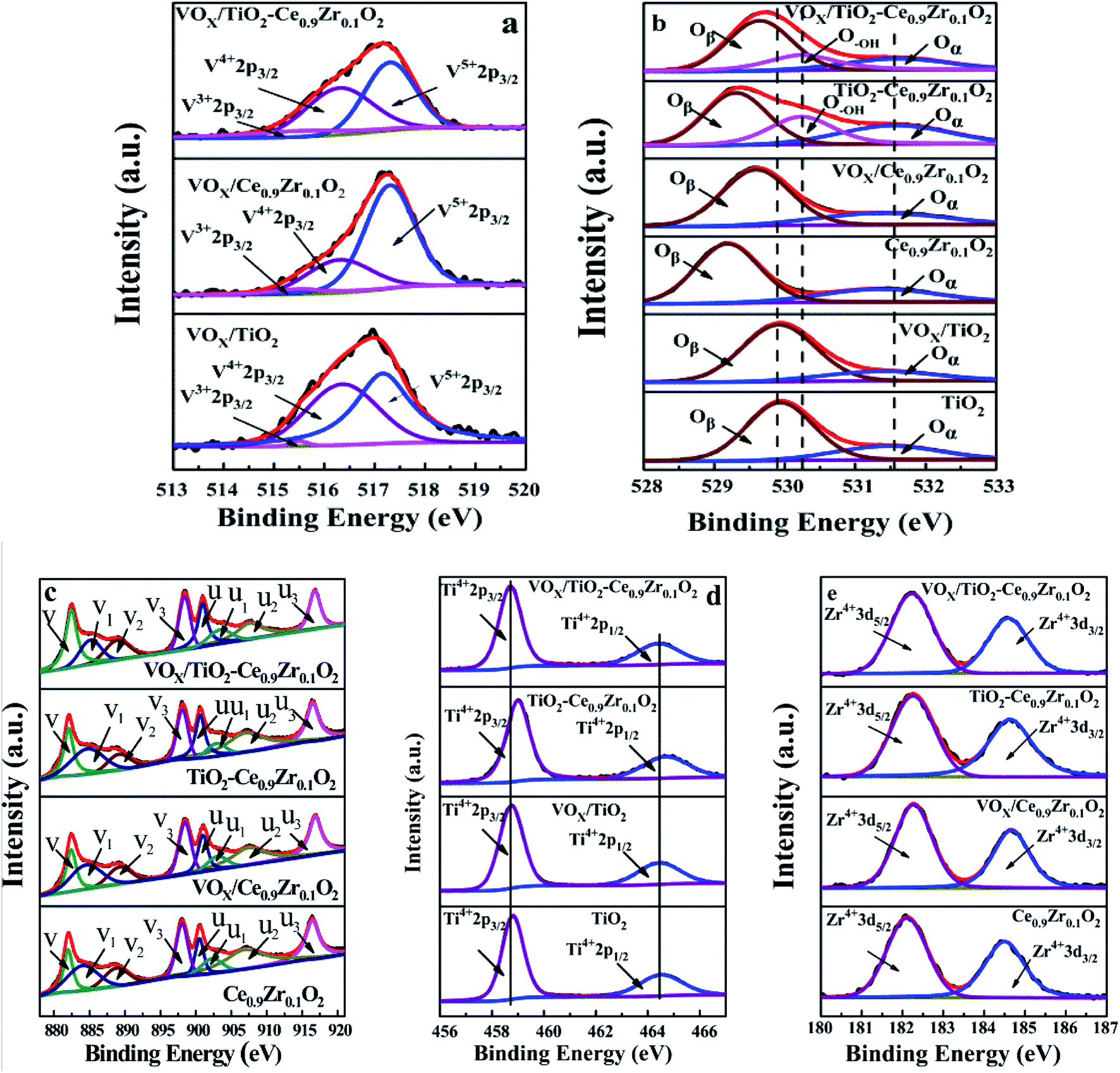 | ||
| Fig. 7 XPS spectra of catalysts ((a) V 2p, (b) O 1s, (c) Ce 3d, (d) Ti 2p, (e) Zr 3d). | ||
| Catalysts | V 2p | O 1s | Ce 3d | ||||
|---|---|---|---|---|---|---|---|
| V3+ + V4+ | V5+ | Oα | Oβ | O–OH | Ce3+ | Ce4+ | |
| VOX/TiO2–Ce0.9Zr0.1O2 | 54.61 | 45.39 | 20.47 | 56.09 | 23.44 | 18.25 | 81.75 |
| TiO2–Ce0.9Zr0.1O2 | — | — | 27.24 | 45.13 | 27.63 | 21.18 | 78.82 |
| VOX/Ce0.9Zr0.1O2 | 31.28 | 68.72 | 27.47 | 72.53 | — | 21.72 | 78.28 |
| Ce0.9Zr0.1O2 | — | — | 33.52 | 66.48 | — | 21.46 | 78.54 |
| VOX/TiO2 | 43.69 | 56.31 | 20.49 | 79.51 | — | — | — |
| TiO2 | — | — | 26.32 | 73.68 | — | — | — |
The results of XPS spectrum fitting of O 1s are shown in Fig. 7b, where the peak corresponding to lattice oxygen (O2−) at 529.7 eV is expressed as Oβ, and the binding energy is 531.4 eV corresponding to chemisorbed oxygen (O22−, O−), expressed as Oα.44 For different carriers, the loading of VOX will reduce the content of Oα, which may be because the loading of VOX occupies the position of the support surface where the oxygen can be chemically adsorbed, resulting in a decrease in the Oα content of the catalyst surface after the loading. It is worth noted that Ce0.9Zr0.1O2 has the lowest Oβ binding energy, indicating that it has the best O migration ability. When VOX is loaded or forms a core–shell structure with TiO2, it will affect the migration ability of Oβ. For TiO2–Ce0.9Zr0.1O2 and VOX/TiO2–Ce0.9Zr0.1O2, a peak of –OH is generated at 530.2 eV, which may be formed by the adsorption of O2 and H2O by oxygen vacancies.45–47 Due to the loading of VOX, a small amount of oxygen vacancies is supplemented by VOX, and the peak belonging to the –OH and Oα were reduced in VOX/TiO2–Ce0.9Zr0.1O2 catalyst.
Fig. 7c shows the XPS spectrum of Ce 3d. According to previous studies,18,48 the XPS spectrum of Ce 3d can be fitted to 8 peaks: where ‘v’ represents the 3d5/2 orbital and ‘u’ represents the 3d3/2 orbital. The peaks of u1 and v1 can be assigned to Ce3+, and the other peaks are related to Ce4+. The presence of Ce3+ causes charge imbalance and forms oxygen vacancies, which is favorable for the formation of surface chemisorbed oxygen.
By comparing the existence of Ce in different samples, it can be found that the content of Ce3+ in VOX/TiO2–Ce0.9Zr0.1O2 catalyst is reduced, and expect for v2, the binding energies of Ce 3d in VOX/TiO2–Ce0.9Zr0.1O2 are higher than those of TiO2–Ce0.9Zr0.1O2. Combined with the change in the valence state of the V element, we speculate that it may be that the loading VOX weaken the interaction between Ce0.9Zr0.1O2 and TiO2 and supplements the oxygen vacancies, resulting in a decrease in the content of Ce3+, but an increase in the content of V3+ and V4+.49
Fig. 7d is the XPS spectrum of Ti 2p, where 464.4 eV is the binding energy of Ti4+ 2p1/2 and 458.7 eV is the binding energy of Ti4+ 2p3/2. For TiO2–Ce0.9Zr0.1O2, the binding energy of Ti is shifted. The reason for this phenomenon is related to the interaction between Ce0.9Zr0.1O2 and TiO2, which may cause lattice distortion of TiO2 (it is reflected in the XRD spectrum).41 It can provide more oxygen vacancies. Due to the load of VOX, while supplementing the oxygen vacancies, the existence of higher binding energy Ti is weakened as well. Therefore, the higher binding energy of Ti in VOX/TiO2–Ce0.9Zr0.1O2 catalyst is not obvious. However, the loaded VOX can provide more active sites.
For the XPS of Zr (Fig. 7e), Zr mainly exists as +4 valence ion, and the addition of Zr4+ is beneficial to the generation of oxygen vacancies. The increase of oxygen vacancies has a great influence on the thermal stability of the catalyst carrier. The strong interaction between the TiO2–Ce0.9Zr0.1O2 support with core–shell structure increases the high temperature stability of the composite supported catalyst, which also explains the higher activity of the catalyst at higher temperatures.50
Combined with the results of the above characterization, we can confirm that after the preparation of TiO2–Ce0.9Zr0.1O2 core–shell structure, the electrons in Ce–O–Ti shift to Ce due to the interaction between TiO2 and Ce0.9Zr0.1O2.49,51 The binding energy of Ti4+ becomes higher and the electron density around Ce increases. The electron cloud density of Ce increase, and the migration ability of O in Ce0.9Zr0.1O2 is enhanced, which is beneficial to the generation of oxygen vacancies. The content of Ce3+ and chemisorbed oxygen indirectly reflects the amount of oxygen vacancies.52 It is generally believed that there are three types of oxygen vacancies (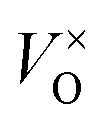 ,
,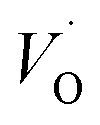 and
and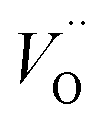 ), which are inseparable from the reactions (1)–(3).45,53–55 Oxygen vacancies adsorb oxygen and dissociate O2 into oxygen atoms. Since oxygen atoms have high activity, they can adsorb substances such as water to form –OH. Due to the presence of oxygen vacancies, Ce can be converted from the +4 valence to the +3 valence (reaction (4)).
), which are inseparable from the reactions (1)–(3).45,53–55 Oxygen vacancies adsorb oxygen and dissociate O2 into oxygen atoms. Since oxygen atoms have high activity, they can adsorb substances such as water to form –OH. Due to the presence of oxygen vacancies, Ce can be converted from the +4 valence to the +3 valence (reaction (4)).
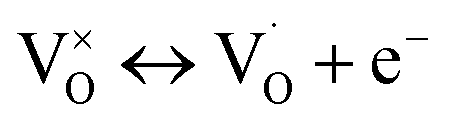 | (1) |
 | (2) |
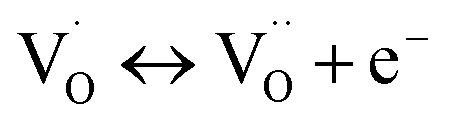 | (3) |
| Ce4+ + e− ↔ Ce3+ | (4) |
When VOX is loaded, an interaction occurs between the active component and the carrier, electron transfer occurs between Ce3+ and VOX, and oxygen vacancies are replenished, and the following reactions may occur during the preparation process (reactions (5)–(8)):
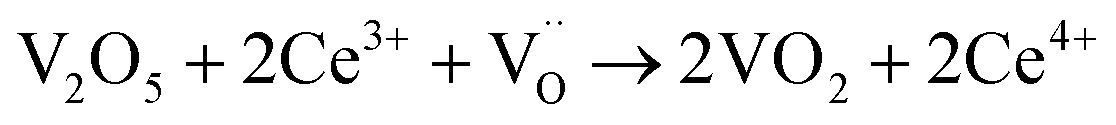 | (5) |
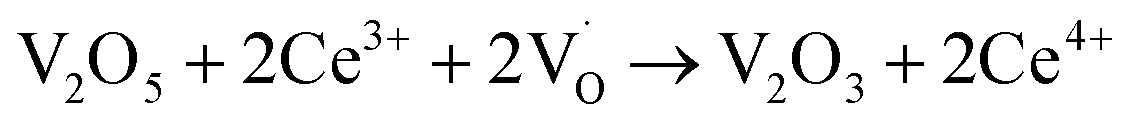 | (6) |
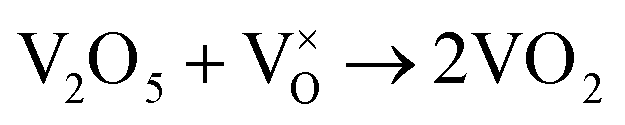 | (7) |
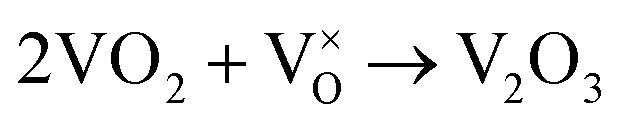 | (8) |
However, this reaction is difficult to accomplish in VOX/Ce0.9Zr0.1O2 catalyst. The oxygen vacancies of Ce0.9Zr0.1O2 coated on the surface of TiO2 were supplemented. The intensity of the diffraction peak in the XRD spectrum of the catalyst is enhanced, and the peak shape is sharper the presence of more low-valence V elements requires higher temperatures due to the increased number of V4+ to V3+ conversions, resulting in a shift in the peak temperature of the H2-TPR (Fig. 3). In summary, the increased V4+ and V3+ content in VOX improves catalytic activity. The interaction between the support and the active component is responsible for the better thermal stability of the catalyst.
Catalyst activity analysis
Fig. 8a is a graph showing the activity of solid solution of different cerium–zirconium ratios. Ce0.9Zr0.1O2 exhibits better catalytic activity, while the catalytic activity of Ce0.1Zr0.9O2 is the worst among all solid solutions at the range of 100–400 °C, although it is better than others at 450 °C. The above results indicate that proper zirconium doping is beneficial to improve the activity and increase the thermal stability of the catalysts. Based on the activity of the cerium–zirconium solid solution catalyst, we selected Ce0.9Zr0.1O2 as the shell structure component to prepare core–shell carrier.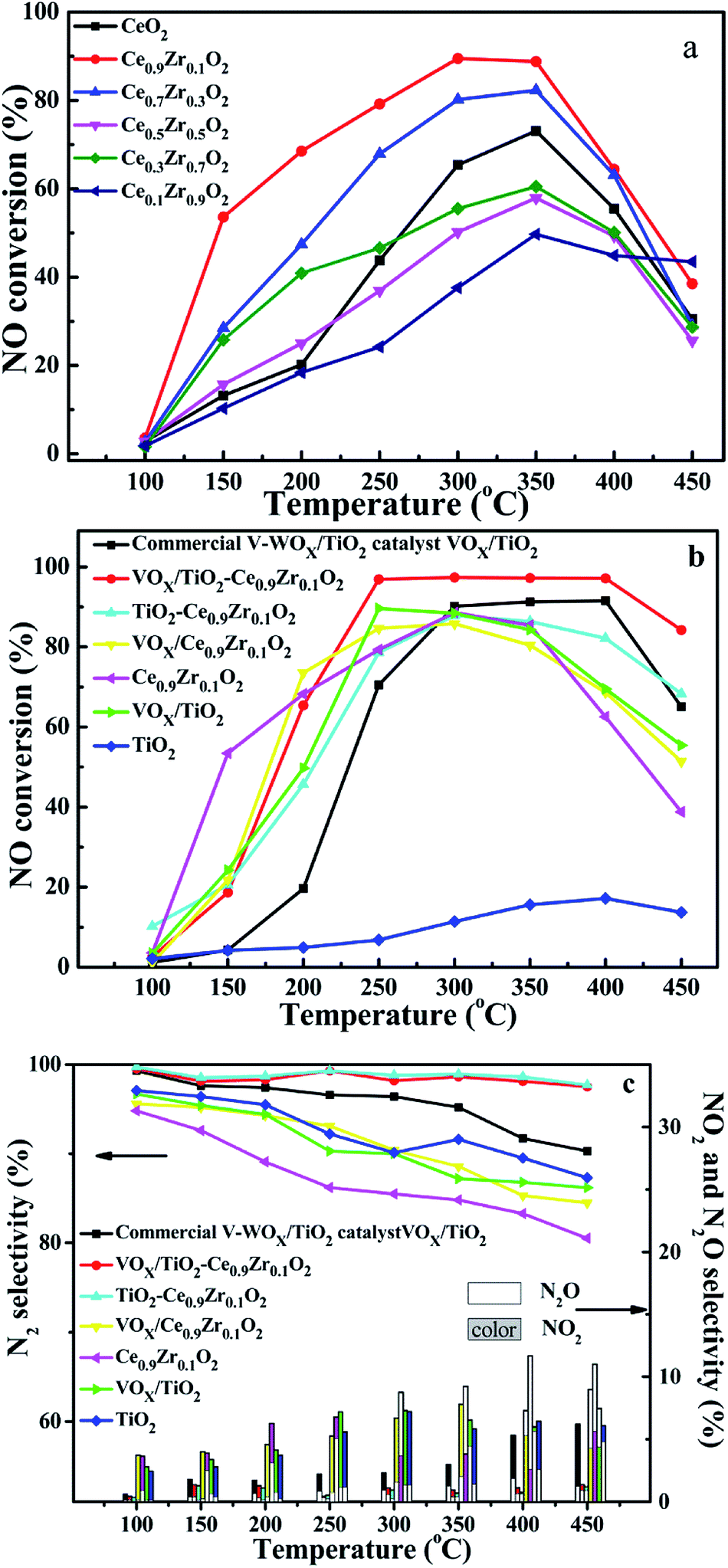 | ||
| Fig. 8 NO conversion curve of CeXZr1−XO2 (a), NO conversion and selectivity of catalysts and carriers (b and c). | ||
Fig. 8b and c show NO conversion curves and selectivity curves for different VOX supported catalysts and their supports. Different colors are used to represent different samples, the line chart represents N2 selectivity, and the histogram shows the selectivity of N2O and NO2 in Fig. 8c. Under the condition of GSHV = 40000 h−1, Ce0.9Zr0.1O2 and VOX/Ce0.9Zr0.1O2 showed higher activity before 250 °C, which may be because the oxygen vacancies and the redox of Ce3+ are the main catalytic sites at low temperature. The VOX site is effectively attached to the surface of the support and the redox properties of the catalysts determine the catalytic activity, rather than the Lewis and Brønsted surface acidic sites in the catalytic system.15,16 After 250 °C, VOX/TiO2–Ce0.9Zr0.1O2 showed excellent catalytic activity and maintained excellent selectivity and thermal stability with NO conversion rate > 97%, N2 selectivity > 97.5% and temperature window is 250–400 °C, which is inseparable from the existence of low-valence V and medium – strong acidic sites. The former is beneficial to increase the activity of the catalyst, and the latter is advantageous for improving the thermal stability of the catalyst. Compared with commercial catalysts, VOX/TiO2–Ce0.9Zr0.1O2 has the higher NO conversion and N2 selectivity, wider temperature window and better thermal stability.31 It is noteworthy that TiO2–Ce0.9Zr0.1O2 also exhibits a high N2 selectivity, although its catalytic properties is not particularly excellent compared to the commercial catalysts and VOX/TiO2–Ce0.9Zr0.1. It indicates that the higher selectivity of N2 is derived from the TiO2–Ce0.9Zr0.1O2 core–shell structure carrier.
Conclusion
In summary, we prepared a TiO2–Ce0.9Zr0.1O2 support with core–shell structure by precipitation method, which may not be completely coated. Owing to the interaction between TiO2 and Ce0.9Zr0.1O2, the carrier has lower lattice oxygen content and a large number of active oxygen vacancies that can adsorb O2 and H2O in the air, which is the reason for the formation of –OH on the surface of the carrier. Due to the presence of Ce0.9Zr0.1O2 coating layer, VOX/TiO2–Ce0.9Zr0.1O2 catalyst exhibits better catalytic properties. After loading VOX, the decrease of Ce3+ and the oxygen vacancies content in the catalyst is due to the supplement of oxygen vacancies and the electron transfer between high valence state V and Ce3+. The presence of the low-valent V is more favourable for the NH3-SCR reaction. At the same time, the VOX/TiO2–Ce0.9Zr0.1O2 catalyst contains more acidic sites, especially medium-strong acid sites. It is beneficial to the adsorption of the reaction gas NH3 on the catalyst surface and increased thermal stability. Compared with the commercial catalysts currently used on the market, both conversion rate and selectivity are superior, as well as better thermal stability and a wider temperature window, which provides ideas for the development of more superior catalysts in the future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Key Research and Development Plan (Grant No. 2017YFE0116200). We are grateful to the teachers of the Harbin Engineering University Analytical Testing Center for their help in testing.References
- X. Liu, X. Wu, T. Xu, W. Duan, Z. Si and R. Ran, Chin. J. Catal., 2016, 37(8), 1340–1346 CrossRef CAS.
- X. T. Zhao, L. Mao and G. J. Dong, Catalysts, 2018, 8(2), 76–89 CrossRef.
- F. Lónyi, H. E. Solt, Z. Pászti and J. Valyon, Appl. Catal., B, 2014, 150–151(18), 218–229 CrossRef.
- Z. Ma, X. Wu, Z. Si, W. Duan, M. Jing and T. Xu, Appl. Catal., B, 2015, 179, 380–394 CrossRef CAS.
- B. X. Shen, F. M. Wang, B. Zhao, Y. W. Li and Y. Y. Wang, J. Ind. Eng. Chem., 2016, 33, 262–269 CrossRef CAS.
- C. E. Quincoces, A. K. D. Figueiredo, A. Lavat and G. G. María, React. Kinet., Mech. Catal., 2000, 71(2), 253–262 CrossRef CAS.
- N. J. Fang, J. X. Guo, S. Shu, H. D. Luo, Y. H. Chu and J. J. Li, Chem. Eng. J., 2017, 325(1), 114–123 CrossRef CAS.
- S. Prodinger, M. A. Derewinski, Y. L. Wang, N. M. Washton, E. D. Walter, J. Szanyi, F. Gao, Y. Wang and C. H. F. Peden, Appl. Catal., B, 2017, 201, 461–469 CrossRef CAS.
- X. Gao, Y. Jiang, Y. C. Fu, Y. Zhong, Z. Y. Luo and K. F. Cen, Catal. Commun., 2010, 11(5), 465–469 CrossRef CAS.
- R. T. Guo, Y. Zhou, W. G. Pan, J. N. Hong, W. L. Zhen, Q. Jin, C. G. Ding and S. Y. Guo, J. Ind. Eng. Chem., 2013, 19(6), 2022–2025 CrossRef CAS.
- T. H. Vuong, J. Radnik, M. Schneider, H. Atia, U. Armbruster and A. Brückner, Catal. Commun., 2016, 84(5), 171–174 CrossRef CAS.
- F. Y. Gao, X. L. Tang, H. H. Yi, J. Y. Li, S. Z. Zhao, J. G. Wang, C. Chu and C. L. Li, Chem. Eng. J., 2017, 317(1), 20–31 CrossRef CAS.
- L. K. Zhao, C. T. Li, J. Zhang, X. N. Zhang, F. M. Zhan, J. F. Ma, Y. E. Xie and G. M. Zeng, Fuel, 2015, 153(1), 361–369 CrossRef CAS.
- P. Li, Y. Xin, Q. Li, Z. P. Wang, Z. L. Zhang and L. R. Zheng, Environ. Sci. Technol., 2012, 46(17), 9600–9605 CrossRef CAS.
- T. H. Vuong, J. Radnik, J. Rabeah, U. Bentrup, M. Schneider, H. Atia, U. Armbruster, W. Grünert and A. Brückner, ACS Catal., 2017, 7(3), 1693–1705 CrossRef CAS.
- T. H. Vuong, J. Radnik, E. Kondratenko, M. Schneider, U. Armbruster and A. Brückner, Appl. Catal., B, 2016, 197(15), 159–167 CrossRef CAS.
- Z. G. Li, J. H. Li, S. X. Liu, X. N. Ren, J. Ma, W. K. Su and Y. Peng, Catal. Today, 2015, 258(2), 11–16 CrossRef CAS.
- S. H. Li, B. C. Huang and C. L. Yu, Catal. Commun., 2019, 98(10), 47–51 Search PubMed.
- S. L. Zhang and Q. Zhong, J. Solid State Chem., 2015, 221, 49–56 CrossRef CAS.
- P. G. W. A. Kompio, A. Brückner, F. Hipler, E. Löffler and W. Grünert, J. Catal., 2012, 286, 237–247 CrossRef CAS.
- D. N. Corra, J. M. D. E. Slilva, E. B. Santos, F. A. Sigoli, A. G. Souza and I. O. Mazali, J. Phys. Chem. C, 2011, 115(21), 10380–10387 CrossRef.
- S. D. Zhao, J. R. Chen, Y. F. Liu, Y. J. Jiang, C. G. Jiang, Z. L. Yin, Y. G. Xiao and S. S. Cao, Chem. Eng. J., 2019, 367(1), 249–259 CrossRef CAS.
- G. N. Li, B. H. Wu and L. Li, J. Mol. Catal. A: Chem., 2016, 424(1), 304–310 CrossRef CAS.
- M. Piumetti, S. Bensaid, N. Russo and D. Fino, Appl. Catal., B, 2016, 180, 271–282 CrossRef CAS.
- Z. H. Ren, P. Wang, J. Kong, M. J. Wang and L. P. Chang, J. Energy Chem., 2017, 26(4), 647–654 CrossRef.
- M. Piumetti, S. Bensaid, D. Fino and N. Russo, Appl. Catal., B, 2016, 197(15), 35–46 CrossRef CAS.
- L. Zhang, J. F. Sun, L. L. Li, S. M. Ran, G. X. Li, C. J. Li, C. Y. Ge and D. Lin, Ind. Eng. Chem. Res., 2018, 57(2), 490–497 CrossRef CAS.
- W. Y. Zhou, J. Y. Liu, J. Y. Song, J. J. Li, J. H. Liu and X. J. Huang, Anal. Chem., 2017, 8(96), 3386–3394 CrossRef.
- J. X. Hao, S. L. Peng, H. Q. Li, S. Dang, T. F. Qin, Y. X. Wen, J. J. Huang, F. Ma, D. Q. Gao, F. Li and G. Z. Cao, J. Mater. Chem. A, 2018, 6, 16094–16100 RSC.
- J. W. Liu, Q. Sun, Y. C. Fu and J. Y. Shen, J. Colloid Interface Sci., 2009, 335(2), 216–221 CrossRef CAS.
- T. Boningari, R. Koirala and P. G. Smirniotis, Appl. Catal., B, 2013, 140–141, 289–298 CrossRef CAS.
- L. Chen, J. H. Li and M. F. Ge, Chem. Eng. J., 2011, 170(2–3), 531–537 CrossRef CAS.
- P. Maitarad, D. S. Zhang, R. H. Gao, L. Y. Shi, H. R. Li, L. Huang, T. Ruangrotmongkol and J. P. Zhang, J. Phys. Chem. C, 2013, 117(19), 9999–10006 CrossRef CAS.
- A. A. H. Elbadawi, M. Y. Khan, M. R. Quddus, S. A. Razzak and M. Hossain, AIChE J., 2017, 63(1), 130–138 CrossRef CAS.
- C. A. Emeis, J. Catal., 1993, 141(2), 347–354 CrossRef CAS.
- E. P. Parry, J. Catal., 1963, 2(5), 371–379 CrossRef CAS.
- Y. Liu, T. T. Gu, Y. Wang, X. L. Weng and Z. B. Wu, Catal. Commun., 2012, 18, 106–109 CrossRef CAS.
- L. C. Li, H. Q. Yue, T. Ji, W. Li, X. J. Zhao, L. Wang, J. She, X. L. Gu and X. B. Li, Appl. Catal., A, 2019, 574(2), 25–32 CrossRef CAS.
- F. Liu, T. F. Wang, Y. Y. Zheng and J. F. Wang, J. Catal., 2019, 355(1), 17–25 CrossRef.
- X. J. Yao, L. Chen, J. Gao, Y. Chen, M. Tian, F. M. Yang, J. F. Sun, C. J. Tang and L. Dong, Chem. Eng. J., 2019, 369(1), 46–56 CrossRef CAS.
- M. A. Eberhardt, A. Proctor, M. Houalla and D. M. Hercules, J. Catal., 1996, 160(1), 27–34 CrossRef CAS.
- M. Demeter, M. Neumann and W. Reichelt, Surf. Sci., 2000, 454(12), 41–44 CrossRef.
- G. Silversmit, D. Depla, H. Poelman, G. B. Marin and R. D. Gryse, J. Electron Spectrosc. Relat. Phenom., 2004, 135(2–3), 167–175 CrossRef CAS.
- M. L. Abel, A. Carley, J. F. Watts, E. Hryha, E. Rutqvist and L. Nyborg, Surf. Interface Anal., 2012, 44(8), 1022–1025 CrossRef.
- Z. Y. Xiu, Z. P. Xing, Z. Z. Li, X. Y. Wu, X. Yan, M. Q. Hu, Y. Gao, S. L. Yang and W. Zhou, Mater. Res. Bull., 2018, 100, 191–197 CrossRef CAS.
- H. U. Khan and I. K. Swati, Ind. Eng. Chem. Res., 2016, 55(23), 6619–6633 CrossRef CAS.
- H. Li, Z. Chen, C. K. Tsang, Z. Li, X. Ran, C. Lee, B. Nie, L. X. Zheng, T. F. Hung and J. Lu, J. Mater. Chem. A, 2013, 2(1), 229–236 RSC.
- R. T. Guo, Q. L. Chen, H. L. Ding, Q. S. Wang, W. G. Pan, N. Z. Yang and C. Z. Lu, Catal. Commun., 2015, 69(5), 165–169 CrossRef CAS.
- T. M. Wandre, P. N. Gaikwad, A. S. Tapase, K. M. Garadkar, S. A. Vanalakar, P. D. Lokhande, R. Sasikala and P. P. Hankare, J. Mater. Sci.: Mater. Electron., 2016, 27(1), 825–833 CrossRef CAS.
- A. Kambolis, H. Matralis, A. Trovarelli and C. Papadopoulou, Appl. Catal., A, 2010, 377(1), 16–26 CrossRef CAS.
- B. Yang, Z. Li, Q. Huang, M. D. Chen, L. L. Xu, Y. S. Shen and S. M. Zhu, Chem. Eng. J., 2019, 360(15), 990–1002 CrossRef CAS.
- L. R. Shah, B. Ali, H. Zhu, W. G. Wang, Y. Q. Song, H. W. Zhang, S. L. Shah and J. Q. Xiao, J. Phys.: Condens. Matter, 2009, 21(48), 486004–486012 CrossRef.
- R. D. Liu, H. Li, L. B. Duan, H. Shen, Q. Zhang and X. R. Zhao, J. Alloys Compd., 2019, 789(15), 1015–1021 CrossRef CAS.
- X. Y. Zhang, J. Q. Qin, Y. N. Xue, P. F. Yu, B. Zhang, L. M. Wang and R. P. Liu, Sci. Rep., 2014, 4, 4596–4603 CrossRef.
- R. Fiorenza, M. Bellardita, T. Barakat, S. Scirè and L. Palmisano, J. Photochem. Photobiol., A, 2018, 352(1), 25–34 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |