
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCarbocatalysed hydrolytic cleaving of the glycosidic bond in fucoidan under microwave irradiation†
Elaine G.
Mission
 *a,
Jonas Karl Christopher N.
Agutaya
a,
Armando T.
Quitain
*b,
Mitsuru
Sasaki
c and
Tetsuya
Kida
d
*a,
Jonas Karl Christopher N.
Agutaya
a,
Armando T.
Quitain
*b,
Mitsuru
Sasaki
c and
Tetsuya
Kida
d
aGraduate School of Science and Technology, Japan. E-mail: elaine_mission@kumadai.jp
bCollege of Cross-Cultural and Multidisciplinary Studies, Japan. E-mail: quitain@kumamoto-u.ac.jp
cInstitute of Pulsed Power Science, Japan
dFaculty of Advanced Science and Technology, Kumamoto University, Kumamoto 860-8555, Japan
First published on 25th September 2019
Abstract
Biomass valorization involves breaking down naturally occurring long chain polysaccharides into their constituent monomers. The polysaccharide chain consists of monomers adjoined via C (carbon)–O (oxygen) glycosidic linkages that are typically cleaved via hydrolytic scission. In this study, we aimed to recover fucose from the polysaccharide fucoidan, which can be extracted from seaweed biomass. We investigated the depolymerisation behavior of fucoidan sourced from two different species of seaweeds, namely Undaria pinnatifida (F-UP) and Fucus vesiculosus (F-FV). Catalytic depolymerisation experiments were performed using four different carbon-based catalysts – graphene, multiwalled carbon nanotubes (MWCNT), graphene oxide (GO), and reduced graphene oxide (rGO) – under microwave (MW) irradiation. Our results showed that the depolymerisation of fucoidan was best achieved using GO, which was attributed to the abundance of oxygen functionalities on its surface. Furthermore, based on gel permeation chromatography analyses, the depolymerisation of fucoidan was found to follow a two-step process: (1) random scission leading to the production of short-chain oligosaccharides and (2) acid-catalysed hydrolysis of the oligosaccharides to fucose. Because of the longer chain length of F-UP (61 kDa), the highest fucose yield of 17.4% using this species was obtained at a higher temperature of 120 °C in a closed vessel. Meanwhile, in the case of F-FV (1.1 kDa), the highest yield of 54.0% was obtained under reflux conditions at a lower temperature of 104 °C. Our mechanistic study based on semi-empirical quantum calculations also revealed that the recovery of fucose from F-FV is more energetically favoured than from F-UP as a result of their structural differences.
1. Introduction
Over the past few years, the global demand for fossil fuels such as oil, coal, and natural gas has been drastically increasing. For instance, oil consumption has more than doubled in the last 50 years at 100 million barrels per day in 2018, with approximately 60% being used for transportation and much of the remaining for the manufacture of plastics.1 This heavy reliance on fossil fuels has resulted in an increase in greenhouse gas emissions, particularly carbon dioxide, which have a negative impact on the environment. Furthermore, owing to their non-renewability, there has also been an upward trajectory in the prices of these resources.2,3 Considering these problems associated with the continued use of fossil fuels, research has focused on searching for alternative feedstocks for the production of fuels and other chemical commodities.Biomass has been widely accepted as a viable alternative to fossil fuels. In particular, terrestrial lignocellulosic biomass, which is largely composed of cellulose and hemicellulose, has attracted much attention because it is a major source of sugars and platform molecules that can be converted to bio-based fuels and chemicals.4–6 In recent years, however, additional challenges on the use of lignocellulosic feedstock have surfaced, which include (1) competition for land use with food production,7 (2) water and fertilizer requirement for cultivation, and (3) pre-treatment for delignification.8 Algal biomass offers most of the benefits derived from lignocellulosic biomass, but its use does not pose the aforementioned challenges. Despite algae typically having a low cellulose content,9 they contain a large amount of other polysaccharides that can be depolymerized to produce a variety of fermentable sugars depending on their species.
An example is the brown algae that is commonly known as wakame (Undaria pinnatifida). While it is a staple food in Japan and Korea, substandard wakame unfit for food or dietary purposes is either discarded as waste or returned to the sea, causing environmental problems. Additionally, it is widely regarded as an invasive species in Europe, North America, and New Zealand where it has caused an ecological imbalance,10 thus enhancing its viability as a renewable feedstock. U. pinnatifida contains fucoidan, a polysaccharide that is composed mainly of L-fucose units, which are connected via 1,3 linkages and are sulphated in the 2- or 4-positions.11
Aside from seaweeds, L-fucose, which is a hexose deoxy sugar, is contained as part of indigestible polysaccharides found in fruits and vegetables. In mammals, this monosaccharide exists predominantly as an O-glycosidically linked component of glycoproteins, glycolipids, and oligosaccharides.12 Similar to glucose, fucose can be used as a renewable feedstock in the production of fuels and platform chemicals. Petit and co-workers reported that the metabolism of fucose by E. coli, S. enterica, and other bacteria produced 1,2-propanediol, which can be converted into propanol and propionate.13 Aside from its various applications as a solvent, propanol has been shown to decrease greenhouse emissions when used as an additive in fuel blends.14,15 Meanwhile, propionate is a precursor to acrylic plastic production.16
Presently, there is a scarcity of research that focuses on the recovery of fucose from fucoidan, indicating that this direction is relatively unexplored. One route is the enzymatic hydrolysis of fucoidan, but this yields mostly oligosaccharides and not the monosaccharide fucose.17,18 In another study, Quitain et al. carried out the microwave-hydrothermal extraction and degradation of de-oiled U. pinnatifida in supercritical carbon dioxide that resulted in the production of low molecular weight products, but they did not specify the amount of fucose in their hydrolysate.19 On the other hand, Rodriguez-Jasso et al. recovered various sugars, majority of which is fucose, from sulphated polysaccharides extracted from Fucus vesiculosus using 2 M trifluoroacetic acid at 121 °C for 2 h.20 Moreover, Malihan et al. utilized hydrochloric acid in combination with an ionic liquid and obtained 147.3 mg of sugars per gram of U. pinnatifida; 10% of this quantity was fucose.21 However, the difficulty in recovering the soluble products such as sugars in a homogeneous aqueous system and the high cost of ionic liquids render these methods impractical for scale-up.
Since the carbohydrates used in energy and chemical conversion systems are mostly polysaccharides, the release of the desired fermentable sugars involves the cleaving of the glycosidic C–O linkage along the polymer chain. A number of deconstruction methods for cellulose have been developed in order to break its structural framework, including mechanical treatment,22 use of ionic liquids,23 and hydrothermal liquefaction.24,25 However, these methods are not applicable in the processing of algae because of its high moisture and oxygen content, and low bulk density. A suitable pathway is the hydrolysis of fucoidan in the liquid phase and in the presence of a catalyst that can effectively break the resilient glycosidic bond in its structure.
In a previous study, we demonstrated that graphene-based catalysts in tandem with microwave irradiation is a promising pre-treatment- and additive-free approach in the depolymerisation of cellulose.26 The in situ de-crystallization, dissolution, and hydrolysis of cellulose were made possible by the synergy between microwave that facilitated the conformational changes in the bulk cellulose structure and graphene oxide (GO) that provided the active sites necessary for the hydrolytic cleaving of the glycosidic bonds in cellulose leading to the production of glucose. However, because of the difference between the structures of cellulose and fucoidan, our key findings for cellulose simply cannot be extrapolated to fucoidan.
In this current work, we employed both an experimental and a theoretical approach to gain mechanistic insights into the hydrolytic cleaving in fucoidan under microwave-carbocatalytic conditions. First, we performed quantum calculations to understand and determine a reaction pathway for the acid-catalysed hydrolysis of fucoidan. Second, we carried out the depolymerisation of fucoidan using various carbon-based catalysts under microwave irradiation. Fucoidan from two different species, namely U. pinnatifida (F-UP) and F. vesiculosus (F-FV), were used as model compounds. The depolymerisation behaviour at several temperatures, reaction time, GO loading and form, and microwave (MW) power were investigated. Lastly, we compared the mechanisms of the depolymerisation of fucoidan and cellulose.
2. Results and discussion
2.1. Characterization of model compounds
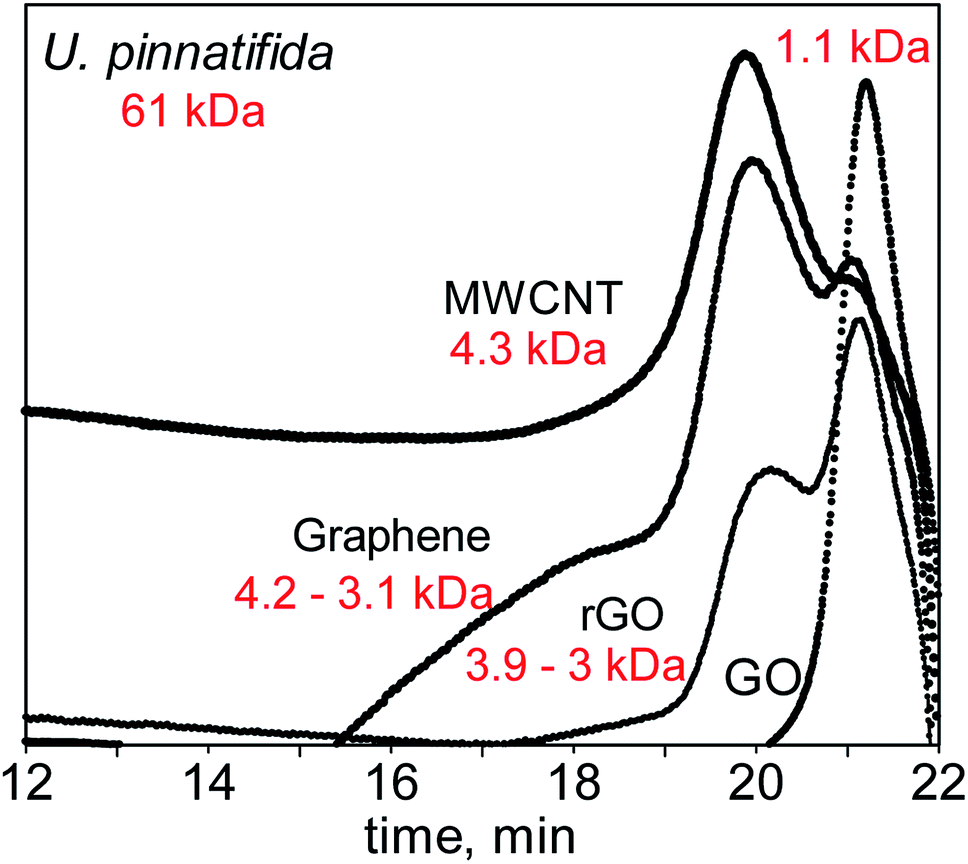
Cellulose and fucoidan both have the simplest structure as polysaccharides since both consist of sugar monomers connected by glycosidic bonds. The major distinction between the two is their structure as revealed by X-ray diffraction (XRD) (Fig. 1). Cellulose is highly crystalline in nature due to the extensive network of hydrogen bonds on its surface. These hydrogen networks make cellulose insoluble in water and protects the β-1,4 glycosidic bonds against cleaving. These hydrogen bonds are practically lacking in fucoidan; thus, it easily dissolves in water. Moreover, fucoidan bears α-(1,3) linkages, which also suggests that it can be depolymerised at a lower temperature than that of cellulose. Using gel permeation chromatography (GPC), the molecular weights for F-UP and F-FV were obtained, which are 61 kDa and 1.1 kDa, respectively (Fig. S1†). Since these compounds were used as received, the variation was potentially a consequence of the difference in their isolation techniques, typically via partial acid hydrolysis, and subsequent purification used in their production.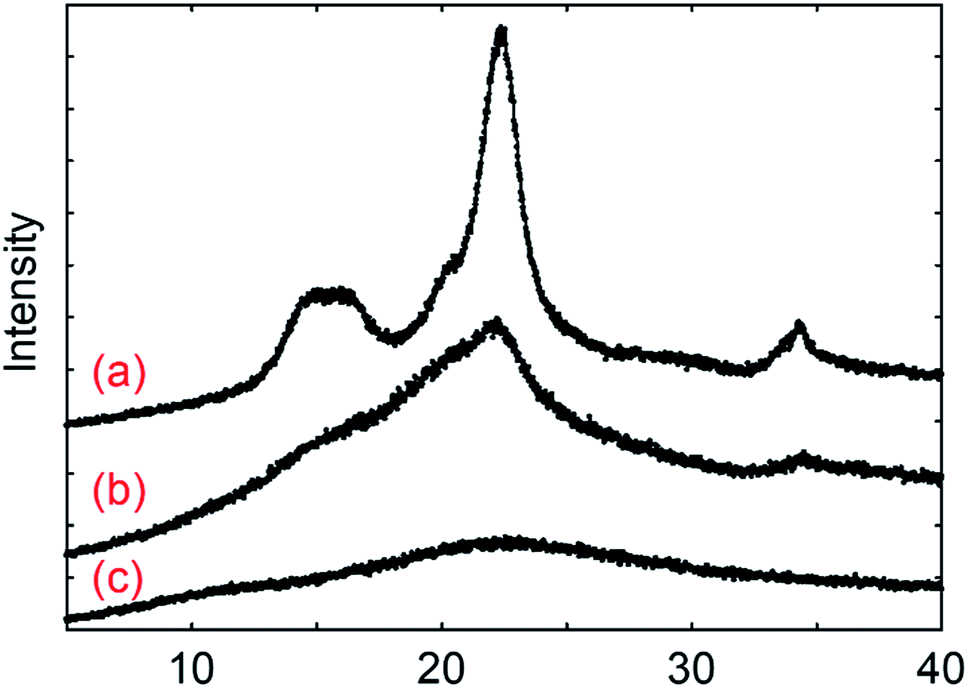 | ||
| Fig. 1 X-ray diffraction spectra for (a) microcrystalline cellulose, (b) amorphous cellulose, and (c) fucoidan from U. pinnatifida. | ||
2.2. Computational deductions
Due to the inherent stability of glycosidic bonds, the hydrolytic cleaving of these linkages is typically achieved using strong acids under elevated temperatures.27–29 With due consideration of the role of the acid in the process, the mechanism of the recovery of fucose from fucoidan was first determined using quantum calculations. The method used was the semi-empirical PM3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 30,31 in the gas state, as implemented in GAMESS (US).32,33 PM3 is third parameterization of the modified neglect of diatomic overlap (MNDO) method. Much like any other semi-empirical method, PM3 significantly reduces the effort in quantum calculations by explicitly considering only the valence electrons of the system. The justification behind this approximation is that the electrons involved in chemical bonding and other related phenomena are those in the valence shell.34 This method effectively neglects some of the integrals, which are calculated in the more robust ab initio Hartree–Fock methods, and thus would make it less accurate. However, given the size of the systems considered in this study (more than 70 atoms), the use of ab initio or even density functional methods is impractical because of the substantial amount of time required to complete the simulations. Therefore, for the purpose of identifying the role of the acid in the hydrolysis of the substrates in this study, the PM3 proved to be a suitable method.
30,31 in the gas state, as implemented in GAMESS (US).32,33 PM3 is third parameterization of the modified neglect of diatomic overlap (MNDO) method. Much like any other semi-empirical method, PM3 significantly reduces the effort in quantum calculations by explicitly considering only the valence electrons of the system. The justification behind this approximation is that the electrons involved in chemical bonding and other related phenomena are those in the valence shell.34 This method effectively neglects some of the integrals, which are calculated in the more robust ab initio Hartree–Fock methods, and thus would make it less accurate. However, given the size of the systems considered in this study (more than 70 atoms), the use of ab initio or even density functional methods is impractical because of the substantial amount of time required to complete the simulations. Therefore, for the purpose of identifying the role of the acid in the hydrolysis of the substrates in this study, the PM3 proved to be a suitable method.
The simulations proceeded as follows: first, a partial geometry optimization at the bond interchange regions was performed for each transition state (TS). Then, the resulting configuration was subjected to a saddle point search, which also computed for the vibrational frequencies. When the structure exhibited a single imaginary frequency, the intrinsic reaction coordinate (IRC)35 was traced from the TS to the energy-minimum geometries or intermediates (Int). The structures were generated using Avogadro36 and the output files were analysed using MacMolPlt.37
The structure of fucose is shown in Fig. 2. As a repeating unit in fucoidan extracted from seaweeds, this sugar is sulphated at different points depending on the order to which the particular species of seaweed belongs. In the case of F-UP, which is from the order Laminariales, the units are sulphated mainly at the O-4 position and some additionally at the O-2 position, and these a-L-fucopyranose residues are α-1,3-linked.38 On the other hand, F-FV of the order Fucales has units that are sulphated at O-2 and, to a lesser extent, at O-3, and the linkages are mainly α-1,3 with a high degree of α-1,4 as well.39Fig. 3 shows the representative models of fucoidan with three repeating units for each species. These structures were adopted from the review paper of Ale and Meyer,40 but with the negatively-charged sulphate functional groups terminated with hydrogen.
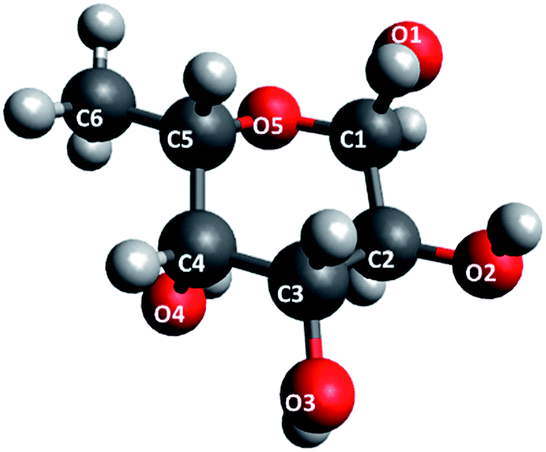 | ||
| Fig. 2 Labelling of fucose. | ||
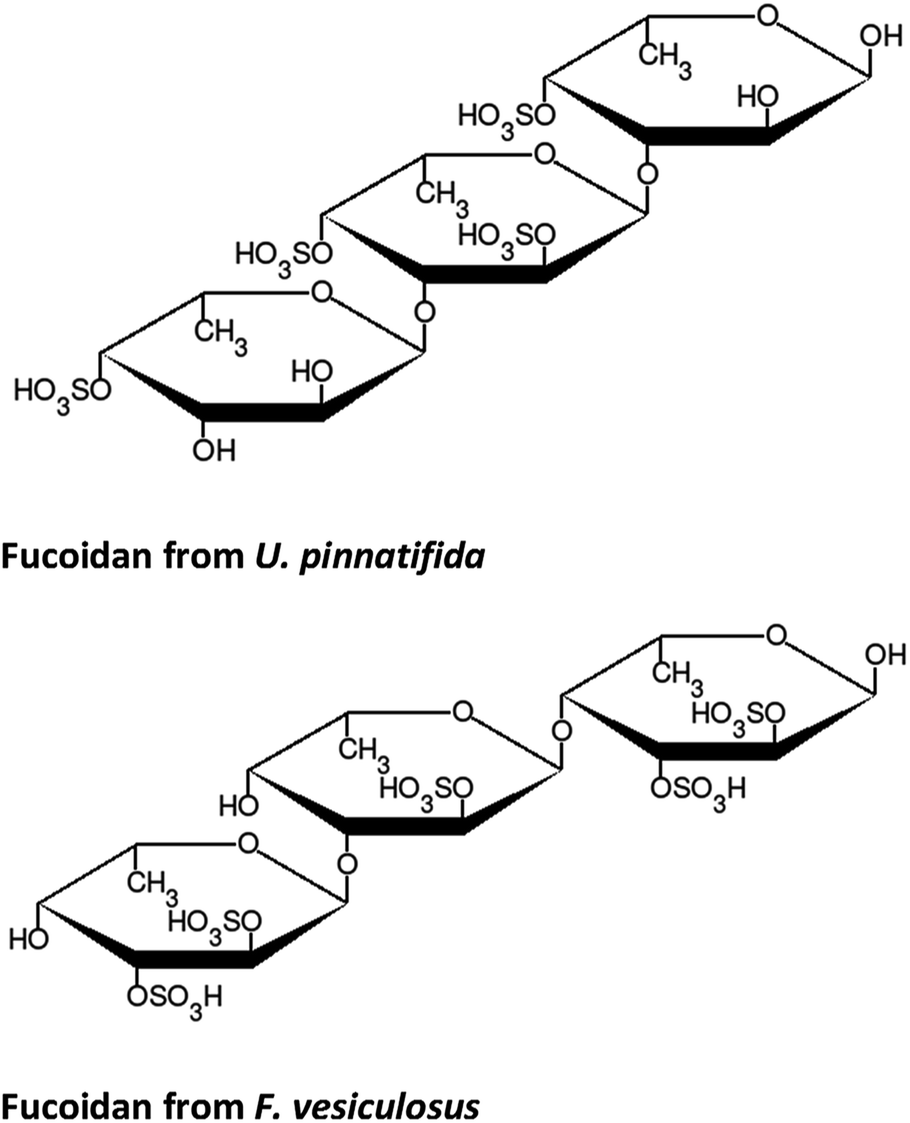 | ||
| Fig. 3 Model structures of fucoidan. | ||
In the simulations, the recovery of a fucose monomer from the non-reducing end of fucoidan was investigated. The simulation revealed that the acid-catalysed hydrolysis of fucoidan involves four steps as shown in Fig. 4. The reaction is initiated by the protonation of the oxygen at the glycosidic linkage (TS1, Int1). While the protonation of the endocyclic oxygen is also possible, this path was no longer explored in this study since it has shown to be less energetically favoured in a study on the disaccharide sucrose.41 Next, the bond between the oxygen and C1 breaks (TS2), leading to the formation of a carbocation (Int2) and a fucoidan with two sugar units. While the bond breakage may also occur between the oxygen and C2, the presence of the more electronegative oxygen bonded to C1 produces a more stable carbocation. The oxygen of a water molecule then attacks the electron-deprived carbon in the intermediate (TS3), resulting in a fucose molecule with a protonated hydroxyl group (Int3). Finally, the proton is recovered (TS4), yielding the desired product. A similar set of calculations was performed in the absence of a proton, but the simulations did not proceed to the fragmentation at the glycosidic bond, which may explain the importance of an acid in initiating the reaction. Performing the same procedure, our simulations shows that the hydrolysis of F-FV follows the same four-step process (Fig. S2†).
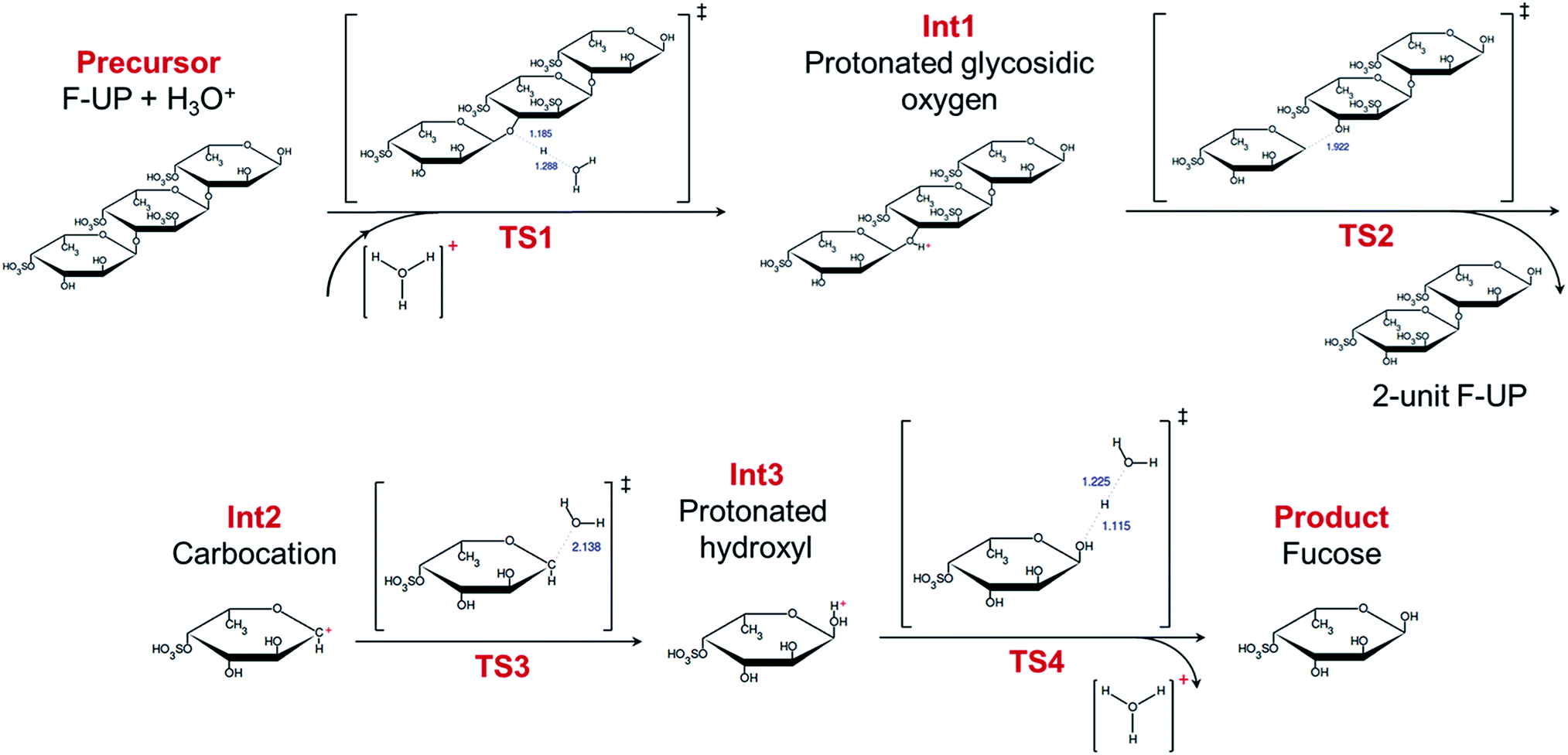 | ||
| Fig. 4 Mechanism of the acid-catalysed hydrolysis of fucoidan from U. pinnatifida. The lengths in ångströms of the bond interchanges in the transition states are shown in blue. | ||
Using cellotriose as the model compound for cellulose, we found that the mechanism for the hydrolysis of cellulose followed the same four-step process from our quantum calculations with fucoidan (Fig. S3†). The energy profiles of the hydrolysis of the three substrates were then generated (Fig. 5). The electronic energies of the structures were computed also at the PM3 level of theory and the values in the graph are relative to the energy of the precursor for each substrate. The activation energies of the four steps are also reflected in the graph. For all three compounds, the first step, which is the protonation of the glycosidic oxygen, was found to be rate-determining because it required the highest activation energy Ea. Considering the values of Ea at this step, the hydrolysis of F-UP demanded the highest energy at 88.11 kJ mol−1, followed by F-FV at 74.78, and cellulose at 70.02. Relative to its precursor, the final product for the depolymerisation of F-UP also possessed the highest electronic energy of 86.69 kJ mol−1. The energies of respective monosaccharides from F-FV and cellulose are much lower and comparable to each other at 38.89 and 41.57, respectively. These comparisons demonstrate that the hydrolysis of F-UP is less favoured via the acid-catalysed hydrolysis route than those of F-FV and cellulose. The trend with respect to the rate of hydrolysis based on the activation energies as determined from the mechanistic studies is in agreement with the yields obtained in the experiments, with cellulose26 having the best values, followed by F-FV, and finally, F-UP. Nevertheless, despite this consistency, the mechanism presented in this current study is limited only to a purely acid-catalysed reaction with the assumption that GO acts as the source of the proton required to initiate the reaction. This assumption is supported by previous studies that have shown that the carboxylic groups42 present in the structure of GO make it effective as an acid catalyst.
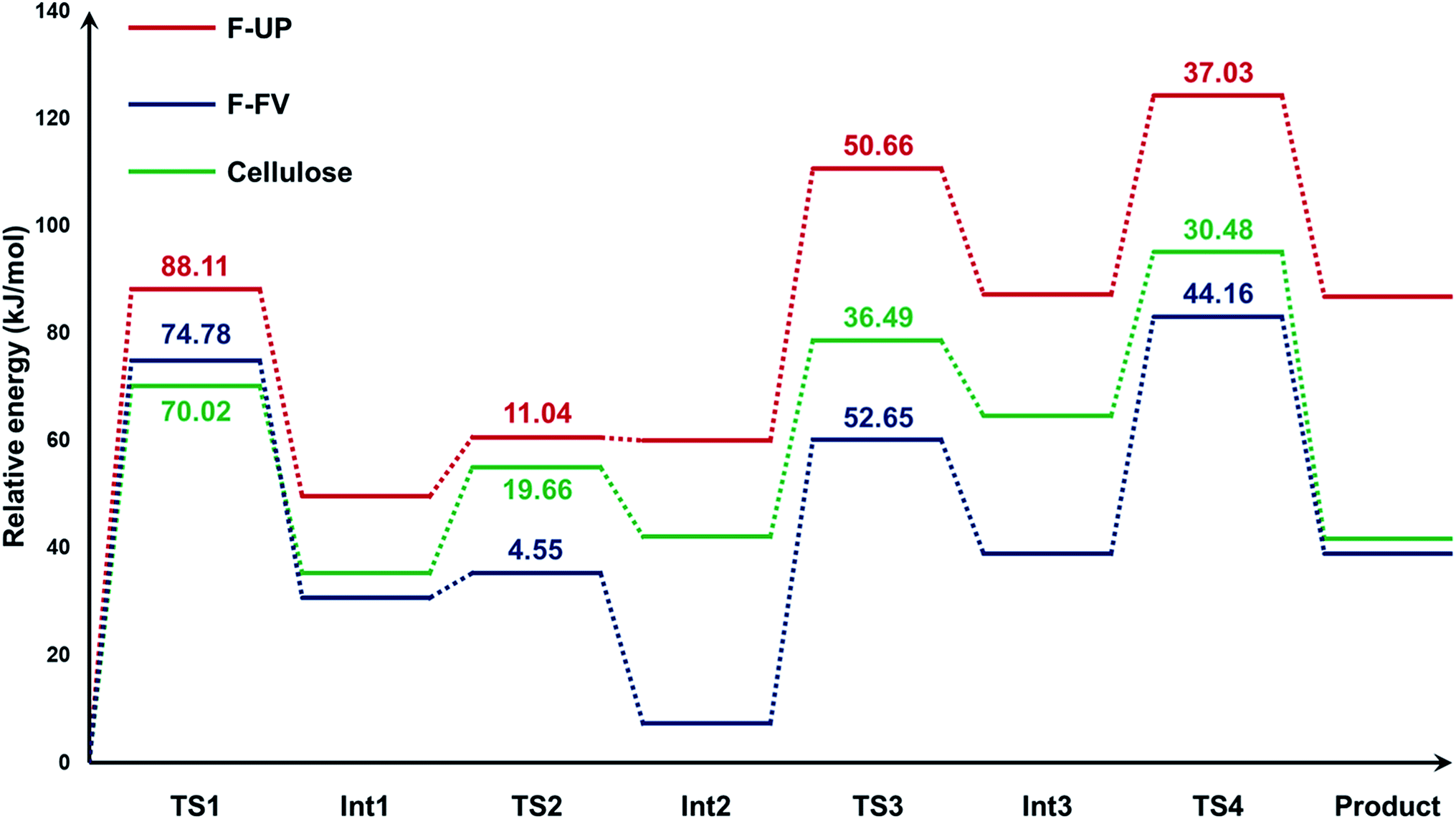 | ||
| Fig. 5 Energy changes of the PM3-optimized geometries in the hydrolysis of fucoidan from U. pinnatifida (F-UP) and F. vesiculosus (F-FV), and cellulose. The activation energies in kJ mol−1 for each step are also shown. | ||
2.3. Depolymerisation of fucoidan using various carbon-based catalysts under microwave irradiation
A number of carbon-based catalysts have been developed for organic reactions, since they are perceived to be more environment friendly and sustainable than their metal-based counterparts.43–45 In this work, we evaluated four of these catalysts, namely, graphene, multiwalled carbon nanotubes (MWCNT), graphene oxide (GO), and thermally-annealed reduced GO (rGO), to compare their performance in the depolymerisation of fucoidan under microwave (MW) irradiation. Their structures are shown in Fig. 6. In principle, graphene is a monolayer of carbon atoms arranged in a hexagonal lattice. Meanwhile, MWCNT is formed by rolling up several layers of graphene in a tubular and concentric manner. On the other hand, GO is synthesized from the oxidation and exfoliation of graphite. Because of the procedure for the synthesis of GO, it is a form of graphene that is heavily decorated with several oxygen functional groups. Finally, when GO is subjected to high temperatures of about 300 °C under argon atmosphere, it loses some of its oxygenated functionalities, leading to the formation of rGO.46,47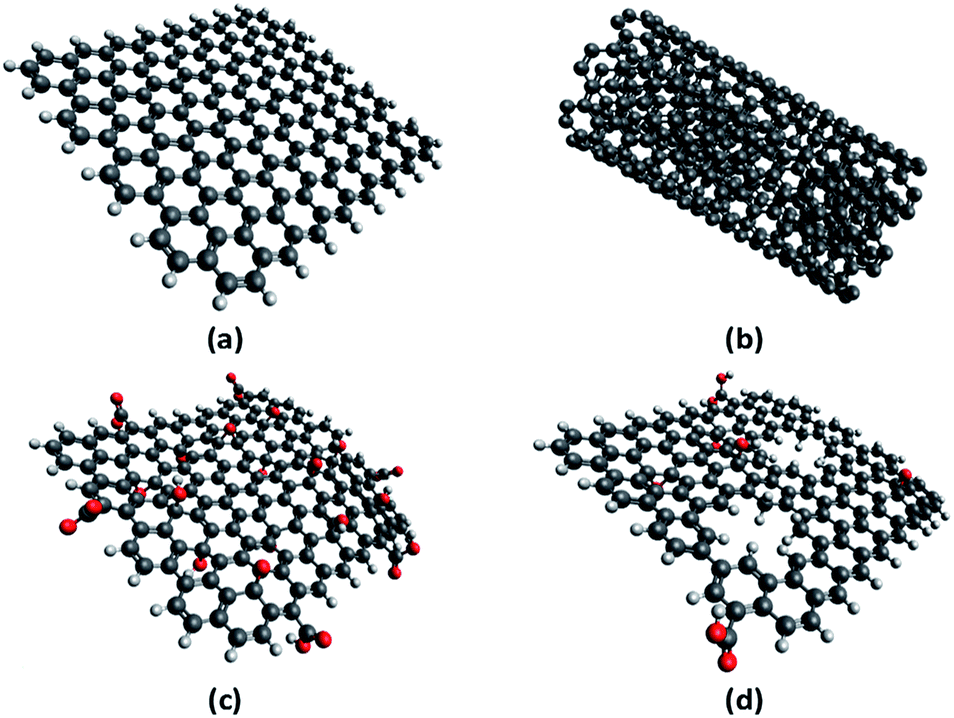 | ||
| Fig. 6 Structures of (a) graphene, (b) multiwalled carbon nanotubes, (c) graphene oxide, and (d) reduced graphene oxide. | ||
In the first set of experiments, F-UP was used as the substrate. Since it has a long chain length of 61 kDa, the MW irradiation of F-UP was carried out in a closed vessel using the Microwave Assisted Reactor System (MARS 6). The Experimental details are in Section 1 of the ESI.† A GPC analysis of the hydrolysates (Fig. 7) revealed that there was a distinct product distribution for each catalyst and that the main products were low molecular weight oligomers of fucoidan. When MWCNT was used, the resulting hydrolysate was found to contain long-chain oligosaccharides with an average molecular weight of 4.3 kDa, represented by a predominantly broad peak. When graphene was used as the catalyst, two peaks were detected representing two sets of oligosaccharides produced that were in the range of 3.1 to 4.2 kDa. In this case, more long-chain oligosaccharides were produced than short ones. Since the main difference between these two catalysts was their structure, it can be inferred that the graphene monolayer provided a larger active surface for the hydrolysis of fucoidan.
 | ||
| Fig. 7 Catalytic performance of graphene, multiwalled carbon nanotubes, graphene oxide, and thermally-annealed reduced graphene oxide in the depolymerisation of fucoidan from U. pinnatifida [reaction conditions: 50 mg fucoidan, 50 mg catalyst, 10 ml water, 200 W, 120 °C, 15 min]. | ||
The chromatogram for the hydrolysate with rGO as the catalyst also exhibited two peaks, but it showed that the mixture is dominated by short chain oligosaccharides. Finally, when GO was used as the catalyst, a single narrow peak corresponding to much shorter oligosaccharides with an average molecular weight of 1.1 kDa reflected in the chromatogram. Since all of the catalysts used in this study have a graphene backbone, they have high MW reception, which allows for the highly localized heating on the surface of these materials.48,49 This phenomenon may have promoted the ionization of water50 at the surface of the catalyst, thus providing the necessary protons to cleave the polymer chain.
Blank experiments were performed to provide further comparison with earlier findings. The results of the GPC analysis are illustrated in Fig. S4.† When the depolymerisation was carried out under microwave irradiation, but without a catalyst, the chain length was reduced moderately from 61 to 58 kDa, indicating that MW alone is not effective in depolymerizing the long chain polymer. On the other hand, when the depolymerisation was carried out with GO under conventional heating, two peaks were observed: the undissociated (61 kDa) and partially dissociated (5.2 kDa) F-UP.
Using high performance liquid chromatography (HPLC), we determined the yield of fucose from the reaction. With the use of MW only, there was very minimal fucose yield production at 0.5%, indicating that this potentially came from the reducing end of the polymer chain and does not necessarily correspond to fucose obtained from the depolymerisation process. With both MWCNT and graphene, no fucose was detected in the hydrolysate. Both graphene and MWCNT are devoid of oxygenated functionalities, therefore, the mechanism for the depolymerisation using these two catalysts can be likened to the hydrothermal treatment of cellulose in the work of Remón et al. where only oligosaccharides were produced at temperatures below 200 °C.51 When rGO was used, a yield of 2.4% was achieved and this value significantly increased to 17.4% when GO was used instead. In contrast, GO possesses a number of functional groups that can catalyse the hydrolysis of fucoidan. For instance, the carboxylic group present in GO52 can serve as the acid necessary to produce the fucose monomer. Finally, the decrease in the yield of fucose in the rGO-catalysed reaction was a result of the elimination of some of the oxygen functionalities in its precursor GO when this catalyst was synthesized. Our evaluation of catalytic performance of four carbon-based materials based on the presence of oxygen functional groups further reinforces the importance of an acid catalyst in effectively recovering monosaccharides from biomass.49,53–55 Moreover, the uniform short chain oligomers produced with GO could have led to the higher fucose yield.
Control experiments employing graphene and MWCNT coupled with trace amounts of acids, which represent the oxygen functionalities in GO, were also performed and the results are available in Section 5 of the ESI.† Results show that the addition of trace acids marginally improved sugar recovery, further highlighting the unique structure GO. Meanwhile, when GO was used under conventional heating, a yield of about 17% was also obtained. However, this required an additional 6 min of preheating at 300 °C over the 15 min reaction time. Because of the higher temperature requirement, longer pre-treatment, and higher energy costs associated with conventional heating, MW irradiation becomes a more practical approach to the depolymerization of fucoidan.
2.4. Reaction conditions for the depolymerisation of fucoidan from U. pinnatifida
In this section, we present the results of the depolymerisation of F-UP using GO under MW irradiation at different temperatures and reaction times. In the chromatogram shown in Fig. 8, pristine F-UP exhibited a single broad peak, which corresponds to an average molecular weight of 61 kDa. Performing a run at 80 °C for 15 min, two peaks were observed in the chromatogram, which correspond to long- and short-chain polymers with an average molecular weight of 2.6 kDa. Because of the wide distribution of molecular weights, random scission56 was suspected to be the mechanism in the depolymerisation at this temperature. Increasing the temperature to 100 °C resulted in a sharp decrease in the peak that corresponds to high molecular weight molecules. Further increasing the temperature to 120 °C completely eliminated this peak. At this temperature, the hydrolysate entirely consists of short-chain oligosaccharides.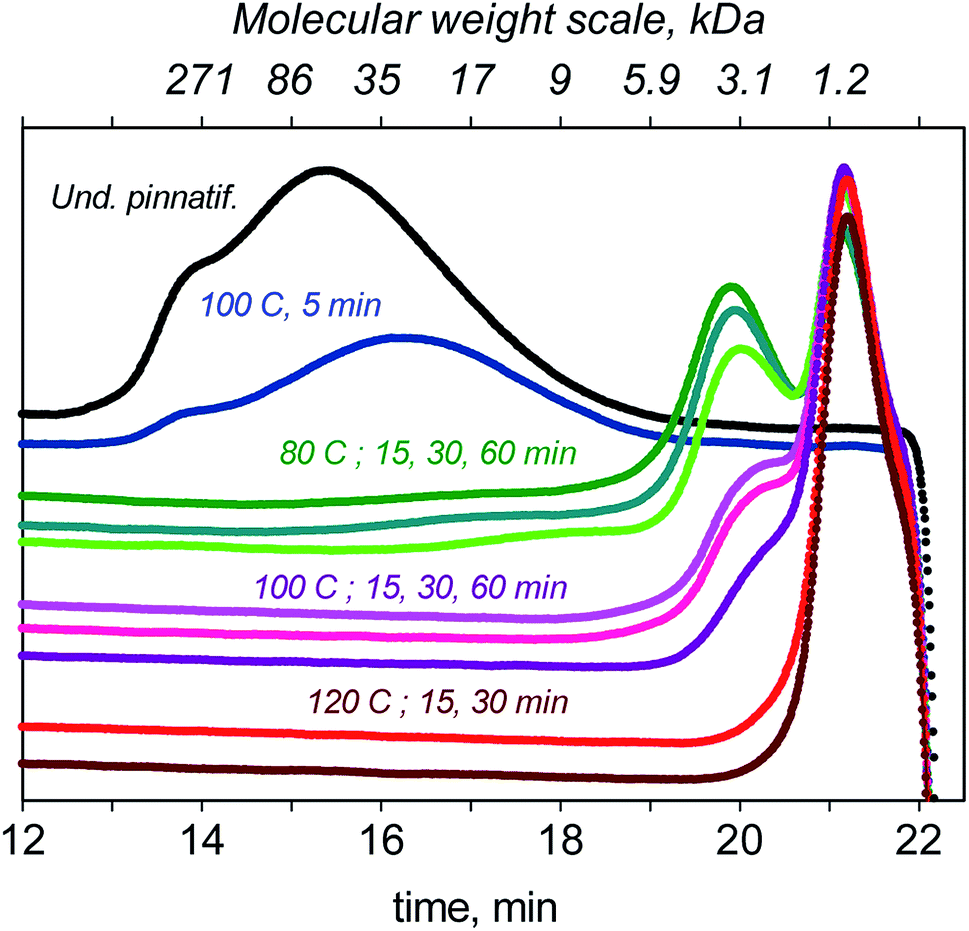 | ||
| Fig. 8 Effect of temperature and reaction time on the reaction on the extent of depolymerisation of fucoidan from U. pinnatifida using graphene oxide as the catalyst and under microwave irradiation [reaction conditions: 50 mg F-UP, 50 mg GO, 10 ml water, 15 min, 200 W]. | ||
The reaction was also carried out for 30 min at the three temperatures. The longer reaction time resulted in taller peaks for short-chain oligosaccharides and shorter peaks for long-chain ones. The oligosaccharides produced after the first time period were speculated to be further broken down to short-chain sugars after the additional 15 min.
Fig. 9 shows the yield of fucose at different temperatures and reaction time. The trend in the graph demonstrates the more significant effect of temperature in improving the yield of fucose. The highest yield of 17.4% was obtained at 120 °C and 30 min. The reaction, especially at this temperature, was no longer extended beyond this period because of the formation of degradation products. Aside from the peaks of GO and fucose, a third peak emerged in the HPLC chromatogram of the mixtures (see Fig. S7†) at 120 °C, which we attributed to the formation of side products. Any further increase in either the temperature or the reaction time may favour the formation of these unwanted compounds and consequently affect the selectivity toward the production of fucose. Under the conditions considered in this study, the recovery of fucose through the hydrolysis of fucoidan cannot be achieved with high yield and selectivity because fucose is highly reactive and forms degradation products, thus affecting hydrolysate quality and selectivity.
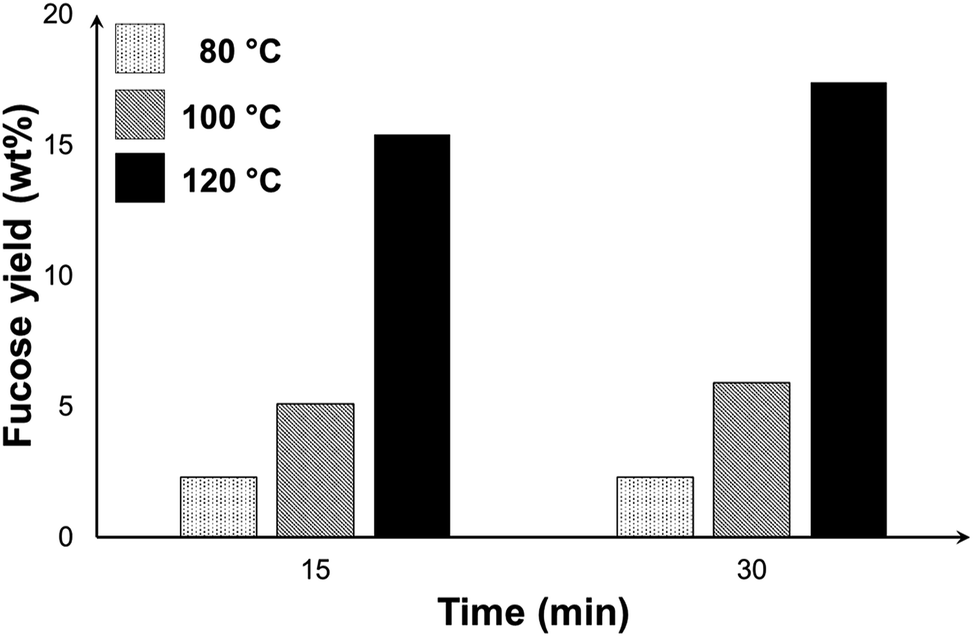 | ||
| Fig. 9 Yield of fucose in the graphene oxide-catalysed depolymerisation of fucoidan under microwave irradiation at different temperatures and reaction time [reaction conditions: 50 mg F-UP, 50 mg GO, 10 ml water, 15 min, 200 W]. | ||
2.5. Reaction conditions for the depolymerisation of fucoidan from F. vesiculosus
We also experimentally carried out the depolymerisation of F-FV. The effect of temperature was investigated by setting the desired operating temperature and allowing the MW to regulate the power. As evident in Fig. 10a, a fucose yield of 5% was constant at 80 and 90 °C, but increased to 8% at 100 °C, which is the normal boiling point of water. As we have also seen in the depolymerisation of F-UP, the hydrolytic cleaving of C–O bonds typically requires elevated temperatures. When the system is operated under reflux conditions where MW power is the controlling factor, the highest temperature recorded was about 104 °C, and fucose yield increased to 14%.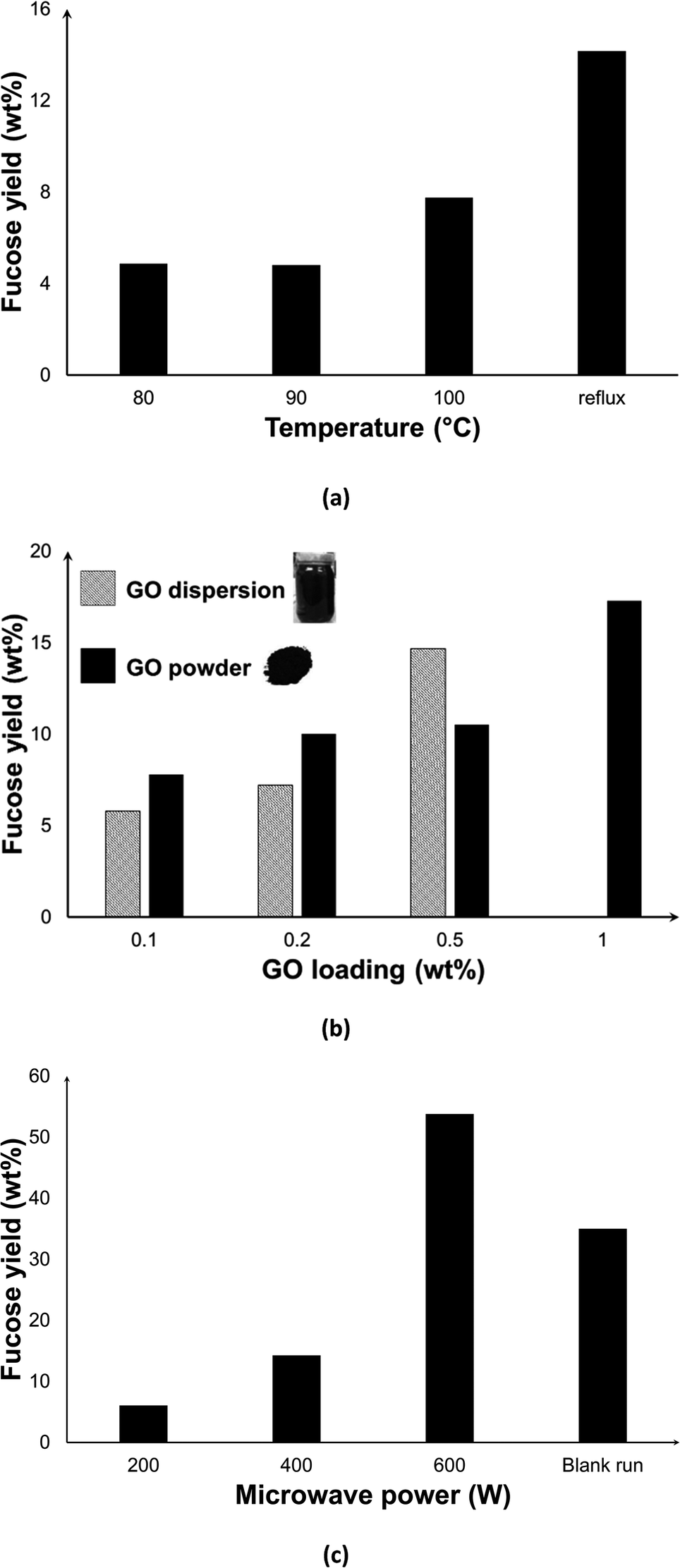 | ||
| Fig. 10 Effect of (a) reaction temperature; (b) graphene oxide form and concentration, and (c) microwave power on fucose yields from the depolymerisation of fucoidan from F. vesiculosus [reaction conditions: 50 mg F-FV in (a) and (c), 100 mg F-FV in (b); 50 mg GO, 10 ml water, 15 min, 200 W except in (c), 300 rpm stirring; blank run in (c) was carried out in 140 °C, 30 min following the protocol in ref. 19]. | ||
We also investigated the effect of the variation in the amount of catalyst dosed into the system and of the form of GO, either powder or dispersed (Fig. 10b). As expected, the increase in the GO loading led to a higher fucose yield due to the increase in the number of active sites. However, some of the GO added to the system simply accumulated on the sides of the vessel, thus any increase in the amount of GO is both impractical and wasteful. At a loading of 0.5 wt%, dispersed GO performed better than the powder form, but a dispersion of higher concentration was too viscous for use which limits the yield.
We achieved higher yields when MW power was increased as seen in Fig. 10c. In contrast to a closed system where the effect of MW power is dominant only during heating up time and then diminishes when the set temperature is reached,26 MW irradiation can be programmed as continuous or intermittent under reflux conditions. At 600 W, the highest fucose yield of 54.0 wt% was achieved. Higher MW power may have caused higher temperatures at the GO surface, facilitating the hydrolysis reaction. Moreover, it has induced rapid rotation of molecules that facilitated molecular collisions leading to a faster hydrolysis process. In conjunction with the thermal effects, incident MW irradiation has also been postulated to increase the collisions between the reacting components in the medium, which in turn promotes the reaction.57 In particular, the increased kinetic energy of water molecules via dielectric polarization58 as a consequence of microwave absorption may have contributed to its improved interaction with fucoidan, leading to its hydrolysis. In order to isolate the contribution of synergistic MW–GO effect on the yields, a blank run (without catalyst) in conventional heating was performed. Despite a much longer reaction duration that includes about 10 min pre-heating and 30 min reaction time, the yield obtained was only about 36%.
3. Summary and future outlook
We have successfully carried out the depolymerisation of fucoidan under microwave-carbocatalytic conditions. By utilizing four carbon-based catalysts, namely, graphene, multiwalled carbon nanotubes (MWCNT), graphene oxide (GO), and reduced graphene oxide (rGO), under microwave irradiation, we were able to observe the depolymerisation behaviour of fucoidan and the recovery of fucose from two species of seaweed, Undaria pinnatifida (F-UP) and Fucus vesiculosus (F-FV). We have observed that short chain oligomers can be produced by using the four catalysts, but only rGO and GO were able to generate fucose. The reduction in oxygen functionalities, specifically the carboxylic group, from GO to rGO to graphene, resulted in a decrease in the yield of the fucose. The further drop in the yield for MWCNT was attributed to the decrease in the surface that facilitated the reaction as compared to graphene. Based on our results, we also concluded that the recovery involves a stepwise process where shorter chains are successively generated from the polysaccharide, ultimately leading to the production of fucose.The highest yield of fucose from F-UP was 17.4%, which was achieved at 120 °C in a closed system. On other hand, the highest yield of fucose from F-FV was 54.0%, which was attained under reflux conditions at a lower temperature of 104 °C. Because of the higher temperature requirement for the depolymerisation of F-UP, the system could not be operated with a reflux using this substrate. The lower temperature needed in the depolymerisation of F-FV was attributed to its lower molecular weight of 1.1 kDa compared to that of F-UP, which is 61 kDa.
To further gain insights into the process, we determined the mechanism for the reaction using quantum calculations, starting with a three-unit fucoidan as a representative model for the short-chain precursor to the recovery of fucose. Our calculations using the semi-empirical PM3 method showed that the acid-catalysed hydrolysis of fucoidan is a four-step process: (1) protonation of the glycosidic oxygen, (2) cleavage of the C–O bond yielding a fucosyl carbocation, (3) nucleophilic attack of water on the resulting carbocation, and (4) deprotonation.
We further extended our mechanistic study to cellulose, which was the substrate used in our previous study. We found that the hydrolysis of cellulose follows the same four-step mechanism. Moreover, our calculation of the activation energies of the reactions showed that cellulose was the easiest to hydrolyse, followed by F-FV, then F-UP. This trend is consistent with our experimental results based on the yield of the respective monosaccharides of these substrates.
As our present work focuses only on the mechanism for the bond cleavage, further studies will be directed to the interaction between graphene oxide and the substrates to fully understand the role of GO as a heterogeneous catalyst in the depolymerisation of carbohydrates. Moreover, since GO serves as a heterogeneous catalyst,59,60 the mechanism of the adsorption of the substrates is also an important factor to consider for future computational work.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support from the Sasakawa Scientific Research Grant (Grant Number 2862-1292) for this research. This work was also partially supported by the Japan Science and Technology Agency via the e-ASIA joint research program in the field of Functional Materials and Bioenergy (Grant Numbers 14528300, 18066393) and JST SICORP (Grant Number JPMJSC18E2).References
- A. Cooper and C. Johnson, Now near 100 million bpd, when will oil demand peak?, https://www.reuters.com/article/us-oil-demand-peak/now-near-100-million-bpd-when-will-oil-demand-peak-idUSKCN1M01TC, accessed 25 March 2019 Search PubMed.
- R. Rapier, Oil Price, 2019 Search PubMed.
- Oil price: Why oil prices will not cool down, but only go up, https://economictimes.indiatimes.com/markets/expert-view/oil-price-is-not-going-to-cool-down-it-will-only-go-up-rs-sharma/articleshow/65765670.cms?from=mdr, accessed 25 March 2019.
- F. H. Isikgor and C. R. Becer, Polym. Chem., 2015, 6, 4497–4559 RSC.
- S. G. Wi, E. J. Cho, D.-S. Lee, S. J. Lee, Y. J. Lee and H.-J. Bae, Biotechnol. Biofuels, 2015, 8, 228 CrossRef.
- Z. Zhou, F. Lei, P. Li and J. Jiang, Biotechnol. Bioeng., 2018, 115, 2683–2702 CrossRef CAS.
- J. Valentine, J. Clifton-Brown, A. Hastings, P. Robson, G. Allison and P. Smith, GCB Bioenergy, 2012, 4, 1–19 CrossRef.
- A. K. Kumar and S. Sharma, Bioresources and Bioprocessing, 2017, 4, 7 CrossRef.
- M. Yanagisawa, S. Kawai and K. Murata, Bioengineered, 2013, 4, 224–235 CrossRef.
- L. Y. Bloch, Invasive Species of the Pacific Northwest, Wakame, 2014 Search PubMed.
- Y. Zhao, Y. Zheng, J. Wang, S. Ma, Y. Yu, W. White, S. Yang, F. Yang, J. Lu, Y. Zhao, Y. Zheng, J. Wang, S. Ma, Y. Yu, W. L. White, S. Yang, F. Yang and J. Lu, Mar. Drugs, 2018, 16, 321 CrossRef.
- S. S. H. Choi, B. S. Lynch, N. Baldwin, E. W. Dakoulas, S. Roy, C. Moore, B. A. Thorsrud and C. H. Röhrig, Regul. Toxicol. Pharmacol., 2015, 72, 39–48 CrossRef CAS PubMed.
- E. Petit, W. G. LaTouf, M. V. Coppi, T. A. Warnick, D. Currie, I. Romashko, S. Deshpande, K. Haas, J. G. Alvelo-Maurosa, C. Wardman, D. J. Schnell, S. B. Leschine and J. L. Blanchard, PLoS One, 2013, 8, e54337 CrossRef CAS.
- S. Pinzi, M. D. Redel-Macías, D. E. Leiva-Candia, J. A. Soriano and M. P. Dorado, Energy Procedia, 2017, 142, 849–854 CrossRef CAS.
- A. Keskin and M. Gürü, Energy Sources, Part A, 2011, 33, 2194–2205 CrossRef CAS.
- S. G. Khokarale and J.-P. Mikkola, Green Chem., 2019, 21, 2138–2147 RSC.
- K. Kitamura, M. Matsuo and Y. Tsuneo, Biosci., Biotechnol., Biochem., 2009, 56, 490–494 CrossRef.
- A. S. Silchenko, M. I. Kusaykin, V. V. Kurilenko, A. M. Zakharenko, V. V. Isakov, T. S. Zaporozhets, A. K. Gazha and T. N. Zvyagintseva, Mar. Drugs, 2013, 11, 2413–2430 CrossRef PubMed.
- A. T. Quitain, T. Kai, M. Sasaki and M. Goto, Ind. Eng. Chem. Res., 2013, 52, 7940–7946 CrossRef CAS.
- R. M. Rodriguez-Jasso, S. I. Mussatto, L. Pastrana, C. N. Aguilar and J. A. Teixeira, Chem. Pap., 2014, 68, 203–209 CAS.
- L. B. Malihan, G. M. Nisola and W. J. Chung, Bioresour. Technol., 2012, 118, 545–552 CrossRef CAS.
- A. Shrotri, H. Kobayashi and A. Fukuoka, J. Jpn. Pet. Inst., 2015, 58, 1–8 CrossRef CAS.
- A. Brandt, J. Gräsvik, J. P. Hallett and T. Welton, Green Chem., 2013, 15, 550 RSC.
- E. Kamio, S. Takahashi, H. Noda, C. Eukuhara and T. Okamura, Ind. Eng. Chem. Res., 2006, 45, 4944–4953 CrossRef CAS.
- M. Möller, F. Harnisch and U. Schröder, RSC Adv., 2013, 3, 11035 RSC.
- E. G. Mission, A. T. Quitain, M. Sasaki and T. Kida, Green Chem., 2017, 19, 3831–3843 RSC.
- K. Grohmann, J. A. Manthey and R. G. Cameron, Carbohydr. Res., 2000, 328, 141–146 CrossRef CAS.
- S. Bower, R. Wickramasinghe, N. J. Nagle and D. J. Schell, Bioresour. Technol., 2008, 99, 7354–7362 CrossRef CAS.
- C. Zhu, C. Krumm, G. G. Facas, M. Neurock and P. J. Dauenhauer, React. Chem. Eng., 2017, 2, 201–214 RSC.
- J. J. P. Stewart, J. Comput. Chem., 1989, 10, 209–220 CrossRef CAS.
- J. J. P. Stewart, J. Comput. Chem., 1989, 10, 221–264 CrossRef CAS.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis and J. A. Montgomery, J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS.
- M. S. Gordon and M. W. Schmidt, in Theory and Applications of Computational Chemistry, Elsevier, 2005, pp. 1167–1189 Search PubMed.
- A. R. Leach, Molecular modelling: principles and applications, Prentice Hall, New York, 2nd edn, 2001 Search PubMed.
- C. Gonzalez and H. B. Schlegel, J. Chem. Phys., 1989, 90, 2154–2161 CrossRef CAS.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 17 CAS.
- B. M. Bode and M. S. Gordon, J. Mol. Graphics Modell., 1998, 16, 133–138 CrossRef CAS.
- A. Cumashi, N. A. Ushakova, M. E. Preobrazhenskaya, A. D'Incecco, A. Piccoli, L. Totani, N. Tinari, G. E. Morozevich, A. E. Berman, M. I. Bilan, A. I. Usov, N. E. Ustyuzhanina, A. A. Grachev, C. J. Sanderson, M. Kelly, G. A. Rabinovich, S. Iacobelli and N. E. Nifantiev, Glycobiology, 2007, 17, 541–552 CrossRef CAS.
- L. Chevolot, A. Foucault, F. Chaubet, N. Kervarec, C. Sinquin, A.-M. Fisher and C. Boisson-Vidal, Carbohydr. Res., 1999, 319, 154–165 CrossRef CAS.
- M. T. Ale and A. S. Meyer, RSC Adv., 2013, 3, 8131–8141 RSC.
- S. Yamabe, W. Guan and S. Sakaki, J. Org. Chem., 2013, 78, 2527–2533 CrossRef CAS.
- A. M. Dimiev, L. B. Alemany and J. M. Tour, ACS Nano, 2013, 7, 576–588 CrossRef CAS.
- O. Mohammadi, M. Golestanzadeh and M. Abdouss, New J. Chem., 2017, 41, 11471–11497 RSC.
- B. Garg, T. Bisht and Y. C. Ling, Molecules, 2014, 19, 14582–14614 CrossRef.
- S. Navalon, A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Chem. Rev., 2014, 114, 6179–6212 CrossRef CAS.
- D. Voiry, J. Yang, J. Kupferberg, R. Fullon, C. Lee, H. Y. Jeong, H. S. Shin and M. Chhowalla, Science, 2016, 353, 1413–1416 CrossRef CAS.
- E. G. Mission, A. T. Quitain, Y. Hirano, M. Sasaki, M. J. Cocero and T. Kida, Catal. Sci. Technol., 2018, 8, 5434–5444 RSC.
- Y. B. Huang and Y. Fu, Green Chem., 2013, 15, 1095–1111 RSC.
- P. H. Blanco, J. B. Lad, A. V. Bridgwater and M. S. Holm, ACS Sustainable Chem. Eng., 2018, 6, 12872–12883 CrossRef CAS.
- W. L. Marshall and E. U. Franck, J. Phys. Chem. Ref. Data, 1981, 10, 295–304 CrossRef CAS.
- J. Remón, F. Santomauro, C. J. Chuck, A. S. Matharu and J. H. Clark, Green Chem., 2018, 20, 4507–4520 RSC.
- A. Lerf, H. He, M. Forster and J. Klinowski, J. Phys. Chem. B, 1998, 102, 4477–4482 CrossRef CAS.
- A. M. Orozco, A. H. Al-Muhtaseb, A. B. Albadarin, D. Rooney, G. M. Walker and M. N. M. Ahmad, RSC Adv., 2011, 1, 839–846 RSC.
- A. Manzi, in Current Protocols in Molecular Biology, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2001, vol. 32, pp. 17.16.1–17.16.11 Search PubMed.
- Y.-B. Huang and Y. Fu, Green Chem., 2013, 15, 1095 RSC.
- A. Tayal and S. A. Khan, Macromolecules, 2000, 33, 9488–9493 CrossRef CAS.
- M. R. Rosana, Y. Tao, A. E. Stiegman and G. B. Dudley, Chem. Sci., 2012, 3, 1240–1244 RSC.
- J. Anwar, U. Shafique, Waheed-uz-Zaman, R. Rehman, M. Salman, A. Dar, J. M. Anzano, U. Ashraf and S. Ashraf, Arabian J. Chem., 2015, 8, 100–104 CrossRef CAS.
- X. Fan, G. Zhang and F. Zhang, Chem. Soc. Rev., 2015, 44, 3023–3035 RSC.
- K. B. Dhopte, R. S. Zambare, A. V. Patwardhan and P. R. Nemade, RSC Adv., 2016, 6, 8164–8172 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: (I) Experimental methods; (II) molecular weights from gel permeation chromatography; (III) depolymerisation of fucoidan from U. pinnatifida using graphene and multiwalled carbon nanotubes coupled with trace amounts of different acids; (IV) effect of temperature and reaction time in the hydrolysis of fucoidan from U. pinnatifida; (V) mechanisms of the hydrolysis of fucoidan and cellulose. See DOI: 10.1039/c9ra03594j |
| This journal is © The Royal Society of Chemistry 2019 |