
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUpgrading biochar via co-pyrolyzation of agricultural biomass and polyethylene terephthalate wastes†
Seok-Young Oh * and
Tae-Cheol Seo
* and
Tae-Cheol Seo
Department of Civil and Environmental Engineering, University of Ulsan, 93 Daehak-ro, Nam-gu, Ulsan 44610, South Korea. E-mail: quartzoh@ulsan.ac.kr; Fax: +82-52-259-2629; Tel: +82-52-259-2752
First published on 9th September 2019
Abstract
Spent polyethylene terephthalate (PETE) bottles were collected and co-pyrolyzed with rice straw (RS) to examine the characteristics and performance of biochar as a sorbent for various types of U.S. EPA priority pollutants, including 2,4-dinitrotoluene (DNT), 2,4-dichlorophenol (DCP), Pb, chromate (CrO42−), and selenate (SeO42−). During sorption of contaminants to PETE/RS-derived biochar, PETE residues from pyrolysis, pH, and pyrolysis temperature greatly affected the sorption process. Depending on the types of contaminants and experimental conditions, co-pyrolysis of PETE and RS may enhance the sorption of contaminants through different sorption mechanisms, including hydrophobicity, electrostatic force, ion exchange, surface complexation, and surface precipitation. Unlike other contaminants, selenate was reductively transformed by delocalized electrons from the graphitic structure in biochar. Our results strongly suggest that co-pyrolysis of PETE and agricultural wastes may be favorable to enhance the properties of biochar. In addition to syn-gas and bio-oil from co-pyrolysis, biochar may be a valuable by-product for commercial use.
1. Introduction
Production, usage, and disposal of plastics are an integral part of modern everyday life. Without plastics, most mass-produced items cannot be packaged for merchandizing. Unfortunately, most plastics revert to waste after use. By 2015, more than 8.3 billion tons of plastics were estimated to have been produced globally, of which only 12% was incinerated and 9% recycled.1 Currently, microplastics pollution affecting ocean plants and animals is being tracked as a hot worldwide issue,2 and disposal and recycling of plastic wastes is a significant environmental concern. According to the Korea Ministry of Environment, about 12.4 million tons of plastic wastes were generated in 2016, but the recycling percentage (such as for refuse plastic fuels) was only 30%. After China banned the import of plastic wastes from South Korea in 2018, the treatment and final disposal of plastic wastes became a huge problem.Polyethylene terephthalate (PETE, (C10H8O4)n) is a popular plastic that is widely used in synthetic textiles, bottles, and films. In 2015, the annual plastic production rate was greater than 56 million tons,3 and PETE bottles have become ubiquitous in daily life. In South Korea, more than 2.5 billion PETE bottles were produced in 2015. PETE, like many other plastics, is an excellent candidate for thermal disposal (incineration), as it is composed of carbon, hydrogen, and oxygen, with only trace amounts of catalyst elements (but no sulfur). PETE has the energy content of soft coal. Therefore, unless collected PETE bottle waste is reprocessed for recycling, incineration is the inevitable disposal option. Since the 1970s, it has been reported that pyrolysis of plastic wastes produces pyrolysis oil and gas, which can be further transformed to commercial products.4,5 Pyrolysis of biomass wastes has also been proposed to dispose of agricultural wastes and to reduce the release of carbon dioxide by converting biomass into char and alternative energy (e.g., syn-gas and bio-oil) in response to climate change.6 Unfortunately, the quality of crude bio-oil is not satisfactory for commercial use as a fuel.7 To improve the quality of bio-oil, several researchers have proposed co-pyrolysis of plastics/polymer wastes, which results in increased carbon and hydrogen contents and improved yield of usable bio-oil with higher heating values and lower acidity, density, and oxygen content.8–11
Char (biochar) is another by-product from co-pyrolysis of biomass and polymers. Biochar has been intensively used as a sorbent for contaminants.12,13 In several studies, the yield of biochar was reduced but the heating values were improved.14–16 Co-pyrolysis also resulted in increased aromaticity via growth of polycyclic aromatic hydrocarbons (PAHs).17,18 Bernardo et al. suggested that upgraded biochar synthesized from co-pyrolysis of biomass and polymers can be used as an adsorbent.19 Recently, we also proposed co-pyrolysis of agricultural and plastic wastes as a disposal option to treat plastic wastes.20 Co-pyrolysis of rice straw (RS) with polypropylene (PP), polyethylene (PE), or polystyrene (PS) increased the carbon content, cation exchange capacity (CEC), surface area, and pH of the biochar. As a result, sorption of 2,4-dinitrotoluene (DNT) and Pb to polymer/RS-derived biochar was markedly enhanced. Increased aromaticity and hydrophobicity may be responsible for enhancing DNT sorption to polymer/RS-derived biochar. In contrast, increased CEC, higher pH, and the newly developed surface area may account for enhancement in Pb sorption. We also found that polymer residues were strongly responsible for enhancement in sorption of nitro explosives.21 Sorption of ionizable halogenated phenols, such as 2,4-dichlorophenol (DCP), 2,4-dibromophenol (DBP), and 2,4-difluorophenol (DFP), onto polymer/RS-derived biochar was also significantly enhanced by changing the properties of biochar due to polymer residues.22 Toxicity characteristic leaching procedures and Microtox® bioassay analyses indicated that polymer/RS-derived biochar showed no harmful effects.21
Although there have been intense studies on the co-pyrolysis of polymer/RS-derived biochar, there have been no attempts to apply pyrolysis for real plastic wastes generated from everyday life. Among the polymer wastes, we selected spent PETE bottles and co-pyrolyzed them with rice straw (RS) to investigate the characteristics and performance of biochar as a sorbent for various types of pollutants. DNT, DCP, Pb, chromate (CrO42−), and selenate (SeO42−) were selected because they are classified as U.S. EPA priority pollutants, and we hypothesized that PETE/RS-derived biochar would enhance sorption of those contaminants. We determined the basic properties of PETE/RS-derived char, and its maximum sorption capacities for those contaminants were determined according to Langmuir sorption isotherm models. We also examined factors affecting sorption of the contaminants to PETE/RS-derived biochar, such as amount of PETE, initial pH, and effect of pyrolysis temperature. Here, the possible sorption mechanisms for each contaminant are also discussed.
2. Materials and methods
2.1. Chemicals and materials
2,4-Dinitrotoluene (DNT, 97%), 2,4-dichlorophenol (DCP, 99%), PbCl2 (98%), Na2CrO4, (98%), and Na2SeO4 (>98%) were purchased from Aldrich Chemical Co. (Milwaukee, WI, USA). Chemicals were used as received without further treatment. All solutions were prepared with deionized water. Spent PETE bottles, mostly commercial 500 mL mineral water bottles, were collected from a recycling collection station in University of Ulsan, South Korea. The labels on the PETE bottles were completely removed, and the bottles were completely dried in the shade. Then they were cut into small pieces with a scissors and ground into small particles (1–5 mm) using an electric mixer.Rice straw collected from rice farms in the city of Ulsan was used as biomass to synthesize PETE/biomass-derived biochar. The sampled RS was dried in an oven at 105 ± 5 °C for at least 2 h. After storage in a desiccator overnight, the dried RS was pulverized into smaller sizes (less than 5 mm) using an electric mixer. Then the PETE particles were mixed with the ground RS at volumetric ratios of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :90 and 20:80. The completely mixed PETE/RS mixtures were co-pyrolyzed at 550 °C for 4 h using a tube-type electrical furnace under N2 at 1000 cm3 min−1. After cooling to room temperature, the co-pyrolyzed PETE/RS-derived biochar was put in a desiccator for additional drying. To determine the effect of pyrolysis temperature, the temperature was changed to 900 °C for another round of co-pyrolyzation.
:90 and 20:80. The completely mixed PETE/RS mixtures were co-pyrolyzed at 550 °C for 4 h using a tube-type electrical furnace under N2 at 1000 cm3 min−1. After cooling to room temperature, the co-pyrolyzed PETE/RS-derived biochar was put in a desiccator for additional drying. To determine the effect of pyrolysis temperature, the temperature was changed to 900 °C for another round of co-pyrolyzation.
Properties of the synthesized PETE/RS-derived biochar, including pH, Brunauer–Emmett–Teller (BET) surface area, cation exchange capacity (CEC), anion exchange capacity (AEC), point of zero charge (PZC), and elemental composition, are summarized in Table 1.23–26 X-ray photoelectron spectroscopy (XPS; K-Alpha™ system, Thermo Scientific, Pittsburgh, PA, USA) and Fourier transform infrared spectroscopy (FT-IR; Nicolet iS5™, ThermoFisher Scientific, Waltham, MA, USA) spectra were obtained to identify the developed surface functional groups. Scanning electron microscopy (SEM; JSM 600F, JEOL Ltd., Tokyo, Japan) images were also obtained to investigate the surface morphology. Thermogravimetric analysis (TGA) was conducted to determine the mass change as a function of pyrolysis temperature under anaerobic conditions using a TGA system (STA409C/3/F, Netzsch Group, Selb, Germany).
| Pyrolysis temperature | pH23 | BET SAa (m2 g−1) | CEC (cmol kg−1)24 | AECb (cmol kg−1)25 | PZC26 | Elemental contentsc,d (%) | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| C | H | O | N | |||||||
| a Brunauer, Emmett, and Teller (BET) surface area was analyzed using a nanoPOROSITY-XQ (Mirae Scientific Instruments, Gwangju, Korea) using N2.b pH = 8.0.c Analyzed using the Vario EL Elemental Analyzer (Elementar Analysensysteme GmbH, Langenselbold, Germany).d Elemental contents of PETE: C 62.5%, H 3.59%, O 38.0%, and N 0.02%. | ||||||||||
| 550 °C | RS | 11.2 | 16.7 | 3.08 | 4.74 | 8.19 | 56.1 | 2.77 | 12.7 | 1.92 |
| RS + PETE (9:1 v/v) |
12.3 | 19.3 | 4.53 | 14.5 | 9.77 | 60.1 | 1.91 | 12.8 | 0.61 | |
| RS + PETE (8:2 v/v) |
11.6 | 35.0 | 7.28 | 13.7 | 10.7 | 63.4 | 1.90 | 9.81 | 0.42 | |
| 900 °C | RS | 11.1 | 44.8 | 1.85 | 14.6 | 9.93 | 63.5 | 0.69 | 0.42 | 0.40 |
| RS + PETE (9:1 v/v) |
10.9 | 32.7 | 3.12 | 6.70 | 10.6 | 59.2 | 1.05 | 3.91 | 0.54 | |
| RS + PETE (8:2 v/v) |
10.7 | 21.3 | 3.62 | 8.87 | 10.7 | 57.7 | 1.03 | 5.11 | 0.55 | |
2.2. Batch sorption experiments
Batch sorption experiments were performed using a 40 mL amber vial containing 20 mL of solution and PETE/RS-derived biochar (0.1–5.0 g) at 25 ± 2 °C. After sealing with screw caps with PTFE–silicon septa, duplicate vials were shaken on an orbital shaker at 180 rpm throughout the experiment, except during sampling. Preliminary experiments indicated that sorption of contaminants onto the biochar reached equilibrium after 24 h. Initial concentrations of the contaminants were 5–600 mg L−1. Equilibrium solution pH was close to biochar's pH (10.7–12.3). Using 0.1 M HCl and 0.1 M NaOH, the effect of pH on sorption of DCP to PETE/RS-derived biochar was evaluated by testing at pH 4, 7, and 10. Though we adjust the initial pH using HCl and NaOH, increasing solution pH at equilibrium was inevitable due to buffering capacity of biochar. Final solution pH for initial pH, 4, 7, and 10 was 8.3–8.7, 8.8–9.8, and 10.3–11.6, respectively. After equilibrium was reached, aliquots were withdrawn using glass syringes and immediately passed through a 0.025 μm cellulose membrane filter (Millipore, Burlington, MA, USA) for analytical determination of contaminants. A set of control vials was set up under identical conditions without PETE/RS-derived biochar to determine possible loss of the contaminants due to sorption onto the inner surface of the vial. Considering the loss of contaminants from batch experiments, the sorbed amounts of contaminants were calculated.2.3. Chemical analysis
DNT and DCP were analyzed using high-performance liquid chromatography (Ultimate® 3000 HPLC; Dionex, Sunnyvale, CA, USA) equipped with a Dionex Acclaim® 120 guard column (4.3 × 10 mm) and an Acclaim® 120 C-18 column (4.6 × 250 mm, 5 μm). The analytical methods and conditions for quantification of DNT and DCP are described in detail elsewhere.20 The concentration of Pb was determined by an atomic absorption spectrophotometer (AAS) (5100 ZL; PerkinElmer, Waltham, MA, USA) equipped with a graphite furnace. Chromate and selenate were analyzed using ion chromatography (Dionex ICS-100). A mixture of Na2CO3 (4.8 mM) and NaHCO3 (1.0 mM) was used as the eluent. The injection volume and flow rate were 10 μL and 1.5 mL min−1, respectively. The suppressor current was set to 40 mA. Analytical duplicates, standards, and blank samples were used for quality control of the data. To identify residues from PETE pyrolysis, qualitative analysis was conducted using gas chromatography-mass spectrometry (GC-MS) (GC-2010 and QP-2010; Shimadzu, Kyoto, Japan) equipped with an Agilent HP-5ms capillary column (30 m × 0.32 mm, 1.0 mm thickness; Agilent, Santa Clara, CA, USA). Analytical conditions are described in detail in our previous paper.203. Results and discussion
3.1. Properties of PETE/RS-derived biochar
After addition of PETE to RS for co-pyrolysis, pH, BET surface area, CEC, and PZC of the PETE/RS-derived biochar significantly increased as the PETE portion increased (Table 1), indicating that sorption of cationic metals to PETE/RS-derived biochar could be favorable. Elemental analysis showed that carbon content increased as hydrogen, oxygen, and nitrogen contents slightly decreased, consistent with previous results in pure polymer/RS-derived biochar.18 This change in elemental contents suggests that residues arising from PETE remain, and that development of surface functional groups may occur. SEM images showed that the morphology of PETE/RS-derived biochar was similar to that of RS-derived biochar (Fig. S1 in ESI†). XPS and FT-IR analyses also supported that development of surface functional groups continued after co-pyrolysis (Fig. S2 and S3†). Additional X-ray diffraction analysis also confirmed maintenance of a graphitic structure (data not shown). These results implied increased sorption of contaminants to PETE/RS-derived biochar. As pyrolysis temperature increased from 550 to 900 °C, BET surface area, PZC, and carbon content of RS-derived biochar increased (Table 1). In contrast to 550 °C, co-pyrolysis with PETE at 900 °C decreased the carbon content, suggesting that carbon residues from PETE are eliminated at elevated temperature. Thermogravimetric analysis showed additional loss of weight at 600–900 °C (Fig. S4†). The addition of PETE also increased the CEC, which could be advantageous for sorption of cationic metals. Overall, our results showed that the properties of PETE/RS-derived biochar were similar to those of pure polymer/RS-derived biochars.203.2. Sorption of DNT and DCP to PETE/RS-derived biochar
Regarding sorption of DNT to RS biochar, the maximum sorption capacity from Langmuir sorption isotherm models was 5.1 mg g−1, as shown in Fig. 1 (hereafter, all maximum sorption capacities are based on Langmuir sorption isotherm models). This sorption capacity was similar to previously reported values (4.6 mg g−1).27 Addition of PETE (by 10 and 20 v%) to RS for co-pyrolysis enhanced the maximum sorption capacity to 9.5 and 10.2 mg g−1, respectively. The enhancement could be attributed to increasing aromaticity, which enhanced π–π electron donor acceptor (EDA) interactions between nitro functional groups and electron-rich parts in graphitic regions.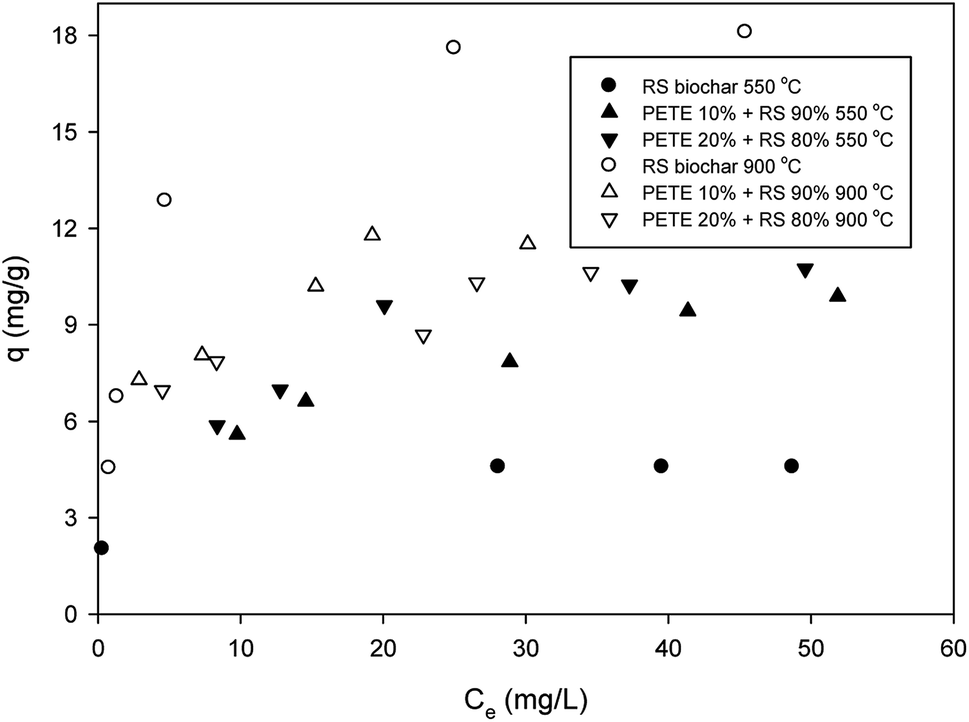 | ||
| Fig. 1 Removal of DNT by PETE/RS-derived biochar. | ||
Carbon residues from PETE pyrolysis may also be involved in enhancement via hydrophobic sorption. The calculated H/C ratios for RS-derived biochar and PETE/RS- (10/90 and 20/80 v/v)-derived biochar were 0.049 and 0.031 and 0.030, respectively (according to Table 1), confirming the increasing hydrophobicity of biochar surface responsible for enhancement in sorption of DNT in the biochar system. GC-MS analysis (Table S1 in ESI†) showed C3–C5 compounds (e.g., 2-propanone, 2-pentene, etc.) as possible residues from PETE pyrolysis. Namely, unlike co-pyrolysis with PP, PE, or PS, smaller molecules remained on the biochar surface, suggesting that hydrophobic sorption may not be markedly dominant. Increasing the pyrolysis temperature to 900 °C, the maximum sorption capacity for RS biochar increased to 19.2 mg g−1, but the maximum sorption capacity for PETE/RS-derived biochar did not change significantly. It is likely that the decrease of sorption due to loss of PETE-originated carbon residues and the increase of sorption due to developed aromaticity at elevated temperature were mutually compensated, consistent with previous results.20
Sorption of DCP to biochar was somewhat different from that of DNT. The maximum sorption capacity of RS biochar for DCP was 11.5 mg g−1 (Fig. 2), consistent with previous results.20 Co-pyrolysis with PETE (10 and 20 v%) increased the sorption capacity to 12.3 and 12.5 mg g−1, respectively. Thus, it appears that the effect of co-pyrolysis with PETE was not significant. In contrast to pure polymer/RS-derived biochar, with which the sorption of DCP was significantly enhanced,18 the sorption to PETE/RS-derived biochar was only slightly enhanced. It is likely that hydrophobic sorption may not be dominant. At equilibrium pH (11.2–12.3), the logKow of DCP was estimated to be 3.16–3.19 according to Nowosielski and Fein's method.28 The DCP was still hydrophobic. Instead, the carbon residues from PETE pyrolysis were mostly C3–C5 (Table S1†) and were less hydrophobic than residues from PP, PE, and PS pyrolysis (mostly C12–C22) under identical conditions.21 Due to the decrease of carbon number in carbon residues from polymers, hydrophobic sorption from PETE/RS-derived biochar may not be as strong as that in RS/PP, PE, or PS-derived biochar systems. Moreover, the DCP was completely deprotonated, negatively charged at equilibrium pH according to the pKa of DCP (7.60). The biochar surface was also slightly negatively charged considering PZC and equilibrium pH. Therefore, a repulsive force between deprotonated DCP and the biochar surface could have existed, and electrostatic sorption was not dominant. Meanwhile, π–π EDA interactions between the chloro functional groups and the electron-rich parts in graphitic regions of the biochar may be a possible mechanism to account for the sorption of DCP. By increasing the pyrolysis temperature to 900 °C, the sorption of DCP to RS biochar increased (to 16.4 mg g−1), probably due to increasing aromaticity. Co-pyrolysis with PETE (10 and 20 v%) also enhanced the maximum sorption capacity (to 19.1 and 20.2 mg g−1, respectively) when the pyrolysis temperature was increased to 900 °C. According to TGA analysis (Fig. S4†), carbon residues were almost completely removed, although small amounts of hydrogen and oxygen remained (Table 1). Accordingly, the aromaticity of PETE/RS-derived biochar may not be greatly different from that of other biochars. Therefore, the slight increase in sorption capacity may be due to change in surface of the biochar from a negative to a neutral charge by changing the PZC and pH after co-pyrolysis with PETE (Table 1). The neutral charge of the biochar surface and the deprotonated DCP having a higher logKow are possible explanations for the slight enhancement of DCP sorption to PETE/RS-derived biochar at a 900 °C pyrolysis temperature. Therefore, we needed to further explore the effect of pH.
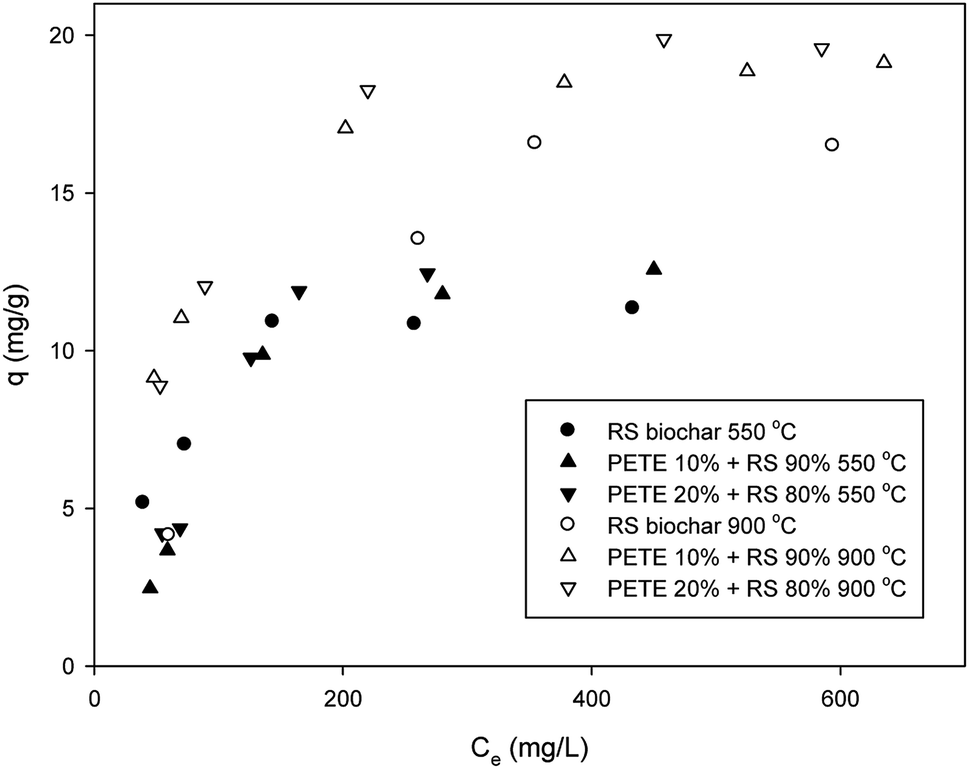 | ||
| Fig. 2 Removal of DCP by PETE/RS-derived biochar. | ||
To determine the effect of pH on sorption of DCP to PETE/RS-derived biochar, we examined the effect of initial pH (Fig. 3). According to Nowosielski and Fein's method,28 as pH decreased, the hydrophobicity of the DCP greatly decreased (estimated logKow = 0.30 and 0.31 for pH 4 and 7, respectively). As pH decreased to 4 and 7, the DCP was not deprotonated (pKa = 7.90), indicating the DCP was neutrally charged. Due to its lower hydrophobicity, π–π EDA interactions between the chloro functional groups and the electron-rich parts in the biochar may be important. At pH 4 and 7, the biochar surface was positively charged according to PZC (Table 1). As shown in Fig. 3(a), at pH 4, addition of PETE (10 and 20 v%) to pyrolysis of RS biochar increased the sorption of DCP (to 18.8 and 22.8 mg g−1, respectively) compared with RS biochar pyrolyzed at 550 °C (12.2 mg g−1). This indicated continuing hydrophobic sorption to the PETE/RS-derived biochar. At 900 °C, the sorption of DCP to the biochar further increased to 21.2 mg g−1 (Fig. 3(a)), indicating that increasing aromaticity at elevated pyrolysis temperature is also responsible for enhanced sorption of DCP. The effect of co-pyrolysis with PETE was not as significant as that at 550 °C, showing a 22–25 mg g−1 sorption capacity, because most of the PETE residues were removed at 900 °C (Fig. 3(a)). At pH 7, very similar results were observed (Fig. 3(b)). However, the sorption trends are different at pH 10 (Fig. 3(c)). At 550 °C, the surface was negatively charged, and the DCP was deprotonated though the estimated logKow of the DCP was 2.72.28 Addition of PETE to RS pyrolysis resulted in change in the surface of the biochar to neutral or less negative. As a result, sorption of DCP to PETE/RS-derived biochar slightly increased. At 900 °C, compared with RS biochar, sorption of DCP to PETE/RS-derived biochar was markedly enhanced. Because the surface charge of the biochar was changed from negative to positive, the maximum sorption capacity of the deprotonated DCP to the biochar surface increased from 31.1 mg g−1 to 52.2 and 53.1 mg g−1, indicating that electrostatic sorption of DCP to biochar was dominant. Overall, the results suggested that electrostatic sorption, hydrophobicity, and π–π EDA interaction sorption mechanisms are involved in the sorption of DCP to PETE/RS-derived biochar, and that the importance of each mechanism differs according to the given conditions (mostly pH).
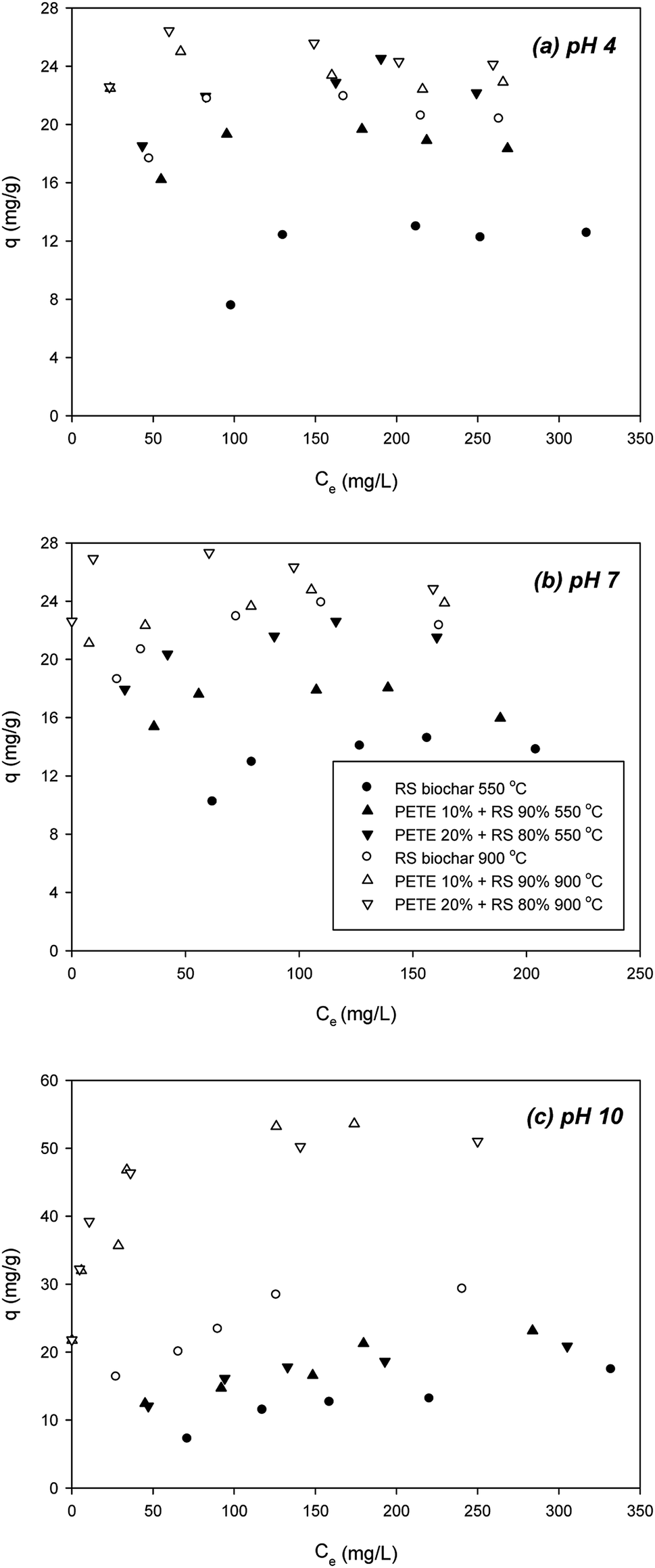 | ||
| Fig. 3 Effect of initial pH on removal of DCP by PETE/RS-derived biochars pyrolyzed at 550 °C and 900 °C, respectively; (a) pH 4, (b) pH 7, and (c) pH 10. | ||
3.3. Sorption of Pb to PETE/RS-derived biochar
Pb sorption to biochar was slightly enhanced after co-pyrolysis with PETE (Fig. 4). By adding PETE (10 and 20 v%), the maximum sorption capacity increased from 95.0 mg g−1 to 111 and 115 mg g−1, respectively, probably due to increasing CEC with intact surface functional groups. Development of strong aromaticity may also be responsible for enhancement of Pb sorption via Cπ (delocalized electrons in biochar)–cation interactions. It also appears that the effect of pH may be dominant via surface precipitation by increasing pH. When increasing the pyrolysis temperature to 900 °C, no significant difference was observed. The maximum sorption capacities of RS biochar and PETE/RS (10/90 and 20/80 v/v)-derived biochar for Pb were 90 mg g−1 and 115–118 mg g−1, respectively (Fig. 4). The negligible effect of co-pyrolysis with PETE may be due to compensation of the two mechanisms. Increasing CEC and aromaticity after co-pyrolysis with PETE (Table 1) may result in an increasing sorption capacity. Simultaneously, the surface charge was changed from negative to positive after co-pyrolysis with PETE, resulting in decreased electrostatic sorption. Thus, due to this compensation, the effect of increasing pyrolysis temperature was not significant. It should be noted that most of the removal of Pb may occur via surface precipitation as a form of hydroxide, oxyhydroxide, or carbonate at elevated pH, which can also account for the negligible differences among PETE/RS-derived biochars at different pyrolysis temperatures. It should be noted that compared with other dried biomass wastes,29 these sorption capacities for Pb were not significantly improved. Therefore, more customized upgrading procedure may be required.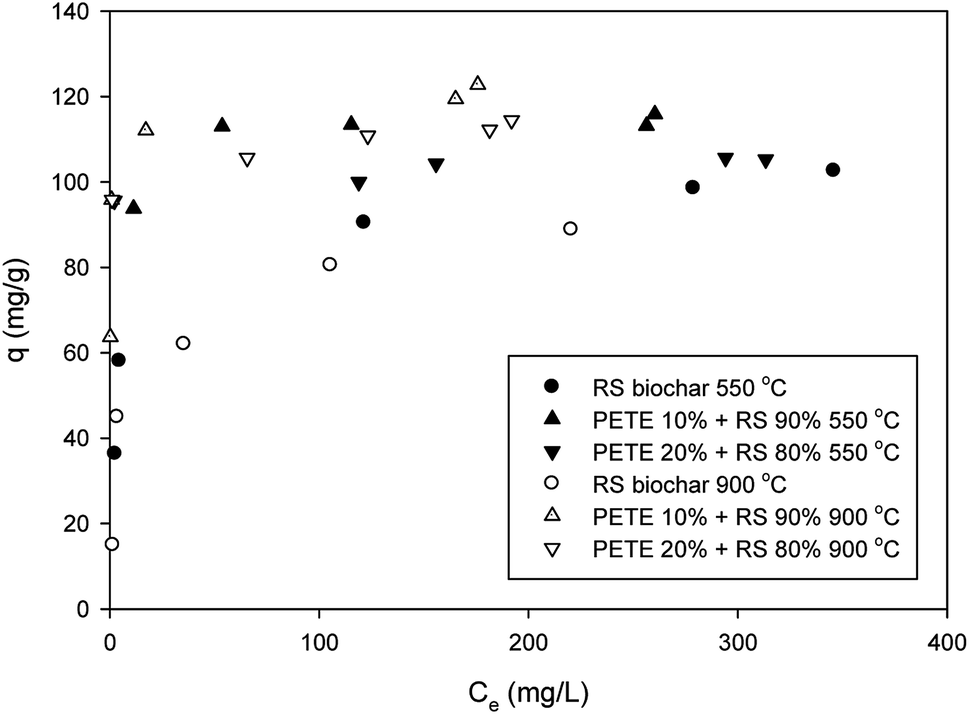 | ||
| Fig. 4 Removal of Pb by PETE/RS-derived biochar. | ||
3.4. Sorption of chromate and selenate to PETE/RS-derived biochar
Sorption of chromate to biochar pyrolyzed at 550 °C was not significant, showing a 1.2 mg g−1 maximum sorption capacity (Fig. 5(a)). Co-pyrolysis with PETE (10 and 20 v%) significantly enhanced the sorption capacity to biochar to 5.5 and 5.8 mg g−1, respectively, probably due to decrease in repulsive force from the surface charge change and increase in AEC. Surface co-precipitation with other cations at elevated pH could not be completely ruled out. Increasing the pyrolysis temperature to 900 °C, chromate sorption to RS biochar was significantly enhanced, showing a 4.9 mg g−1 maximum sorption capacity (Fig. 5(a)). Increasing AEC could be responsible for the enhancement (Table 1). Meanwhile, increasing aromaticity may also be an explanation of this enhancement. Interaction between anions and electron-deficient parts of the graphitic regions in biochar may account for the increase of sorption capacity. Co-pyrolysis with PETE (10 and 20 v%) slightly enhanced the maximum sorption capacity to 5.9 and 6.1 mg g−1, respectively, possibly due to surface charge change from negative to neutral.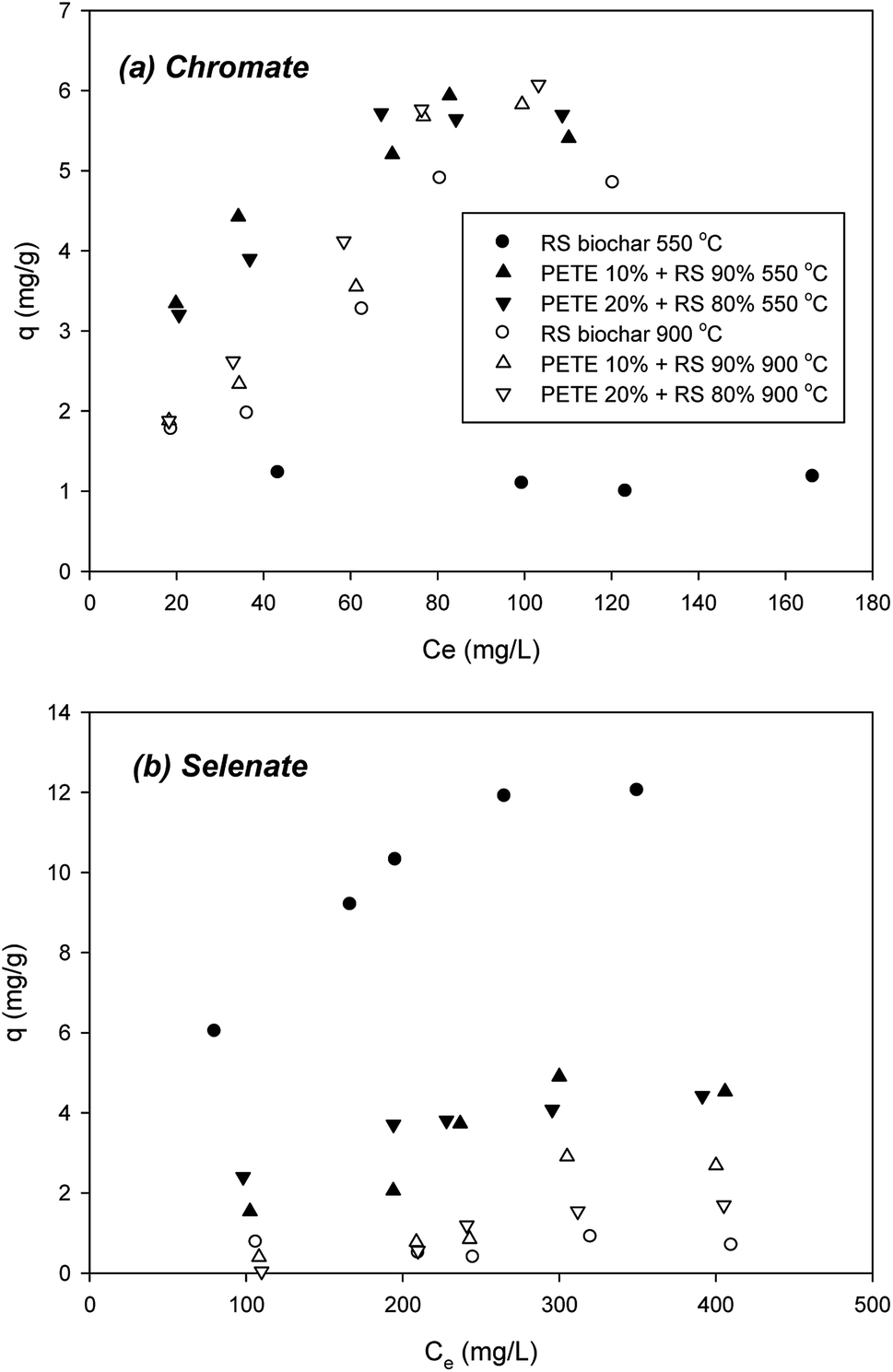 | ||
| Fig. 5 Removal of (a) chromate and (b) selenate by PETE/RS-derived biochar. | ||
Selenate sorption was very different (Fig. 5(b)), showing a 12.0 mg g−1 maximum sorption capacity to RS biochar. However, by adding PETE to RS pyrolysis (10 and 20 v%), the maximum sorption capacity greatly decreased to 4.5 and 4.2 mg g−1, respectively. This result indicated that the sorption mechanisms were different from chromate sorption to biochar. Neither electrostatic sorption between anions and biochar surface nor increasing AEC of PETE/RS-derived biochar explained this steep decrease in selenate sorption. It appears that removal of selenate may involve other mechanisms. With the assumption that biochar transforms selenate over time, we conducted controlled experiments with selenate in the presence of PETE/RS-derived biochar. The results showed that the selenate was transformed into selenite (SeO32−) with PETE/RS-derived biochar (Fig. S5†). Thus, removal of selenate by biochar was due to sorption to biochar as well as reductive transformation by delocalized electrons generated by graphitic structures in the biochar. Even at a 900 °C pyrolysis temperature, sorption of selenate to biochar remained unexplainable. The increasing aromaticity at elevated temperature did not increase the sorption capacity of selenate. It appears that development of aromaticity may enhance the production of delocalized electrons to reduce selenate, and that the reductive transformation of selenate was more dominant at a 900 °C pyrolysis temperature. It should be noted that selenite was also reductively transformed to more reduced forms, specifically elemental selenium (Se0) and selenide (Se2−), in the presence of biochar (data not shown). The kinetics and pathways of selenate reduction with PETE/RS-derived biochar remain to be explored. We will report on this result in the near future.
4. Conclusions
Overall, during sorption of contaminants to PETE/RS-derived biochar, the pH and PETE residues from pyrolysis strongly affect the sorption of contaminants. Depending on the types of contaminants, co-pyrolysis of PETE and RS may enhance the sorption of contaminants through different sorption mechanisms, including hydrophobicity, electrostatic force, ion exchange, surface complexation, and surface precipitation. Unlike other contaminants, selenate was reductively transformed by delocalized electrons from the graphitic structures in the biochar. Our results suggest that co-pyrolysis of PETE and agricultural wastes may be favorable to enhance the properties of syn-gas, bio-oil, and biochar. Energy properties and values of syn-gas and bio-oil, the toxicity of biochar, and the effects of impurities in various types of plastic waste will be relevant factors in the development of new biochar products through co-pyrolysis with plastic waste.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2016R1D1A1B03931048).References
- R. Geyer, J. R. Jambeck and K. L. Law, Production, use, and fate of all plastics ever made, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- S. Rezania, J. Park, M. F. M. Din, S. M. Taib, A. Talaiekhozani, K. K. Yadav and H. Kamyab, Microplastics pollution in different aquatic environments and biota: a review of recent studies, Mar. Pollut. Bull., 2018, 133, 191–208 CrossRef CAS PubMed.
- S. Yoshida, K. Hiraga, T. Takehana, I. Taniguchi, H. Yamaji, Y. Maeda, K. Toyohara, K. Miyamoto, Y. Kimura and K. Oda, A bacterium that degrades and assimilates poly(ethylene terephthalate), Science, 2016, 351(6278), 1196–1199 CrossRef CAS.
- F. Ates, N. Miskolcczi and N. Borsodi, Comparison of real waste (MSW and MPW) pyrolysis in batch reactor over different catalysts. Part 1: product yields, gas and pyrolysis oil, Bioresour. Technol., 2013, 133, 443–454 CrossRef CAS PubMed.
- C. Muhammad, J. A. Onwudili and P. T. Williams, Thermal degradation of real-world waste plastics and simulated mixed plastics in a two-stage pyrolysis – catalysis reactor for fuel production, Energy Fuels, 2015, 29, 2601–2609 CrossRef CAS.
- J. Lehmann, Bio-energy in the black, Front. Ecol. Environ., 2007, 5, 381–387 CrossRef.
- S. Czernik and A. V. Bridgwater, Overview of applications of biomass fast pyrolysis oil, Energy Fuels, 2004, 18, 590–598 CrossRef CAS.
- P. Bhattacharya, P. H. Steele, E. B. M. Hassan, B. Mitchell, L. Ingram and C. U. Pittman Jr, Wood/plastic copyrolysis in an augur reactor: chemical and physical analysis of the products, Fuel, 2009, 88, 1251–1260 CrossRef CAS.
- F. Paradela, F. Pinto, I. Gulyurtlu, I. Cabrita and N. Lapa, Study of the slow batch pyrolysis of mixtures of plastics, tyres and forestry biomass wastes, J. Anal. Appl. Pyrolysis, 2009, 85, 392–398 CrossRef CAS.
- J. D. Martínez, A. Veses, A. M. Mastral, R. Murillo, M. V. Navarro, N. Puy, A. Artigues, J. Bartrolí and T. García, Co-pyrolysis of biomass with waste tyres: upgrading of liquid bio-fuel, Fuel Process. Technol., 2014, 119, 263–271 CrossRef.
- E. Önal, B. B. Uzun and A. E. Pütün, Bio-oil production via co-pyrolysis of almond shell as biomass and high density polyethylene, Energy Convers. Manage., 2014, 78, 704–710 CrossRef.
- S. Wu, H. He, X. Inthapanya, C. Yang, L. Lu, G. Zeng and Z. Han, Role of biochar on composting of organic wastes and remediation of contaminated soils – a review, Environ. Sci. Pollut. Res., 2017, 24, 16560–16577 CrossRef CAS PubMed.
- X. Wu, H. He, W. L. Yang, J. Yu and C. Yang, Efficient removal of atrazine from aqueous solutions using magnetic Saccharomyces cerevisiae bionanomaterial, Appl. Microbiol. Biotechnol., 2018, 102, 7597–7610 CrossRef CAS PubMed.
- M. Brebu, S. Ucar, C. Vasile and J. Yanik, Co-pyrolysis of pine cone with synthetic polymers, Fuel, 2010, 89, 1911–1918 CrossRef CAS.
- M. Sajdak and R. Muzyka, Use of plastic waste as a fuel in the co-pyrolysis of biomass. Part 1: the effect of the addition of plastic waste on the process and products, J. Anal. Appl. Pyrolysis, 2014, 107, 267–275 CrossRef CAS.
- Y. Xue, S. Zhou, R. C. Brown, A. Kelkar and X. Bai, Fast pyrolysis of biomass and waste plastic in a fluidized bed reactor, Fuel, 2015, 156, 40–46 CrossRef CAS.
- K. H. Ko, V. Sahajwalla and A. Rawal, Specific molecular structure changes and radical evolution during biomass-polyethylene terephthalate co-pyrolysis detected by 13C and 1H solid-state NMR, Bioresour. Technol., 2014, 170, 248–255 CrossRef CAS PubMed.
- D. V. Suriapparao, D. K. Ojha, T. Ray and R. Vinu, Kinetic analysis of co-pyrolysis of cellulose and polypropylene, J. Therm. Anal. Calorim., 2014, 117, 1441–1451 CrossRef CAS.
- M. Bernardo, N. Lapa, M. Goncalves, B. Mendes, F. Pinto, I. Fonseca and H. Lopes, Physico-chemical properties of chars obtained in the co-pyrolysis of waste mixtures, J. Hazard. Mater., 2012, 219–220, 196–202 CrossRef CAS PubMed.
- S. Y. Oh and Y. D. Seo, Polymer/biomass-derived biochar for use as a sorbent and electron transfer mediator in environmental applications, Bioresour. Technol., 2016, 218, 77–83 CrossRef CAS PubMed.
- S. Y. Oh, Y. D. Seo, T. Y. Jeong and S. D. Kim, Sorption of nitro explosives to polymer/biomass-derived biochar, J. Environ. Qual., 2018, 47, 353–360 CrossRef CAS PubMed.
- S. Y. Oh and Y. D. Seo, Factors affecting the sorption of halogenated phenols onto polymer/biomass-derived biochar: effects of pH, hydrophobicity, and deprotonation, J. Environ. Manage., 2019, 232, 145–152 CrossRef CAS PubMed.
- H. H. Rump and H. Krist, Laboratory Manual for the Examination of Water, Wastewater and Soil, VCH, New York, 1988 Search PubMed.
- P. R. Hesse, A Textbook of Soil Chemical Analysis, John Murry, London, 1971 Search PubMed.
- M. Lawrinenko and D. A. Laird, Anion exchange capacity of biochar, Green Chem., 2015, 17, 4628–4636 RSC.
- P. Faria, J. Órfão and M. Pereira, Adsorption of anionic and cationic dyes on activated carbons with different surface chemistries, Water Res., 2004, 38, 2043–2052 CrossRef CAS PubMed.
- S. Y. Oh and Y. D. Seo, Sorptive removal of nitro explosives and metals using biochar, J. Environ. Qual., 2014, 43, 1663–1671 CrossRef PubMed.
- B. E. Nowosielski and J. B. Fein, Experimental study of octanol-water partition coefficients for 2,4,6-trichlorophenol and pentachlorophenol: derivation of an empirical model of chlorophenol partitioning behavior, Appl. Geochem., 1998, 13, 893–904 CrossRef CAS.
- Y. Cheng, C. Yang, H. He, G. Zeng, K. Zhao and Z. Yan, Biosorption of Pb(II) ions from aqueous solutions by waste biomass from biotrickling filters: kinetics, isotherms, and thermodynamics, J. Environ. Eng., 2016, 142, C4015001 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: SEM images, XPS and FT-IR spectra, and TGA curves of PETE/RS-derived biochar, identified compounds in PETE pyrolysis residues by GC-MS, and transformation of selenate and selenite with PETE/RS-derived biochar. See DOI: 10.1039/c9ra05518e |
| This journal is © The Royal Society of Chemistry 2019 |